

Abstract
The recent recognition of the importance of transforming growth factor beta (TGF-β) in mediating many cellular immune functions has popularized the use of mouse models where TGF-β signaling is blocked in specific cell types. However, there are some caveats affiliated with each model. Here we describe and show evidence for some of the limitations of these models and provide insight into ways to improve the utility of these animals for in vivo studies.
Keywords: TGF-beta, animal model, immunological rejection, genetic blockade
1. Introduction
TGF-β is a pleiotropic cytokine with potent regulatory activity. TGF-β binds to TGF-β receptor II (TGF-βRII), triggering the kinase activity of the cytoplasmic domain that in turn activates TGF-βRI. The activated receptor complex leads to nuclear translocation of Smad molecules, and transcription of target genes (Li et al., 2006b). The multi-faceted effects of TGF-β on numerous immune functions are cellular and environmental context dependent (Li and Flavell, 2008a). Therefore, much emphasis has been put toward studying the effects of TGF-β in vivo.
Several mouse models have been developed to specifically block TGF-β signaling in T cells. The first two models used the CD2, and the CD4 promoter without the CD8 silencer, to express a dominant-negative form of TGFβRII, lacking the kinase domain, specifically in T cells (Gorelik and Flavell, 2000; Lucas et al., 2000). The CD2-DNR mice exhibit deregulated growth of the CD8+ memory T cell population that eventually transforms into lymphoma (Lucas et al., 2004; Lucas et al., 2006). By contrast, the CD4-DNR mice develop an autoimmune inflammatory phenotype associated with increased numbers of CD4+ Th1 and Th2 cells, as well as activated CD8+ T cells that produce effector cytokines (Gorelik and Flavell, 2000). As a result, the CD4-DNR mice develop spontaneous inflammatory bowel disease (IBD), and die within 6–8 months (Table 1) (Gorelik and Flavell, 2000). The exact reason for the differences seen between the two DNR mice is not known, but it may be related to differential expression of the DNR transgene. Nonetheless, both DNR mouse models show less pathology than either the TGFβ1−/− or T-cell specific TGF-βRII deletion (4cre-RII/RII) mice (Shull et al., 1992; Li et al., 2006a; Marie et al., 2006). More aggressive than the CD4-DNR mice, the peripheral T cells of the 4cre-RII/RII mice become activated spontaneously, with the skewing of CD4+ T cells into a Th1 phenotype (Li et al., 2006a; Marie et al., 2006). These mice develop an inflammatory autoimmune wasting disease with lymphocyte infiltration into all organs, and they die within 5 weeks of age (Li et al., 2006a; Marie et al., 2006).
Table 1.
Comparison of mouse models with genetic blockade of TGF-β signaling.
| CD4-DNR | 4cre-RII/RII | CD11C-DNR | |
|---|---|---|---|
| Breeding Strategy | ♀ B6 X ♂ CD4-DNR | ♀ RIIflx/flx X ♂ 4cre-RII+/flx ♀ 4cre-RII+/flx X ♂ RIIflx/flx | ♀ B6 X ♂ CD11c-DNR ♀ CD11c-DNR X ♂ B6 |
| Expected Progeny | 50% Tg+, 50% Tg− | 25% 4cre-RIIflx/flx, 25% 4cre-RII+/flx | 50% Tg+, 50% Tg− |
| Expected survival age | 6–B months | 3–5 weeks | 1–2 years |
| Spontaneous Phenotype | IBD | Systemic Inflammatory Autoimmunity | None |
| Medicated Diet | *4-drug Helicobacter | None | None |
| Inhibition of signaling | 50–80% | 95–100% | 50–80% |
Amoxicillin, Clarithromycin, Metronidazole, and Omeprazole
TGF-β also plays an important role in suppressing the cells of the innate immune system. Thus, another animal model was developed where TGF-β signaling was blocked in Natural Killer (NK) and dendritic cells (DC) by expressing the TGFβRII dominant negative receptor under the control of the CD11c promoter (CD11c-DNR) (Laouar et al., 2005). These mice do not develop spontaneous disease. However, blockade of TGF-β signaling in NK cells caused the accumulation of a large number of NK cells capable of secreting large amounts of IFN-γ, which resulted in enhanced skewing of CD4+ cells into a Th1 phenotype in response to Leishmania infection (Laouar et al., 2005). However, blockade of TGF-β signaling in DCs from these mice did not affect DC homeostasis or IL-12 production (Laouar et al., 2005). Instead, when CD11c-DNR mice were crossed to MOG TCR transgenic mice, they showed spontaneous EAE-like disease as a result of inactivation of TGF-β signaling in DCs (Laouar et al., 2008). In addition, when these mice were crossed to the Tg2576 Alzheimer's disease mouse model, the aged double-transgenic mice showed complete mitigation of Tg2576-associated hyperactivity and partial mitigation of defective spatial working memory (Town et al., 2008).
2. Materials and methods
2.1. Mice
CD4-TGF-βRII-DNR mice (Gorelik and Flavell, 2000), also available through JAX lab, were bred on to OTI Vα2/Vβ5 TCR transgenic specific for OVA257–264 that recognizes SIINFEKL epitope in the context of MHC class I H-2Kb. These mice were then crossed to RAG1−/− (JAX) and CD45.1 (NCI) congenic markers. CD11c-TGF-β RII-DNR transgenic mice (Laouar et al., 2005), also available through JAX lab, were maintained on a C57BL/6 background.
2.2. Helicobacter treatment
Mice were fed 4-drug Helicobacter medicated chow diet (BIO-SERV), containing Amoxicillin, Clarithromycin, Metronidazole, and Omeprazole for 9 consecutive weeks. After the completion of the treatment, mice were handled first and kept separate from untreated animals. This treatment maintains mice and their progeny helicobacter-free for 6–12 months.
2.3. Adoptive co-transfer and LM-OVA infection
Every co-transfer experiment consisted of 2.5×104 naïve sorted OTI and OTI-DNR cells each on a different congenic marker mixed at a 1:1 ratio. One to two days after adoptive transfer, mice were infected with 1×105 recombinant Listeria monocytogenes, expressing OVA (LM-OVA), which was a generous gift from Hao Shen (U. Penn). 2×106 naïve sorted cells were co-transferred for steady-state experiments.
2.4. Isolation of peripheral blood lymphocytes
100–150μl of blood from tail vein of infected mice was added to 30μl of 1× heparin (500 units/ml). 500μl of 1× ACK lysis buffer (Lonza) was added directly to the cells and incubated at room temperature for 2–3 minutes. Cells were centrifuged at 4000 rpm for 5 minutes. The top layer was aspirated and another 500μl of 1× ACK lysis buffer was added followed by centrifugation. Cells were resuspended in FACS buffer (PBS + 0.5% FBS) and stained with surface antibodies.
3. Results and Discussion
There is a tremendous wealth of information that can be learned from using genetic mouse models to address the in vivo role of TGF-β signaling under various conditions. However, there are several caveats affiliated with each model, some of which have already been described and are only briefly discussed here and summarized in Table 1. The 4cre-RII/RII model provides the most complete blockade of TGF-β signaling in T cells, resulting in a much more severe phenotype compared to the CD4-DNR mice (Gorelik and Flavell, 2000; Lucas et al., 2000; Li et al., 2006a; Marie et al., 2006). However, major disadvantages of using this model is that the majority of the T cells from these mice display an activated phenotype, the animals only survive for 3–5 weeks, and autoimmune disease can develop as early as 2 weeks after birth (Li et al., 2006a; Marie et al., 2006). It is therefore crucial to breed RII+/flx heterozygote mice when crossed to Lck- or CD4-cre mice (Table 1). The use of inducible cre lines can be extremely useful for isolating naïve precursor cells as well as for temporal deletion of TGF-β RII. In addition, crossing the 4cre-RII/RII mice to specific TCR transgenic lines and further placing them on a RAG−/− background eliminates the autoimmune phenotype. T cells can then be isolated and adoptively transferred into wild type hosts for further studies.
The CD4-DNR mice provide a significant advantage over the 4cre-RII/RII mice because they survive much longer (Table 1). In fact, the lifespan and health of these mice can be improved if they are kept under helicobacter-free conditions. Once crossed to TCR transgenic lines on a RAG−/− background, the autoimmune phenotype is completely diminished in these mice, and large numbers of naïve T cells can be isolated from adult animals. However, two major disadvantages exist in the CD4-DNR model. First, these mice only display about 50–100% blockade of TGF-β signaling, which can vary depending on cellular state and experimental conditions (Gorelik and Flavell, 2000). The level of endogenous TGF-β receptor II is a good indication of the level of blockage that can be expected in the CD4-DNR mice under specific conditions. Obviously, a reciprocal relationship would be expected; the higher the level of endogenous receptor, the lower the expected blockage through the dominant negative receptor.
The second and more problematic disadvantage comes from our findings that T cells isolated from CD4-DNR mice cannot be adoptively transferred into wild type C57Bl6 (B6) recipients, because they are immunologically rejected (Fig. 1). When OTI-DNR double transgenic mice were co-transferred along with wild type OTI cells into B6 mice and stimulated using recombinant Listeria monocytogenes expressing OVA (LM-OVA), the OTI-DNR cells could not be detected by day 6 post infection (Fig. 1a). This phenomenon was specific to the DNR carrying cells, regardless of the congenic markers used. We considered the possibility that the DNR cells were being rejected in the wild type host, since the dominant negative construct that was used to generate the C4-DNR mice contains human rather than mouse TGF-βRII sequence, and therefore antigenic amino acids may have been introduced. To test this hypothesis, we performed similar adoptive co-transfer experiments using the CD4-DNR mice as recipients. We reasoned that these mice must be tolerant to the human DNR sequence as this transgene is expressed in the hematopoietic compartment, and thus rejection would be unlikely to occur. As we had predicted, the OTI-DNR cells were not rejected in CD4-DNR hosts (Fig. 1b). In fact, the OTI-DNR cells survived the expansion and contraction much better than the wild type cells, an important biological phenomenon that has been extensively described elsewhere (Sanjabi et al., 2009).
Figure 1.

T cells from CD4-DNR mice are immunologically rejected in immunocompetent hosts. Longitudinal comparison of the difference in the fraction of co-transferred OTI and OTI-DNR cells after infection with LM-OVA when transferred into (a) C57Bl6, (b) CD4-DNR, and (c) CD11c-DNR hosts. Data is representative of 3–5 independent experiments with similar results and 3–5 recipients each.
Although DNR-bearing cells cannot be adoptively transferred into B6 animals, they can be transferred into other DNR-bearing hosts. However, the use of CD4-DNR mice as recipients can become problematic because these mice have an activated/memory repertoire of both CD4+ and CD8+ T cells (Gorelik and Flavell, 2000). In addition, they tend to develop autoimmunity and can potentially develop disease during the course of a long-term study. Instead, the use of the CD11c-DNR mice is recommended under circumstances where T cell activation must be achieved under immunogenic conditions. Since these mice express the TGF-β DNR transgene in the hematopoietic compartment, they too are tolerant to the human dominant negative TGF-β RII sequence (Fig. 1c). Unlike the CD4-DNR mice, the CD11c-DNR mice do not develop spontaneous disease and have a normal T cell repertoire (Table 1), but they do have a larger compartment of NK cells capable of producing high levels of IFN-γ (Laouar et al., 2005). Thus, to control for any untoward effects of the CD11c-DNR transgene on the results of adoptive transfer experiments, we recommend co-transfer of test and control cells into the same host, and the adoptive transfer and analysis of the control cells into both the CD11c-DNR transgene positive and negative mice as has recently been done in a study where the role of TGF-β signaling on effector CD8+ T cells was being studied (Sanjabi et al., 2009). An alternative strategy to using DNR-bearing mice as hosts would be to first generate WT:DNR mixed bone marrow chimeras in B6 animals and study the effect of TGF-β signaling in T cells by the use of congenic markers and MHC-tetramers (Tinoco et al., 2009).
To address whether cells bearing the DNR transgene could be used in short-term studies not involving immunization, we co-transferred OTI and OTI-DNR cells into wild type B6 mice and monitored T cell rejection over time (Fig. 2a). No rejection in OTI-DNR cells was detected up to 6 days post adoptive transfer. However, by about two weeks post transfer, only half as many OTI-DNR cells were left, and by about three weeks post transfer, almost all the OTI-DNR cells had been rejected (Fig. 2a). In addition, we also tested the engraftment of OTI and OTI-DNR cells in RAG1−/− recipients (Fig. 1e), and did not detect any rejection in these mice. In fact, over time, the OTI-DNR cells showed a higher capacity for lymphopenia-induced proliferation, which is related to the inhibitory effects of TGF-β on this process. Interestingly, when DNR-bearing T cells were adoptively transferred into immunosuppressed B6 mice due to a chronic viral infection, the rejection phenomenon was also delayed, presumably due to immunosuppression of the corresponding cytolytic T cells (Tinoco et al., 2009). Collectively, these results suggest that immunosuppressed or RAG−/− mice can be used as recipients for DNR-bearing cells for long term experiments, but only short-term experiments may be performed if immunocompetent B6 mice are used as recipients.
Figure 2.
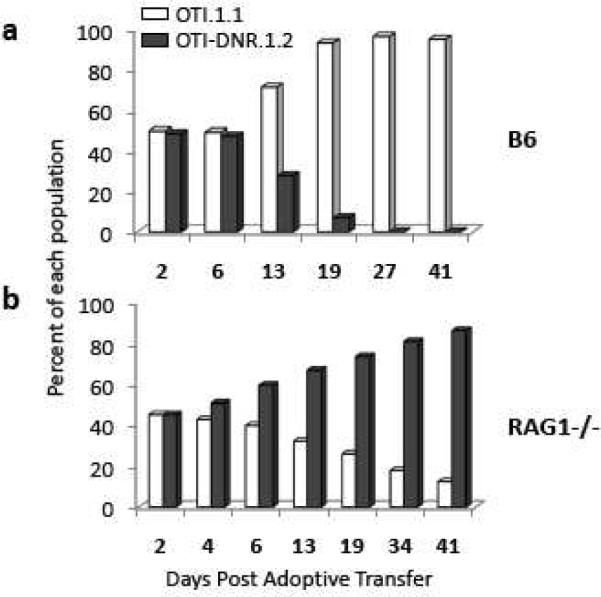
T cells from CD4-DNR mice are not immunologically rejected in immunocompromised hosts. Longitudinal comparison of the difference in the fraction of co-transferred OTI and OTI-DNR cells transferred into (a) C57Bl6, and (b) RAG1−/− hosts under steady state conditions. Data is representative of 3–5 independent experiments with similar results and 3–5 recipients each.
In the thymus, development of natural killer T (NKT) cells, natural regulatory T (nTreg) cells, and CD8+ T cells is dependent on TGF-β signaling (Li and Flavell, 2008b; Li and Flavell, 2008a). In the periphery, TGF-β is necessary for the survival of naïve T cells, and it also maintains peripheral tolerance by inhibiting the proliferation and differentiation of self-reactive CD4+ and CD8+ T cells (Li and Flavell, 2008b; Li and Flavell, 2008a). In addition, TGF-β maintains the survival of naturally occurring Treg cells, and in combination with IL-2 and retinoic acid it promotes the differentiation of induced Treg cells (Li and Flavell, 2008b; Li and Flavell, 2008a). Under inflammatory conditions, TGF-β in the presence of IL-6 drives the differentiation of Th17 cells; whereas in combination with IL-4, it promotes the differentiation of IL-9- and IL-10-producing T cells (Dardalhon et al., 2008; Veldhoen et al., 2008). Thus, animal models are an essential component of the immune arsenal. In addition, it is imperative to study the role of TGF-β in a cellular and context dependent manner, using the animal models described above, but keeping in mind the advantages and disadvantages of each model.
Acknowledgements
R.A.F is an investigator of the Howard Hughes Medical Institute. This work is supported by a post-doctoral fellowship grant from Cancer Research Institute (S.S.), with additional support from NIH grants CA121974 and DK051665 (R.A.F.) and JDRF grant 32-2008-352 (R.A.F.). The authors declare that they have no competing financial interests.
Abbreviations
- TGF-β
Transforming growth factor beta
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Dardalhon V, Awasthi A, Kwon H, Galileos G, Gao W, Sobel RA, Mitsdoerffer M, Strom TB, Elyaman W, Ho IC, Khoury S, Oukka M, Kuchroo VK. IL-4 inhibits TGF-beta-induced Foxp3+ T cells and, together with TGF-beta, generates IL-9+ IL-10+ Foxp3(−) effector T cells. Nat Immunol. 2008;9:1347–55. doi: 10.1038/ni.1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorelik L, Flavell RA. Abrogation of TGFbeta signaling in T cells leads to spontaneous T cell differentiation and autoimmune disease. Immunity. 2000;12:171–81. doi: 10.1016/s1074-7613(00)80170-3. [DOI] [PubMed] [Google Scholar]
- Laouar Y, Sutterwala FS, Gorelik L, Flavell RA. Transforming growth factor-beta controls T helper type 1 cell development through regulation of natural killer cell interferon-gamma. Nat Immunol. 2005;6:600–7. doi: 10.1038/ni1197. [DOI] [PubMed] [Google Scholar]
- Laouar Y, Town T, Jeng D, Tran E, Wan Y, Kuchroo VK, Flavell RA. TGF-beta signaling in dendritic cells is a prerequisite for the control of autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2008;105:10865–70. doi: 10.1073/pnas.0805058105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li MO, Flavell RA. Contextual regulation of inflammation: a duet by transforming growth factor-beta and interleukin-10. Immunity. 2008a;28:468–76. doi: 10.1016/j.immuni.2008.03.003. [DOI] [PubMed] [Google Scholar]
- Li MO, Flavell RA. TGF-beta: a master of all T cell trades. Cell. 2008b;134:392–404. doi: 10.1016/j.cell.2008.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li MO, Sanjabi S, Flavell RA. Transforming growth factor-beta controls development, homeostasis, and tolerance of T cells by regulatory T cell-dependent and -independent mechanisms. Immunity. 2006a;25:455–71. doi: 10.1016/j.immuni.2006.07.011. [DOI] [PubMed] [Google Scholar]
- Li MO, Wan YY, Sanjabi S, Robertson AK, Flavell RA. Transforming growth factor-beta regulation of immune responses. Annu Rev Immunol. 2006b;24:99–146. doi: 10.1146/annurev.immunol.24.021605.090737. [DOI] [PubMed] [Google Scholar]
- Lucas PJ, Kim SJ, Mackall CL, Telford WG, Chu YW, Hakim FT, Gress RE. Dysregulation of IL-15-mediated T-cell homeostasis in TGF-beta dominant-negative receptor transgenic mice. Blood. 2006;108:2789–95. doi: 10.1182/blood-2006-05-025676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas PJ, Kim SJ, Melby SJ, Gress RE. Disruption of T cell homeostasis in mice expressing a T cell-specific dominant negative transforming growth factor beta II receptor. J Exp Med. 2000;191:1187–96. doi: 10.1084/jem.191.7.1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas PJ, McNeil N, Hilgenfeld E, Choudhury B, Kim SJ, Eckhaus MA, Ried T, Gress RE. Transforming growth factor-beta pathway serves as a primary tumor suppressor in CD8+ T cell tumorigenesis. Cancer Res. 2004;64:6524–9. doi: 10.1158/0008-5472.CAN-04-0896. [DOI] [PubMed] [Google Scholar]
- Marie JC, Liggitt D, Rudensky AY. Cellular mechanisms of fatal early-onset autoimmunity in mice with the T cell-specific targeting of transforming growth factor-beta receptor. Immunity. 2006;25:441–54. doi: 10.1016/j.immuni.2006.07.012. [DOI] [PubMed] [Google Scholar]
- Sanjabi S, Mosaheb MM, Flavell RA. Opposing effects of TGF-beta and IL-15 cytokines control the number of short-lived effector CD8+ T cells. Immunity. 2009;31:131–44. doi: 10.1016/j.immuni.2009.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, Yin M, Allen R, Sidman C, Proetzel G, Calvin D, et al. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature. 1992;359:693–9. doi: 10.1038/359693a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinoco R, Alcalde V, Yang Y, Sauer K, Zuniga EI. Cell-intrinsic transforming growth factor-beta signaling mediates virus-specific CD8+ T cell deletion and viral persistence in vivo. Immunity. 2009;31:145–57. doi: 10.1016/j.immuni.2009.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Town T, Laouar Y, Pittenger C, Mori T, Szekely CA, Tan J, Duman RS, Flavell RA. Blocking TGF-beta-Smad2/3 innate immune signaling mitigates Alzheimer-like pathology. Nat Med. 2008;14:681–7. doi: 10.1038/nm1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veldhoen M, Uyttenhove C, van Snick J, Helmby H, Westendorf A, Buer J, Martin B, Wilhelm C, Stockinger B. Transforming growth factor-beta `reprograms' the differentiation of T helper 2 cells and promotes an interleukin 9-producing subset. Nat Immunol. 2008;9:1341–6. doi: 10.1038/ni.1659. [DOI] [PubMed] [Google Scholar]