

Abstract
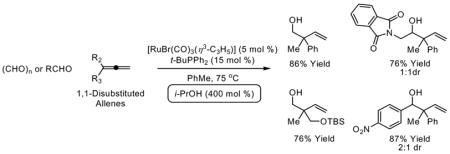
Upon exposure to the conditions of ruthenium catalyzed transfer hydrogenation employing isopropanol as the terminal reductant, 1,1-disubstituted allenes 1a-1h engage in reductive coupling to paraformaldehyde to furnish homoallylic alcohols 2a-2h. Under identical transfer hydrogenation conditions, 1,2-disubstituted allenes engage in reductive coupling to aldehydes 3a-3f to furnish homoallylic alcohols 4a-4n. In all cases, reductive coupling occurs with branched regioselectivity to deliver homoallylic alcohols bearing all-carbon quaternary centers.
Although ruthenium catalyzed transfer hydrogenation ranks among the foremost methods for enantioselective ketone reduction, 1 reductive C-C bond formation catalyzed by ruthenium is highly uncommon.2,3,4 We have developed a family of reductive C-C bond formations employing elemental hydrogen as the terminal reductant.5,6 More recently, we discovered that reductive C-C bond formation could be achieved under the conditions of iridium catalyzed transfer hydrogenation, wherein allenes or cyclohexadiene are coupled to aldehydes and, remarkably, alcohols.7 In an effort to expand the scope of such “transfer hydrogenative C-C bond formations,” ruthenium catalysts were found to promote the coupling of acylic 1,3-dienes or 1,3-enynes to both aldehydes and alcohols to furnish products of carbonyl allylation and propargylation, respectively. 8 For these transfer hydrogenative processes, rather than using elemental hydrogen as reductant, hydrogen embedded in isopropanol or an alcoholic substrate is redistributed among reactants to generate nucleophile-electrophile pairs, enabling carbonyl addition from the aldehyde or alcohol oxidation level (Scheme 1).
Scheme 1.

Carbonyl allylation from the aldehyde or alcohol oxidation level via transfer hydrogenative C-C coupling of allenes.
As part of a continuing effort to broaden this emergent class of C-C couplings, we investigated the coupling of 1,1-disubsituted allenes to paraformaldehyde and higher aldehydes under the conditions of ruthenium catalysis. Here we disclose that allenes 1a-8a engage in branch-selective reductive coupling to paraformaldehyde and higher aldehydes under the conditions of ruthenium catalyzed transfer hydrogenation employing isopropanol as the terminal reductant to furnish homoallylic alcohols 2a-2h and 4a-4n, respectively, bearing all-carbon quaternary centers. Additionally, we report isotopic labeling studies that provide further insight into key mechanistic features of these processes.
In an initial set of experiments, structurally diverse ruthenium complexes were assayed for their ability to catalyze the reductive coupling of allene 1a to paraformaldehyde using isopropanol as the terminal reductant (Table 1, entries 1–7). It was found that the complex prepared in situ from [RuBr(CO)3(η3-C3H5)] (5 mol%) and PPh3 (15 mol%) in toluene (0.5 M) at 75°C catalyzes the reductive C-C coupling of 1a to paraformaldehyde to provide the desired adduct 2a in 55% isolated yield along with small quantities of the corresponding formate ester, which is hydrolytically cleaved upon isolation (Table 1, entry 7).9 Notably, only trace quantities of adduct are formed in the absence of PPh3 (Table 1, entry 8), suggesting that appropriate selection of ligand is crucial. Indeed, screening the precatalyst [RuBr(CO)3(η3-C3H5)] against a series of phosphine ligands (Table 1, entries 9–13) reveals that monodentate ligands promote greater conversion than bidentate or tridentate ligands (Table 1, entries 9 and 10). Under optimal conditions employing [RuBr(CO)3(η3-C3H5)] (5 mol%) as precatalyst in combination with t-BuPPh2 (15 mol%) as ligand in toluene (0.5 M) at 75°C, the coupling product 2a is obtained in 81% isolated yield (Table 1, entry 12). By increasing the concentration of the reaction mixture (1.0 M in toluene), the isolated yield was increased to 86% (Table 1, entry 13). These optimized reaction conditions proved to be quite general, as demonstrated by the coupling of paraformaldehyde to 1,1-disubstituted allenes 1a-1h to furnish the homoallylic alcohols 2a-2h (Figure 1).
Table 1.
Optimization of ruthenium catalyzed coupling of paraformaldehyde to 1-methyl-1-phenylallene (1a) via transfer hydrogenation.a
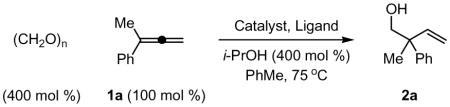 | ||||
|---|---|---|---|---|
| entry | catalystb | ligand (mol%) | concn (M) | yield% |
| 1 | [Cp(p-cymene)Ru]PF6 | PPh3 (15) | 0.5 | --- |
| 2 | CpRuCl(PPh3)3 | --- | 0.5 | --- |
| 3 | Ru3(CO)12 | PPh3 (15) | 0.5 | --- |
| 4 | RuHCl(PPh3)3.Toluene | --- | 0.5 | Trace |
| 5 | [Ru(cod)Cl2]n | PPh3 (15) | 0.5 | 18 |
| 6 | RuHCl(CO)(PPh3)3 | --- | 0.5 | 20 |
| 7 | [RuBr(CO)3(η3-C3H5)] | PPh3 (15) | 0.5 | 55 |
| 8 | [RuBr(CO)3(η3-C3H5)] | --- | 0.5 | Trace |
| 9 | [RuBr(CO)3(η3-C3H5)] | BINAP (7.5) | 0.5 | 49 |
| 10 | [RuBr(CO)3(η3-C3H5)] | MeC[CH2PPh2]3 (5) | 0.5 | Trace |
| 11 | [RuBr(CO)3(η3-C3H5)] | CatacXium A (15) | 0.5 | 72 |
| 12 | [RuBr(CO)3(η3-C3H5)] | t-BuPPh2 (5) | 0.5 | 81 |
| 13 | [RuBr(CO)3(η3-C3H5)] | t-BuPPh2 (5) | 1.0 | 86 |
Cited yields are of isolated material.
5 mol % with respect to ruthenium content. See Supporting Information for detailed experimental procedures.
Figure 1. Ruthenium catalyzed reductive coupling of paraformaldehyde to 1,1-disubstituted allenes 1a-1h via transfer hydrogenation.a.

aCited yields are of isolated material. Standard conditions employ 1 equivalent of allene and 4 equivalents of paraformaldehyde. See Supporting Information for detailed experimental procedures.
Under identical conditions, the coupling of allenes to higher aldehydes was explored. It was found that aromatic aldehydes (Table 2, entries 1 and 2), heteroaromatic aldehydes (Table 2, entry 3), α, β-unsaturated aldehydes (Table 2, entry 4), and aliphatic aldehydes (Table 2, entries 5 and 6) couple to a range of 1,1-disubstituted allenes in good yields but with low diastereoselectivity. Under standard conditions, electron rich aldehydes couple less efficiently, as demonstrated by the coupling of allene 1a to hexanal, which occurs in 38% yield to provide a 4:1 diastereomeric ratio of adducts.
Table 2.
Ru-catalyzed reductive coupling of aldehydes 3a-3f to 1,1-disubstituted allenes via transfer hydrogenation.a
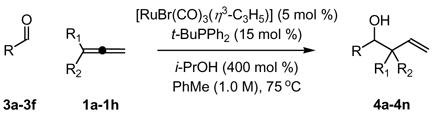 | |||
|---|---|---|---|
| 3a, R = p-NO2Ph | 3c, R = 2-(4-NO2-Furyl) | 3e, R = CH2OBn | |
| 3b, R = p-(CO2Me)Ph | 3d, R = o-NO2-Cinnamyl | 3f, R = CH2NPhth | |
| entry | RCHO, allene | product | yield % (dr) |
| 1 | 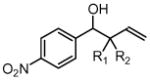 |
||
| 3a, 1a | 4a, R1 = Me, R2 = Ph | 87% (2:1 dr) | |
| 3a, 1b | 4b, R1 = Me, R2 = p-FPh | 86% (2:1 dr) | |
| 3a, 1d |
4c, R1 = CH2OMe R2 = p-MeOPh |
76% (1:1 dr) | |
| 3a, 1f | 4d, R1 = Me, R2 = CH2Ph | 83% (1:1 dr) | |
| 3a, 1g | 4e, R1 = Et, R2 = n-Bu | 82% (1:1 dr) | |
| 2 | 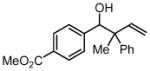 |
||
| 3b, 1a | 4f | 69% (2:1 dr) | |
| 3 | 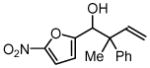 |
||
| 3c, 1a | 4g | 74% (1:1 dr) | |
| 4 | 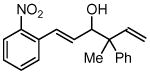 |
||
| 3d, 1a | 4h | 61% (2:1 dr) | |
| 5 | 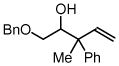 |
||
| 3e, 1a | 4i, R1= Me, R2 = Ph | 71% (1:1 dr) | |
| 3e, 1d |
4j, R1= CH2OMe R2 = p-MeOPh |
72% (1:1 dr) | |
| 3e, 1g | 4k, R1 = Et, R2 = n-Bu | 70% (1:1 dr) | |
| 6 | 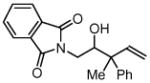 |
||
| 3f, 1a | 4l, R1= Me, R2 = Ph | 76% (1:1 dr) | |
| 3f, 1b | 4m, R1 = Me, R2 = p-FPh | 77% (1:1 dr) | |
| 3f, 1g | 4n, R1 = Et, R2 = n-Bu | 74% (1:1 dr) | |
Cited yields are of pure isolated material. See Supporting Information for detailed experimental procedures.
To gain insight into the catalytic mechanism, the coupling of paraformaldehyde and allene 1a was conducted employing d8-isopropanol as the terminal reductant. As revealed by 1H NMR analysis, deuterium is incorporated solely at the vinylic position (30% 2H). Similary, coupling of allene 1a to p-nitrobenzaldehyde 3a employing d8-isopropanol as the terminal reductant also results in deuterium incorporation exclusively at the vinylic position, but to a far greater extent (80% 2H). Notably, the isolated yield of coupling products in reactions employing d8-isopropanol as the terminal reductant are substantially lower than the isolated yields obtained in the parent reactions (Scheme 2). 10 A plausible catalytic cycle accounting for the relatively low levels of deuterium incorporation in the reaction of paraformaldehyde and allene 1a is indicated (Scheme 3). Reaction of the precatalyst [RuBr(CO)3(η3-C3H5)] with formaldehyde in the presence of isopropanol generates the homoallylic alcohol I and active catalyst II, 11 which hydrometallates allene 1a to furnish the allyl ruthenium complexes IIIa and IIIb.12 Allyl transfer to the aldehyde delivers the ruthenium alkoxide IV. The intermediate IV may react with isopropanol to provide the coupling product V and ruthenium isopropoxide VI, which upon β-hydride elimination regenerates the active catalyst II (Cycle A). Alternatively, the intermediate IV may react with another molecule of aldehyde to form ruthenium complex VII,13 which delivers ester VIII upon β-hydride elimination and regenerates the active catalyst II (Cycle B). The latter catalytic cycle suggests that the aldehyde may serve as a hydride donor. Indeed, when the coupling of paraformaldehyde and 1a is carried out in the absence of isopropanol, substantial quantities of the formate ester VIII (R = H) are detected. Mechanisms involving ruthenium catalyzed allene hydroacylation with subsequent isopropanol-mediated reduction of the resulting aldehyde are inconsistent with the results of deuterium incorporation. Upon resubjecting diastereomerically enriched adduct 4a to standard coupling conditions, a 2:1 ratio of diastereomeric ratio is reestablished, suggesting redox equilibration of the homoallylic alcohol under coupling conditions. That is, the diastereomeric ratio of adducts appears to be thermodynamically controlled.
Scheme 2. Ruthenium catalyzed coupling of allene 1a to paraformaldehyde and p-nitrobenzaldehyde employing d8-isopropanol as the terminal reductant.a.

aCited yields are of isolated material. See Supporting Information for detailed experimental procedures.
Scheme 3.

Plausible catalytic mechanisms as supported by deuterium labeling studies.
In summary, ruthenium catalyzed transfer hydrogenation ranks among the most powerful methods available for the reduction of polar functional groups, yet reductive C-C couplings under the conditions of ruthenium catalysis are highly uncommon. Recent studies from our laboratory are aimed at addressing this deficiency, with the ultimate goal of developing a broad new family of “transfer hydrogenative C-C bond formations.” In prior work, ruthenium catalyzed transfer hydrogenation was applied to the coupling of acyclic 1,3-dienes (butadiene, isoprene and 2,3-dimethylbutadiene) and 1,3-enynes to aldehydes or alcohols. Here, we demonstrate the ruthenium catalyzed reductive coupling of 1,2-disubstituted allenes 1a-1h to paraformaldehyde and higher aldehydes 3a-3f to furnish homoallylic alcohols 2a-2h and 4a-4n, respectively. Stereoselective variants of these transformations and the development of new transfer hydrogenative couplings, including imine additions from the amine oxidation level, are currently underway.
Supplementary Material
Acknowledgments
Acknowledgment is made to the Robert A. Welch Foundation, the ACS-GCI Pharmaceutical Roundtable, Merck, and the NIH-NIGMS (RO1-GM69445) for partial support of this research.
Footnotes
Supporting Information Available. Experimental procedures and spectral data for all new compounds (1H NMR, 13C NMR, IR, HRMS). This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.For selected reviews on ruthenium catalyzed transfer hydrogenation, see: Zassinovich G, Mestroni G, Gladiali S. Chem Rev. 1992;92:1051.Noyori R, Hashiguchi S. Acc Chem Res. 1997;30:97.Noyori R, Ohkuma T. Angew Chem. 2001;113:40.Angew Chem Int Edit. 2001;40:40.Noyori R, Yamakawa M, Hashiguchi S. J Org Chem. 2001;66:7931. doi: 10.1021/jo010721w.Noyori R. Angew Chem. 2002;114:2108.Angew Chem Int Edit. 2002;41:2008.Noyori R. Adv Synth Catal. 2003;345:15.Noyori R. Chem Commun. 2005:1807. doi: 10.1039/b502713f.Gladiali S, Alberico E. Chem Soc Rev. 2006;35:226. doi: 10.1039/b513396c.Ikariya T, Blacker AJ. Acc Chem Res. 2007;40:1300. doi: 10.1021/ar700134q.
- 2.Ruthenium complexes catalyze alkene hydroformylation. For a review, see: Kalck P, Peres Y, Jenck J. Adv Organomet Chem. 1991;32:121.
- 3.For ruthenium catalyzed reductive C-C bond formations beyond alkene hydroformylation, see: Tsuji Y, Mukai T, Kondo T, Watanabe Y. J Organomet Chem. 1989;369:C51.Kondo T, Ono H, Satake N, Mitsudo T, Watanabe Y. Organometallics. 1995;14:1945.Yu CM, Lee S, Hong YT, Yoon SK. Tetrahedron Lett. 2004;45:6557.
- 4.For selected reviews of ruthenium catalyzed C-C coupling, see: Trost BM, Toste FD, Pinkerton AB. Chem Rev. 2001;101:2067. doi: 10.1021/cr000666b.Kondo T, Mitsudo T-a. Curr Org Chem. 2002;6:1163.Dérien S, Monnier F, Dixneuf PH. Top Organomet Chem. 2004;11:1.
- 5.For reviews on hydrogenative C-C coupling, see: Iida H, Krische MJ. Top Curr Chem. 2007;279:77.Ngai MY, Kong JR, Krische MJ. J Org Chem. 2007;72:1063. doi: 10.1021/jo061895m.Skucas E, Ngai MY, Komanduri V, Krische MJ. Acc Chem Res. 2007;40:1394. doi: 10.1021/ar7001123.
- 6.For recent examples, see: C=X Vinylation: Kong JR, Ngai MY, Krische MJ. J Am Chem Soc. 2006;128:718. doi: 10.1021/ja056474l.Skucas E, Kong J-R, Krische MJ. J Am Chem Soc. 2007;129:7242. doi: 10.1021/ja0715896.Barchuk A, Ngai MY, Krische MJ. J Am Chem Soc. 2007;129:8432. doi: 10.1021/ja073018j.Ngai MY, Barchuk A, Krische MJ. J Am Chem Soc. 2007;129:12644. doi: 10.1021/ja075438e.Aldol and Mannich addition: Jung CK, Garner SA, Krische MJ. Org Lett. 2006;8:519. doi: 10.1021/ol052859x.Jung CK, Krische MJ. J Am Chem Soc. 2006;128:17051. doi: 10.1021/ja066198q.Garner SA, Krische MJ. J Org Chem. 2007;72:5843. doi: 10.1021/jo070779w.Bee C, Han SB, Hassan A, Iida H, Krische MJ. J Am Chem Soc. 2008;130:2746. doi: 10.1021/ja710862u.C=O Allylation: Skucas E, Bower JF, Krische MJ. J Am Chem Soc. 2007;129:12678. doi: 10.1021/ja075971u.
- 7.(a) Bower JF, Skucas E, Patman RL, Krische MJ. J Am Chem Soc. 2007;129:15134. doi: 10.1021/ja077389b. [DOI] [PubMed] [Google Scholar]; (b) Bower JF, Patman RL, Krische MJ. Org Lett. 2008;10:1033. doi: 10.1021/ol800159w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Shibahara F, Bower JF, Krische MJ. J Am Chem Soc. 2008;130 doi: 10.1021/ja801213x. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Patman RL, Williams VM, Bower JF, Krische MJ. Angew Chem Int Ed. 2008;47 doi: 10.1002/anie.200801359. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.See supporting information for detailed experimental procedures.
- 10.Couplings employing d8-isopropanol as terminal reductant were conducted twice. The isolated yields and levels of deuterium incorporation .for deuterio-2a and deuterio-4a were reproducible within experimental error.
- 11.When α phthalimidoacetaldehyde was coupled to one equivalent of [RuBr(CO)3(η3-C3H5)], the corresponding homoallylic alcohol and its isomers were detected. Also, see reference 3b.
- 12.For hydrometallation of allenes with Ru complexes, see: Hill AF, Ho CT, Wilton-Ely JDET. Chem Commun. 1997:2207.Nakanishi S, Sasabe H, Takata T. Chem Lett. 2000:1058.Sasabe H, Nakanishi S, Takata T. Inorg Chem Commun. 2002;5:177.Sasabe H, Nakanishi S, Takata T. Inorg Chem Commun. 2003;6:1140.Xue P, Bi S, Sung HHY, Williams ID, Lin Z, Jia G. Organometallics. 2004;23:4735.Sasabe H, Kihara N, Mizuno K, Ogawa A, Takata T. Chem Lett. 2006;35:212.Bai T, Zhu J, Xue P, Sung HHY, Williams ID, Ma S, Lin Z, Jia G. Organometallics. 2007;26:5581.
- 13.For Ru-catalyzed oxidative transformation of alcohols and aldehydes to esters, see: Blum Y, Shvo Y. Isr J Chem. 1984;24:144.Blum Y, Shvo Y. J Organomet Chem. 1984;263:93.Blum Y, Shvo Y. J Organomet Chem. 1985;282:C7.Murahashi SI, Naota T, Ito K, Maeda Y, Taki H. J Org Chem. 1987;52:4319.Zhang J, Leitus G, Ben-David Y, Milstein D. J Am Chem Soc. 2005;127:10840. doi: 10.1021/ja052862b.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.