

Abstract
Receptor-associated late transducer (RALT) is a feedback inhibitor of epidermal growth factor receptor signaling. RALT has been shown previously to be induced in the ischemic heart and to promote cardiomyocyte apoptosis in vitro. However, the role of RALT in cardiac hypertrophy remains unclear. We hypothesized that forced expression of RALT in the murine heart would protect the heart against cardiac hypertrophy in vivo. We investigated the effect of cardiac overexpression of rat RALT on cardiac hypertrophy induced by angiotensin II and isoproterenol in RALT transgenic mice and wild-type littermates. The extent of cardiac hypertrophy was assessed by 2D and M-mode echocardiography as well as by molecular and pathological analyses of cardiac samples. Constitutive expression of rat RALT in cardiac myocytes of murine heart attenuated both hypertrophic and inflammatory responses and preserved cardiac function. These beneficial effects were associated with the attenuation of the epidermal growth factor receptor–dependent cascade that was triggered by angiotensin II and isoproterenol stimulation. Additional evidence demonstrated that RALT expression blocked fibrosis in vivo and collagen synthesis in vitro. Therefore, cardiac overexpression of RALT improves cardiac function and inhibits maladaptive hypertrophy, inflammation, and fibrosis through attenuating epidermal growth factor receptor–dependent signaling.
Keywords: RALT, EGFR, ERK1/2, cardiac hypertrophy, heart failure, fibrosis
Cardiac hypertrophy is a response of the myocardium to increased workload, characterized by increased myocardial mass with extracellular matrix accumulation.1,2 Although initially a beneficial adaptive response, prolonged hypertrophy may result in ventricular dilatation and heart failure.3,4 One evolving concept is that the underlying signaling mechanism, rather than the presence of hypertrophy alone, may determine the functional outcome of cardiac hypertrophy. Thus, it is important to define and modulate the specific signaling mechanism activated by each hypertrophic stimulus and its effect on the cardiac phenotype.
Epidermal growth factor receptor (EGFR) transactivation is an important step in the activation of downstream tyrosine kinases and serves as a scaffold for various signaling molecules in cardiac myocytes.5 Inhibition of EGFR activation by its inhibitor AG1478 and the metalloproteinase inhibitor BB94 significantly attenuated cardiac hypertrophy in vitro and in vivo.6,7 However, neither systemic effects of these interventions nor nonspecific effects of the chemical inhibitors can be excluded. Indeed, Kagiyama et al8 showed that inhibition of EGFR by antisense oligonucleotides in mice caused a significant reduction of blood pressure, which could secondarily affect the extent of cardiac hypertrophy. These observations prompted us to investigate a molecular target that specifically blocks EGFR transactivation for inhibiting cardiac hypertrophy and heart failure. One such protein is receptor-associated late transducer (RALT; also named mitogen-inducible gene 6 [Mig6] or Gene 33), which is a feedback inhibitor of EGFR.9 RALT inhibits EGFR-mediated signals in mouse skin, and deletion of the RALT gene leads to hyperactivation of EGFR.10 Recently, Xu et al11,12 demonstrated that RALT expression induces apoptosis in cardiac myocytes through suppressing EGFR and EGFR-dependent downstream signaling pathways. Although RALT has been shown to inhibit EGFR-dependent signaling, little is known about the role of this regulation in cardiac dysfunction. Therefore, the aim of this study was to determine whether RALT attenuates cardiac hypertrophy in vivo by impairing EGFR-dependent signaling pathway.
Methods and Materials
Transgenic Mice and Animal Models
The study protocol was approved by the Animal Care and Use Committee of our hospital. The details for transgenic (TG) mice production, angiotensin II (Ang II), and isoproterenol (ISO) infusion can be found in an online supplement available at http://www.hypertensionaha.org. Hearts and lungs of the euthanized mice were dissected and weighed to compare heart weight to body weight (mg/g) and lung weight to body weight (mg/g) ratios in TG mice and littermate controls.
Echocardiography and Cardiac Catheterization
Echocardiography was performed using SONOS 5500 ultrasound (Philips Electronics; Amsterdam) with a 15-MHz linear array ultrasound transducer. Hemodynamic measurements were recorded at baseline and after injection of dobutamine (2.5 μg/g body weight), as described in the online supplement.
Histological Analysis and Apoptosis
Hearts were excised, washed with saline solution, and placed in 10% formalin. Several sections of heart (4 to 5 μm thick) were prepared and stained with hematoxylin and eosin for histopathology or Picrosirius red for collagen deposition and visualized by light microscopy. The outline of 200 myocytes was traced in each group. Apoptosis was evaluated by a TUNEL assay that was performed in sections using the CardiaoTACS in situ Apoptosis Detection Kit (R&D Systems) according to manufacturer recommendations. Apoptosis was confirmed by the detection of caspase-3/8 activation.
Western Blot Analysis and Quantitative Real-Time RT-PCR
Quantitative real-time RT-PCR and Western blot analysis is described in the online supplement.
Recombinant Adenoviral Vectors and Cultured Neonatal Rat Cardiac Myocytes and Fibroblasts
We used replication-defective adenoviral vectors expressing the full-length rat RALT gene under control of the cytomegalovirus promoter and a control adenoviral vector expressing the green fluorescent protein. We screened 3 lines of adenovirus (Ad) generated from the shRNA constructs against rat RALT from SuperArray (catalog No. KR54214G) and selected one that led to the greatest decrease in RALT levels for additional experiments. The control Ad-shRNA virus from SuperArray was used as control. We infected cardiac myocytes with Ad-RALT, Ad-green fluorescent protein, Ad-shRALT, or Ad-shRNA at a multiplicity of infection of 100, resulting in 95% to 100% of cells expressing the transgenes without toxicity. Primary cultures of cardiac myocytes and fibroblasts were prepared as described previously,13–15 the details of which are available in the online supplement.
Reporter Assays and Analysis of Collagen Synthesis
The luciferase activity was examined as described previously.13 The effects of RALT on DNA synthesis and collagen synthesis in cultured cardiac fibroblasts were evaluated by measuring the cell incorporation of [3H]-proline, respectively, as described previously,14 the details of which are in the online supplement.
Statistical Analysis
All values are expressed as mean±SEM. The probability of statistical differences between 2 groups was determined by a Student t test. Comparisons between multiple groups were assessed by 1-way ANOVA, followed by Bonferroni correction for multiple hypothesis testing. A value of P<0.05 was considered statistically significant.
Results
Characterization of Cardiac-Specific Rat RALT TG Mice
To examine the effect of constitutive rat RALT expression on cardiac hypertrophy, we generated TG mouse lines with a construct expressing full-length rat RALT under the control of the α-myosin heavy chain promoter. Four lines of TG mice were confirmed by PCR (data not shown) and Western blot analysis (Figure 1A). All the lines were viable and fertile, and there were no detectable differences in cardiac size and structure between TG and wild-type (WT) mice, either grossly or microscopically. We analyzed RALT protein levels in various tissues in TG mice by Western blot analysis using anti-RALT antibody and found that the TG rat RALT was expressed strongly in the heart but not in other organs, suggesting the specificity of the TG expression (Figure 1B). Among 4 established lines of TG mice, Tg2 was used for additional experiments. Cardiac hypertrophy was induced by Ang II or ISO infusion, and the induction of RALT expression in the hypertrophic hearts of WT and TG mice was analyzed by Western blotting. The expression of RALT protein in the normal heart was very low but markedly induced 6 hours after Ang II and ISO infusion, peaked after 24 hours, and then decreased (Figure 1C). However, the TG RALT protein levels in the TG mice were not significantly affected by Ang II or ISO infusion during the whole time course examined (Figure 1C).
Figure 1.
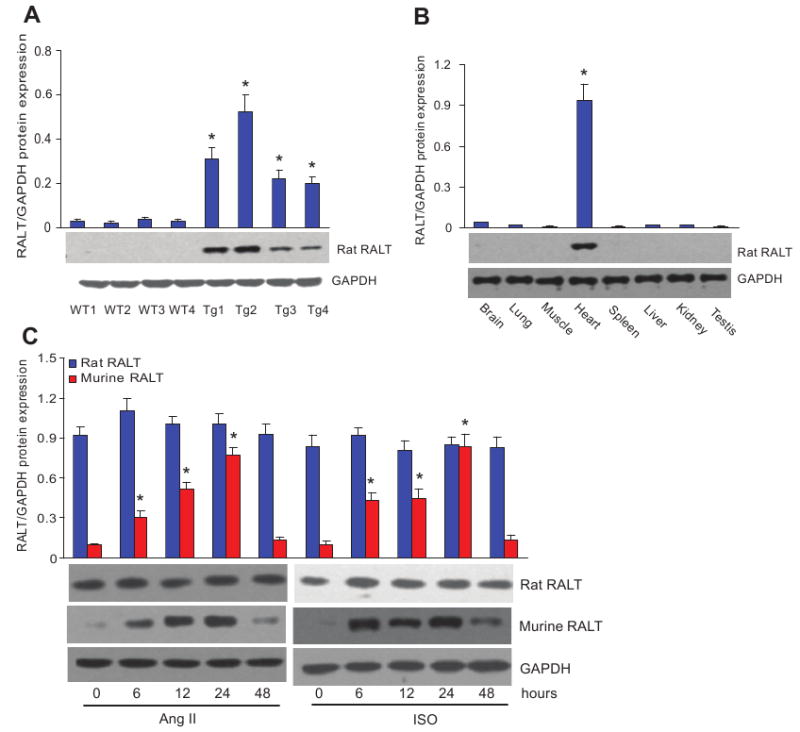
Characterization of rat RALT TG mice. A, Rat RALT protein in the heart tissue from 4 lines of both TG and WT mice. Top, Statistical result. Bottom, Representative Western blots. Data represent typical results of 4 different experiments as mean±SE (n=5 mice per group). *P<0.01 was obtained for the WT1 values. B, Endogenous RALT protein from different tissue of TG mice as indicated. Top, Statistical result. Bottom, Representative Western blots. Data represent typical results of 3 different experiments as mean±SE (n=5 mice per group). C, TG RALT and endogenous RALT protein levels in the heart after infusion of Ang II and ISO in indicated time. Top, Statistical result. Bottom, Representative Western blots. Data represent typical results of 4 different experiments as mean±SE (n=6 mice per group). *P<0.01 was obtained for the untreated group values.
Gravimetric Data and Cardiac Function of RALT TG Mice Under Basal Conditions
All experiments were performed with 8- to 9-week-old male TG and WT littermate mice. As shown in Table 1, body weight, blood pressure, and heart rate were not significantly different between TG and WT mice. There was no evidence of fibrosis in either group by microscopic examinations of multiple histological sections (data not shown). The ratios of heart weight to body weight and lung weight to body weight were not different between 2 groups, and echocardiography demonstrated normal LV chamber dimensions, wall thickness, and fractional shortening in TG mice (Table 1). To test whether rat RALT expression influences heart morphology and function in aging mice, we examined TG and WT mice at 1.5 years old. No significant change in echocardiographic indices was observed in aging TG mice (data not shown). Real-time PCR analysis of the heart in 8- to 9-week-old and 1.5-year-old mice showed no difference in the expression of hypertrophy markers, including ANP, BNP, myosin, heavy polypeptide 7, cardiac muscle, β (Myh7), and actin, α1, skeletal muscle (Acta1) in TG mice compared with WT mice (data not shown). These results suggested that the expression of the rat RALT gene in cardiac myocytes does not cause spontaneous cardiac dysfunction in mice.
Table 1. Anatomic and Cardiac Function Data for 8-Week-Old WT and RALT TG Mice Under Basal Condition.
| Parameter | WT Mice | TG Mice |
|---|---|---|
| No. | n=13 | n=15 |
| Anatomic analysis | ||
| BW, g | 24.7±1.2 | 24.5±1.3 |
| HW/BW, mg/g | 4.56±0.14 | 4.52±0.11 |
| LW/BW, mg/g | 5.13±0.03 | 5.22±0.08 |
| CSA, μm2 | 267±30.5 | 255±31.1 |
| Echocardiography analyses | ||
| HR, bpm | 472±32 | 467±28 |
| PWT, mm | 1.21±0.03 | 1.24±0.05 |
| LVEDD, mm | 3.68±0.02 | 3.71 ±0.04 |
| LVESD, mm | 2.25±0.01 | 2.33±0.04 |
| IVSd, mm | 0.64±0.05 | 0.64±0.02 |
| LVPWd, mm | 0.61 ±0.05 | 0.62±0.03 |
| Homodynamic analyses | ||
| FS, % | 55.3±2.3 | 56.4±3.2 |
| MAP, mm Hg | 85.1 ±3.4 | 87.5±2.4 |
| SBP, mm Hg | 115.1 ±3.0 | 116.6±4.2 |
| LVEDP, mm Hg | 9.6±1.2 | 9.4±1.4 |
| LV dP/dTmax, mm Hg/s | 8785.8±466.8 | 8794.9±452.5 |
| LV dP/dTmin, mm Hg/s | −7655.7±653.5 | −7734.2±463.3 |
HR indicates heart rate; BW, body weight; HW, heart weight; CSA, cardiomyocyte cross-sectional area; PWT, posterior wall thickness; LVEDD, LV end-diastolic diameter; LVESD, LV end-systolic diameter; IVSd, LV septum, diastolic; LVPWd, LV posterior wall, diastolic; FS, fractional shortening; MAP, mean aortic pressure; SBP, systolic blood pressure; LVEDP, LV end-diastolic pressure.
All values are mean±SEM.
RALT Expression Inhibits Cardiac Hypertrophy
To investigate the role of RALT in the heart under biomechanical stress, Ang II and ISO infusion models using implanted osmotic mini-pumps were adopted. Cardiac morphology and function were examined after 4 weeks of infusion. The heart weight to body weight and lung weight to body weight ratios as well as the size of the cross-sectional area of cardiac myocytes were significantly lower in TG mice than in WT mice, whereas the ratio of liver to body weight was similar in all 4 groups of mice after Ang II or ISO infusion (Figure 2A and 2B). The increases in LV chamber dimensions and wall thickness induced by Ang II and ISO were also markedly reduced in TG mice compared with WT littermates (Table 2). Gross hearts and hematoxylin and eosin staining further confirmed the inhibitory effect of RALT on cardiac remodeling in response to Ang II and ISO stimulation (supplemental Figure 1A and 1B). We compared the expression level of several cardiac hypertrophy markers in TG and WT mice after 4 weeks of Ang II or ISO infusion and found that the expression levels of ANP, BNP, and Myh7 were increased significantly in WT mice after infusion and that these increases were markedly attenuated in TG mice (Figure 2C and 2D). Expression of Myh6 was unchanged under all tested conditions. These results indicate that RALT expression in cardiac myocytes decreases the expression of cardiac hypertrophic markers and inhibits cardiac hypertrophy induced by sustained stimulation of G-protein–coupled receptors (GPCRs).
Figure 2.
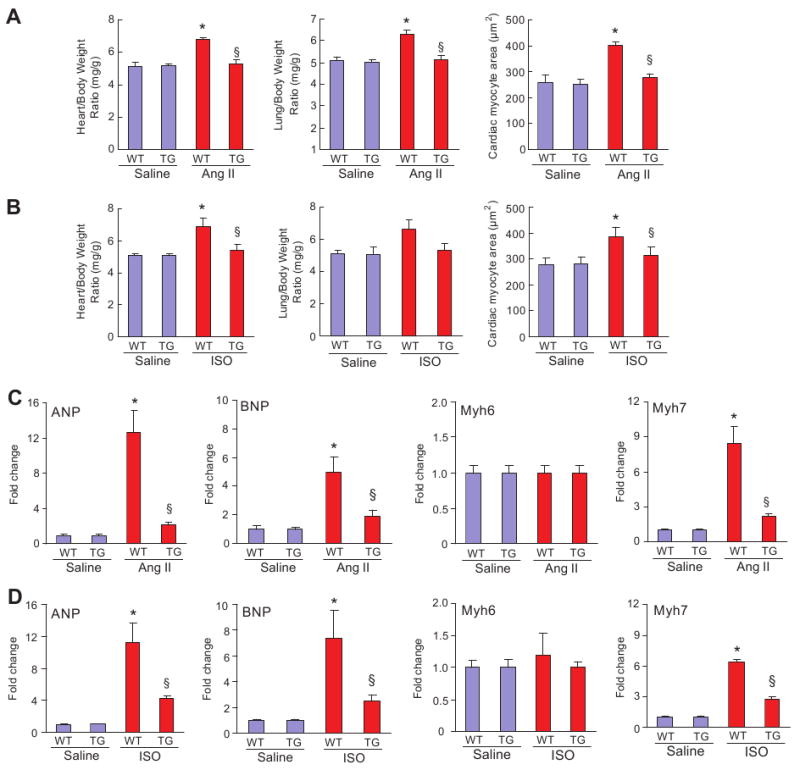
Effects of RALT on cardiac hypertrophy in vivo. A, Statistical results of heart rate to body weight ratio, lung weight to body weight ratio, and the size of myocyte cross-sectional areas (n=200 cells per group) after 4-week infusion of Ang II or ISO (n=8). Heart and lung tissues were freshly isolated from each group, and heart weight/body weight and lung weight/body weight ratios were determined. C and D, Analysis of hypertrophic markers. Total RNA was isolated from hearts of mice of the indicated groups, and expression of transcripts for ANP, BNP, Myh-7, and Myh-6 induced by Ang II or ISO infusion was determined by real-time PCR analysis. Data represent typical results of 3 to 4 different experiments as mean±SE (n=5 mice per group). *P<0.01 vs WT/saline; §P<0.01 vs WT after Ang II or ISO infusion.
Table 2. Echocardiographic and Hemodynamic Data Showed the Effects of RALT on Cardiac Hypertrophy Induced by 4 Weeks of Ang II or ISO Infusion.
| Parameter | WT-Saline | TG-Saline | WT-Ang II | TG-Ang II | WT-ISO | TG-ISO |
|---|---|---|---|---|---|---|
| No. | n=12 | n=11 | n=12 | n = 10 | n=12 | n=10 |
| BW, g | 26.7±1.5 | 27.3±1.8 | 26.9±1.5 | 26.7±1.6 | 27.3±1.6 | 27.4±1.3 |
| HR, bpm | 464 ±25 | 471 ±23 | 478±31 | 479±33 | 478±25 | 459±35 |
| PWT, mm | 1.22±0.03 | 1.23±0.04 | 2.56±0.02* | 1.47±0.03† | 2.64±0.02* | 1.55±0.03† |
| LVEDD, mm | 3.67±0.02 | 3.66±0.04 | 4.66±0.02* | 3.82±0.04† | 4.76±0.05* | 3.77±0.03† |
| LVESD, mm | 2.45±0.04 | 2.44±0.03 | 3.54±0.05* | 2.61 ±0.04† | 3.67±0.07* | 2.73±0.06† |
| IVSd, mm | 0.64±0.02 | 0.65±0.03 | 1.44±0.05* | 0.75±0.04† | 1.51±0.05* | 0.79±0.04† |
| LVPWd, mm | 0.65±0.06 | 0.65±0.04 | 1.44±0.01* | 0.91±0.05† | 1.50±0.07* | 0.88±0.06† |
| FS, % | 55.6±4.7 | 53.8±5.2 | 41.1±2.5* | 49.5±1.2† | 38.5±1.4* | 48.4±3.1† |
| SBP, mm Hg | 115.3±5.1 | 118.1±5.7 | 147.1±5.8* | 141.6±6.3* | 148.4±6.1* | 144.2±6.1* |
| MAP, mm Hg | 87.3±2.5 | 84.5±2.8 | 96.1±3.1* | 97.1±4.5* | 95.7±4.6* | 97.2±3.5* |
| LVEDP, mm H | 9.7±1.1 | 9.5±1.3 | 19.8±1.5* | 12.8±1.3† | 21.5±1.4* | 13.3±1.2† |
| LV dP/dTmax, mm Hg/s | 8765.4±449.3 | 8853.5±512.3 | 7125.1±366.1* | 8298.4±444.7† | 7045.3±496.1* | 8188.3±464.3† |
| LV dP/dTmin, mm Hg/s | −7675.4±553.7 | −7839.4±457.5 | −6002.5±503.9* | −7331.6±474.6† | −6153.9±403.6* | −7414.4±352.6† |
HR indicates heart rate; BW, body weight; PWT, posterior wall thickness; LVEDD, LV end-diastolic diameter; LVESD, LV end-systolic diameter; IVSd, LV septum, diastolic; LVPWd, LV posterior wall, diastolic; FS, fractional shortening; MAP, mean aortic pressure; SBP, systolic blood pressure; LVEDP, LV end-diastolic pressure.
P<0.01: Ang II or ISO infusion vs WT-Saline infusion;
P<0.01: TG mice vs WT mice after Ang II or ISO infusion.
RALT Expression Improves Cardiac Performance
To further determine the in vivo functional consequences of RALT overexpression, hemodynamic parameters were measured under basal conditions. TG mice showed enhanced contraction and relaxation by ejection fraction, end-systolic pressure, the first derivative of left ventricular (LV) systolic pressure rise (+dP/dt), as well as the decline (−dP/dT) compared with WT littermates (Table 2). Importantly, loading parameters such as heart rate, mean aortic pressure, and LV diastolic pressure were indistinguishable between TG and WT mice (Table 2). Thus, RALT not only inhibited the development of cardiac hypertrophy but also improved ventricular function in vivo.
RALT Expression Blocks EGFR Signaling
We next determined whether RALT is also capable of repressing EGFR signaling in cardiac myocytes in vivo. Five autophosphorylation sites have been identified in the EGFR: 3 major sites (Tyr1173, Tyr1068, and Tyr1148) and 2 minor sites (Tyr992 and Tyr1086). Using EGFR phosphorylation site-specific antibodies (Cell Signaling), we found that Ang II or ISO infusion markedly increased phosphorylation of Tyr1173 and Tyr1068 but not Tyr1086, Tyr1148, and Tyr992 (supplemental Figure 2A and 2B). Importantly, on Ang II and ISO infusion, phosphorylation of Tyr1173 and Tyr1068 was significantly inhibited (by 76% and 81% for Ang II infusion; by 72% and 78% for ISO infusion) in the hearts of TG mice compared with WT mice supplemental Figure 2A). Total EGFR level is not affected by RALT expression. To further examine the effects of RALT on EGFR activation, acute infusion of Ang II was performed. Similar results were found for the EGFR phosphorylation (78±2% and 82±3% inhibition for Tyr1173 and Tyr1068 phosphorylation, respectively). For in vitro confirmation, cardiac myocytes were infected with Ad-RALT and Ad-shRALT for 24 hours and then treated with Ang II for the indicated time. We screened 3 shRALT and chose shRALT No. 2 for further experiments because it markedly knocked down RALT expression in cardiac myocytes in response to Ang II treatment (Figure 3A). RALT overexpression almost completely blocked the phosphorylation of Tyr1173 and Tyr1068 of EGFR, whereas knockdown of RALT significantly promoted the phosphorylation of Tyr1173 and Tyr1068 of EGFR mediated by Ang II (Figure 3B).
Figure 3.
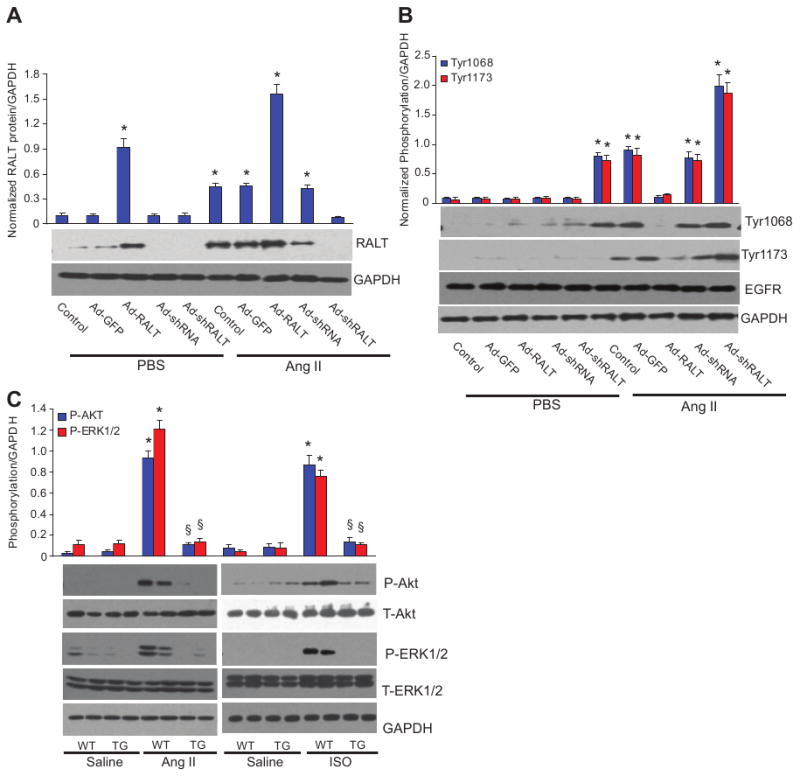
Effect of RALT on EGFR signaling pathway. A, Protein expression level of RALT after infection with Ad-RALT or Ad-shRALT (n=4). Quantification results of the protein bands were shown above the representative blots. Values are mean±SEM. *P<0.01 vs control/PBS. B, Effect of RALT on EGFR phosphorylation in cultured cardiac myocytes (n=5). Cardiac myocytes were infected with different Ad for 24 hours, cultured in serum-free medium for 24 hours, and treated with 1 μmol/L Ang II for 12 hours. Quantification results of the protein bands are shown above the representative blots. Values are mean±SEM. *P<0.01 vs control/PBS. C, Effect of RALT on ERK1/2 and AKT phosphorylation in response to chronic Ang II or ISO infusion in TG and WT mice (n=5). Results were reproducible in 3 separate experiments as mean±SEM. *P<0.01 vs WT/saline; §P<0.01 vs WT after Ang II or ISO infusion.
EGFR transactivation by Ang II has also been shown to cause activation of downstream targets mitogen-activated protein kinase (MAPK) and AKT in cardiac myocytes. Therefore, we examined the activation of MAPK and AKT signaling. Our data revealed that Ang II or ISO also induced significant activation of extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK), p38, and AKT in WT mice. However, ERK1/2 and AKT activation were blocked in TG mice (Figure 3C), and p38 and JNK1/2 phosphorylation levels were unchanged in WT and TG mice (supplemental Figure 2C). The reason for selective inhibition of ERK1/2 and AKT activity is not clear and needs further investigation. The total levels of ERK, p38, JNK, or AKT are unchanged in all groups. Ang II or ISO treatment led to a significant increase of phosphorylation of ERK downstream targets glycogen synthase kinase-3β, mTOR, and FOXO1, as well as AKT downstream targets ELK and p90RSK phosphorylation in WT mice but not in mice with RALT overexpression (supplemental Figure 2D and 2E).
RALT Expression Protects Against Fibrosis In Vivo and Inhibits Collagen Synthesis In Vitro
Heart sections from both TG and WT mice with or without Ang II or ISO infusion were stained with Picrosirius red to detect fibrosis. In both groups, collagen accumulated in the heart after 4 weeks of Ang II or ISO infusion. The interstitial fibrosis observed in the hearts of WT mice was conspicuously reduced in TG mice (Figure 4A and 4B). Quantitative analysis also showed markedly decreased collagen volume in TG mice compared with WT mice. Consistent with this observation, we found that protein and mRNA levels of transforming growth factor-β1 (Tgfβ1), procollagen type I α1 (Col1α1), procollagen type III α1 (Col3α1), and connective tissue growth factor (Ctgf) were significantly higher in WT than TG mice after Ang II or ISO infusion (Figure 4C and 4D). To further assess the role of RALT on fibrosis, we next examined the role of RALT on collagen synthesis by measuring cellular incorporation of [3H]-proline in cultured cardiac fibroblasts. Cells were infected with Ad-RALT or Ad-shRALT for 24 hours and then serum-starved for 12 hours in 0.5% FCS and treated with 1 μmol/L Ang II for the indicated time. Overexpression of RALT inhibited Ang II–induced [3H]-proline incorporation as well as COL1A2 and CTGF promoter activities and protein expressions, whereas knockdown of RALT promoted these effects (Figure 4E). Further, pretreatment with the EGFR inhibitor AG1478 completely abolished Ang II–induced collagen synthesis and COL1A2 promoter activity (Figure 4F). More important, AG1478 completely blocked the stimulating effects of Ad-RALT shRNA infection on collagen synthesis and COL1A2 promoter activity, indicating that the inhibitory effect of RALT on fibrosis is through specifically blocking EGFR transactivation.
Figure 4.
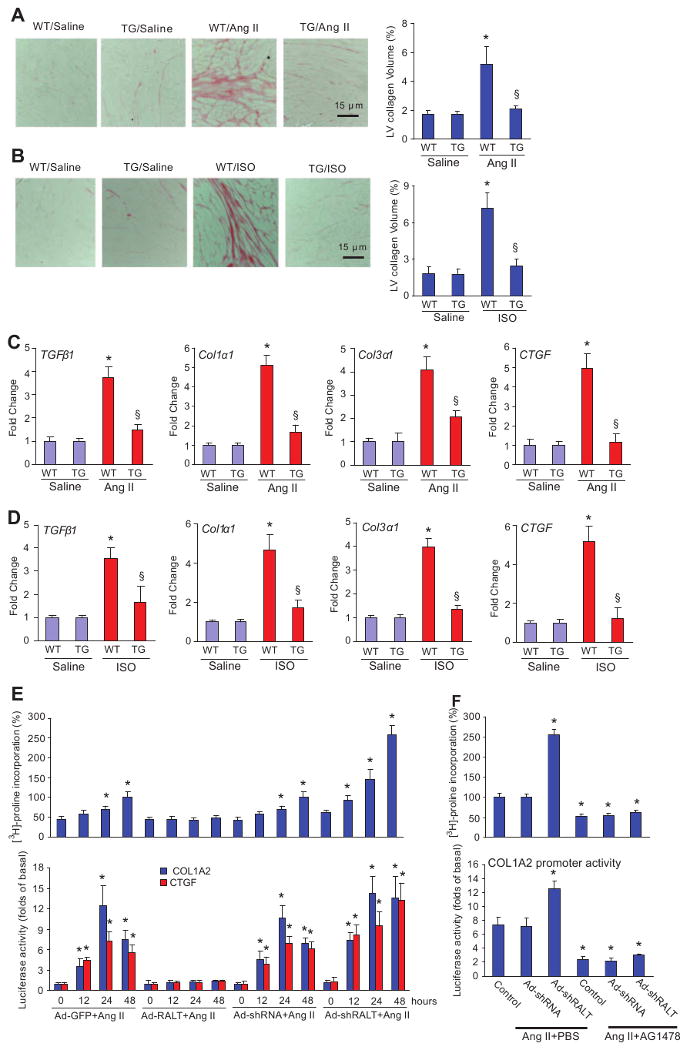
Effects of RALT on fibrosis in vivo and in vitro. A and B, Picosirius red staining on histological sections of the left ventricle was performed on indicated groups 4 weeks after Ang II or ISO infusion. Fibrotic areas from histological sections were quantified using an image analyzing system (n=5). *P<0.01 vs WT/saline; §P<0.01 vs WT after Ang II or ISO infusion. C and D, Real-time PCR analyses of Tgfβ1, Col1α1, Col3α1, and Ctgf were performed to determine mRNA expression levels in indicated groups. GAPDH was used as the normalization control. Data represent typical results of 3 different experiments as mean±SE (n=4 to 5 mice per group). *P<0.01 vs WT/saline; §P<0.01 vs WT after Ang II or ISO infusion. E, Effects of RALT on Ang II–induced [3H]-proline incorporation and the promoter activities of COL1A2 and CTGF. Cardiac fibroblasts were infected with Ad-RALT, Ad-shRALT, Ad–green fluorescent protein (Ad-GFP), or Ad-shRNA for 24 hours and then incubated with 1 μmol/L Ang II for the indicated time to observe [3H]-proline incorporation and promoter activity. *P<0.01 vs Ad-GFP infection alone. F, Effects of AG1478 on Ang II–induced [3H]-proline incorporation and the promoter activity of COL1A2. Cardiac fibroblasts were infected with Ad-shRALT or Ad-shRNA for 24 hours, treated with 25 μmol/L AG1478 for 60 minutes, and then incubated with 1 μmol/L Ang II for the indicated time to observe [3H]-proline incorporation and promoter activity. *P<0.01 vs control/Ang II+PBS. [3H]-proline incorporation and luciferase assay were performed as described in Materials and Methods.
RALT Expression Inhibits Inflammatory Response but not Apoptosis
To determine whether expression of RALT prevents the inflammatory responses in the hearts, macrophage infiltrates was characterized by immunohistochemical analyses. LV sections from TG and WT mice were immunostained with Mac-3 antibody. The numbers of Mac-3–positive cells were increased in the hearts of WT mice after 4 weeks of Ang II or ISO infusion compared with the WT-saline mice, and such increase was markedly inhibited in TG mice (Figure 5A), indicating that RALT overexpression decreases the increase of the number of macrophages mediated by Ang II or ISO. Additional studies showed that TG mice have significantly lower tumor necrosis factor-α, interleukin-1β, monocyte chemoattractant protein-1, and mRNA levels than WT mice (supplemental Figure 3A; Figure 5B and 5C). Our study also demonstrated that forced expression of RALT blocked cytokines tumor necrosis factor-α and interleukin-1β protein expression induced by Ang II in cultured cardiac myocytes, whereas knockdown of RALT promoted Ang II effects (supplemental Figure 3B). Interestingly, AG1478 also significantly inhibited tumor necrosis factor-α and interleukin-1β protein expression mediated by Ang II and blocked the effects of Ad-RALT shRNA infection on these cytokines (supplemental Figure 3B). These results indicate that RALT attenuates cytokine induction and abolishes the inflammatory response by specifically blocking EGFR signaling. In addition, RALT has been shown to promote apoptosis in cardiac myocytes. Therefore, we performed TUNEL assays on sections of cardiac tissues from TG and control mice stimulated with Ang II or ISO. The fraction of apoptotic cells versus total cells was comparable in TG and control mice (supplemental Figure 4A and 4B). In addition, the amounts of cleaved caspase-3/8 as well as the protein expression of Bax and Bcl-2 were not different between TG mice and WT mice (supplemental Figure 4C). This suggested that the decreased cardiac fibrosis and hypertrophy in the TG mice was not caused by decreased cardiomyocyte apoptosis.
Figure 5.

Effect of RALT on inflammation in vivo and in vitro. A, Quantitative analysis showing the number of Mac-3–positive cells in the hearts of WT and TG mice (n=5). *P<0.01 vs WT/saline. §P<0.01 vs WT after Ang II or ISO infusion. B and C, Real-time PCR analysis of tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), and monocyte chemoattractant protein-1 (MCP-1) mRNA expression in the myocardium obtained from indicated groups after 4-week Ang II or ISO infusion (n=5). Values are mean±SE. Results were reproducible in 3 separate experiments. Each assay was performed in triplicate. *P<0.01 vs WT/saline; P<0.01 vs WT after Ang II or ISO infusion.
Discussion
The present study demonstrates that cardiac-specific expression of RALT protects against LV dysfunction, maladaptive hypertrophy, dilatation, inflammation, and fibrosis in response to stimulation of GPCRs through attenuating EGFR transactivation and EGFR-dependent ERK and AKT pathways. These novel findings suggest that cardiac-inhibiting EGFR is critically important in protecting the heart from GPCR stimulation. To our knowledge, these data provide the first direct evidence that RALT exerts a critical role in protecting the heart against hypertrophy and contractile dysfunction.
The GPCR superfamily comprises 7 transmembrane-spanning receptors that transduce extracellular stimuli into cellular responses via classical G-protein signaling mechanisms.16,17 Stimulation of certain GPCRs such as β-adrenoceptors, Ang II, and endothelin-1 can induce cardiomyocyte growth and accumulation of extracellular matrix in the heart.18 Transactivation of EGFR is a major mechanism by which GPCRs influence growth-related signaling pathways. Once activated, EGFRs serve as a docking site for Grb2/Shc/Sos complexes and transducer 2 pathways: the phosphatidylinositol 3-kinase/AKT cascade, which leads to cellular metabolism, growth, and survival, and the Ras/Raf/ERK pathway, which leads to cellular growth, hypertrophy, and inflammation.19 Kagiyama et al8 reported that EGFR activation is required for Ang II–mediated hypertension and LV, both of which are attenuated when rats are treated with antisense oligodeoxynucleotide to EGFRs. These studies suggest that molecular targets that block EGFR signaling would inhibit cardiac growth and improve cardiac function.
RALT was cloned while searching for hepatic genes induced by glucocorticoids.20 Expression of RALT is regulated by a variety of mitogenic stimuli and stresses.21 Recent studies demonstrated that RALT has a negative effect on EGFR signaling, suggesting that RALT may attenuate cardiac hypertrophy. Consistent with this notion, we found that GPCR-induced cardiac hypertrophy and dysfunction were attenuated by cardiac forced expression of RALT. The biochemical mechanisms by which RALT functions in the heart remain unclear. Recent evidence demonstrated that RALT binds directly to EGFR Tyr kinases and reduces their autophosphorylating activity.11,12 Consistent with these findings, we found that cardiac-forced expression of RALT markedly attenuated EGFR autophosphorylating activity. Five autophosphorylation sites have been identified in the EGFR: 3 major sites (Tyr1173, Tyr1068, and Tyr1148) and 2 minor sites (Tyr992 and Tyr1086). Using EGFR site-specific and phosphospecific antibodies, we further demonstrated that RALT specifically attenuated Tyr1173 and Tyr1068 phosphorylation sites of EGFR. This may account for the RALT-dependent suppression of EGFRs and EGFR-dependent ERK and AKT activation because these pathways depend on the binding of Src homology 2 domain–containing proteins to autophosphorylated receptor Tyr kinases.
EGFRs are required for cardiac growth under basal conditions.22,23 Forced cardiac RALT expression has a negligible effect on the normal heart but not under chronic stimulation of GPCRs. In the adult heart, we did not find an appreciable increase in cell death with RALT overexpression, which argues for a different role of EGFRs in the regulation of postnatal cardiac growth compared with its role during early cardiac morphogenesis, when it appears to be essential. We propose that in the context of the postnatal adult myocardium, EGFRs may play an important role in either the maintenance or induction of cardiac growth. EGFRs may maintain basal function in the normal heart but promote cardiac growth under stress. Importantly, in contrast to the complete depletion of EGFR in knockout mice, elevated phosphorylation levels of EGFR were reduced by ≈80% (but not eliminated) in RALT TG mice in this study. The lack of physiological levels of EGFR activity during embryonic cardiac development is not comparable to the suppression of elevated EGFR activity in adult animals with cardiac hypertrophy. In this study, RALT is thought to correct the disease-related increase in EGFR activity.
Another major finding of this study is that forced cardiac expression of RALT attenuates fibrosis and inflammation in vitro and in vivo. These effects are also mediated by blocking EGFR transactivation in cardiac fibroblasts and myocytes. GPCR-mediated EGFR activation plays an important role in the proliferation in cardiac fibroblasts and fibrosis in the heart.7,24 Our data confirmed that the EGFR inhibitor AG1478 abrogated Ang II–induced collagen synthesis and collagen protein expression as well as rescued the promoted effects of inhibition of RALT, indicating that RALT inhibits fibrosis through attenuating EGFR signaling. These results are consistent with previous findings that demonstrated that GPCR-mediated stimulation of collagen synthesis requires EGFR signaling.25 Inflammation plays an important role in the progression to cardiac hypertrophy and fibrosis.26 We observed that the expression of cytokines in response to GPCR stimulation was attenuated by cardiac forced expression of RALT, indicating that RALT regulates the inflammatory response in the heart.
The MAPK and AKT signaling pathways are often activated in response to extracellular stresses such as inflammation or oxidative stress and have been shown to contribute to cardiac hypertrophy and heart failure.27,28 The increased myocardial stress after Ang II or ISO infusion in the present study, associated with activating phosphorylation of p38, JNK1/2, ERK1/2, AKT, and FOXOs and inactivating phosphorylation of glycogen synthase kinase-3β, is consistent with previous reports.29 The phosphorylation decreases of ERK1/2 and AKT as well as ERK1/2- and AKT-dependent signaling as a result of RALT probably contributed to the lesser degrees of inflammation, fibrosis, and cardiac myocyte hypertrophy in the RALT TG mice.
In conclusion, our data demonstrate that cardiac-forced expression of RALT protects against cardiac dysfunction, dilatation, inflammation, and fibrosis in response to hypertrophic stimuli by antagonizing EGFR signaling in vitro and in vivo. These observations may have significant implications for development of novel strategies for treatment of cardiac hypertrophy through targeting of the EGFR signaling pathway. Additional studies are necessary to attain a better understanding of the effects of RALT on cardiac function and the effects of EGFR inhibition in animal models of cardiac hypertrophy.
Perspectives
Heart disease is one of the most common causes of death in the world. Cardiac hypertrophy is an adaptive response to compensate for a decreased cardiac output and may occur as a result of a variety of stimuli, including myocardial infarction, hypertension, and endocrine disorders. Because cardiac hypertrophy is a highly complex disease that results from an interaction of genetic, physiological, and environmental factors, many genes and their products participated in its pathogenesis through different signaling. Therefore, the discovery of antihypertrophic novel genes is important for preventing cardiac hypertrophy and heart failure. In this study, we demonstrated that cardiac-specific expression of rat RALT protects against ventricular hypertrophy, fibrosis, and dysfunction that occur in the heart exposed to chronic Ang II or ISO infusion. This study also identifies that EGFR-dependent signaling is targeted by RALT, and that may explain the antihypertrophic effects of RALT. Based on the results of the present study, we propose that cardiac-specific overexpression of RALT protein level should be considered a therapeutic target for the treatment or prevention of cardiac hypertrophy and heart failure worthy of additional validation and investigation.
Supplementary Material
Supplemental Figure 1. The effects of RALT on cardiac hypertrophy in vivo (A) Gross hearts and H&E staining of 4 weeks of Ang II-infused mice. (B) Gross hearts and H&E staining of 4 weeks of ISO-infused mice.
Supplemental Figure 2. The effect of RALT on EGFR signaling pathway (A) The effect of RALT on the autophosphorylation of EGFR in Thr 1068 and Thr 1173 sites in response to chronic Ang II or ISO stimulation in TG and WT mice (n=5). Upper, Statistical result; bottom, representative Western blots. *P<0.01 vs WT/saline. §P<0.01 vs WT after Ang II or ISO infusion. (B) The effect of RALT on the autophosphorylation of EGFR in Tyr 1086, Tyr 1148 and Tyr 992 sites in response to chronic Ang II or ISO stimulation in TG and WT mice (n=5). (C) The effect of RALT on p38 and JNK phosphorylation in response to chronic Ang II or ISO infusion in TG and WT mice (n=5). (D) The effect of RALT on GSK3β, mTOR and FOXO1 as well as ELK, p90RSK (n=5). The results were reproducible in three separate experiments. (E) The statistical results for GSK3β, mTOR and FOXO1 as well as ELK, p90RSK phosphorylation. The results were reproducible in three separate experiments as mean±SEM. *P<0.01 vs WT/saline. §P<0.01 vs WT after Ang II or ISO infusion.
Supplemental Figure 3. The effects of RALT on inflammation (A) TNF-α, IL-1β and MCP-1 protein expression in response to chronic infusion of Ang II and ISO (n=4). Upper, Statistical result; bottom, representative Western blots. *P<0.01 vs WT/saline. §P<0.01 vs WT after Ang II or ISO infusion. (B) The effects of AG1478 on TNF-α and IL-1β protein levels in cultured cardiac myocytes from indicated groups (n=4). The results were reproducible in three separate experiments. *P<0.01 vs control/Ang II+PBS.
Supplemental Figure 4. The effects of RALT on apoptosis (A) Representative images of TUNEL staining from indicated groups. (B) TUNEL positive cells from histological sections were quantified (n=5). *P<0.01 vs WT/saline. (C) Western blot analysis of cleaved caspase-3 and caspase-8, Bcl-2 and Bax protein expression in response to chronic infusion of Ang II and ISO (n=5).
Acknowledgments
Sources of Funding: This work was supported by grants 30600337 and 30770875 from National Natural Science Foundation of China (X.-C.Y.).
Footnotes
Disclosures: None.
References
- 1.Dorn GW., II The fuzzy logic of physiological cardiac hypertrophy. Hypertension. 2007;49:962–970. doi: 10.1161/HYPERTENSIONAHA.106.079426. [DOI] [PubMed] [Google Scholar]
- 2.Reudelhuber TL, Bernstein KE, Delafontaine P. Is angiotensin II a direct mediator of left ventricular hypertrophy? Time for another look. Hypertension. 2007;49:1196–1201. doi: 10.1161/HYPERTENSIONAHA.106.075085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Epstein JA, Parmacek MS. Recent advances in cardiac development with therapeutic implications for adult cardiovascular disease. Circulation. 2005;112:592–597. doi: 10.1161/CIRCULATIONAHA.104.479857. [DOI] [PubMed] [Google Scholar]
- 4.Frey N, Katus HA, Olson EN, Hill JA. Hypertrophy of the heart: a new therapeutic target? Circulation. 2004;109:1580–1589. doi: 10.1161/01.CIR.0000120390.68287.BB. [DOI] [PubMed] [Google Scholar]
- 5.Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signaling pathways. Nat Rev Mol Cell Biol. 2006;7:589–600. doi: 10.1038/nrm1983. [DOI] [PubMed] [Google Scholar]
- 6.Mehta PK, Griendling KK. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol. 2007;292:C82–C97. doi: 10.1152/ajpcell.00287.2006. [DOI] [PubMed] [Google Scholar]
- 7.Zhai P, Galeotti J, Liu J, Holle E, Yu X, Wagner T, Sadoshima J. An angiotensin II type 1 receptor mutant lacking epidermal growth factor receptor transactivation does not induce angiotensin II–mediated cardiac hypertrophy. Circ Res. 2006;99:528–536. doi: 10.1161/01.RES.0000240147.49390.61. [DOI] [PubMed] [Google Scholar]
- 8.Kagiyama S, Eguchi S, Frank GD, Inagami T, Zhang YC, Phillips MI. Angiotensin II–induced cardiac hypertrophy and hypertension are attenuated by epidermal growth factor receptor antisense. Circulation. 2002;106:909–912. doi: 10.1161/01.cir.0000030181.63741.56. [DOI] [PubMed] [Google Scholar]
- 9.Kagiyama S, Qian K, Kagiyama T, Phillips MI. Antisense to epidermal growth factor receptor prevents the development of left ventricular hypertrophy. Hypertension. 2003;41:824–829. doi: 10.1161/01.HYP.0000047104.42047.9B. [DOI] [PubMed] [Google Scholar]
- 10.Ferby I, Reschke M, Kudlacek O, Knyazev P, Pantè G, Amann K, Sommergruber W, Kraut N, Ullrich A, Fässler R, Klein R. Mig6 is a negative regulator of EGF receptor-mediated skin morphogenesis and tumor formation. Nat Med. 2006;12:568–573. doi: 10.1038/nm1401. [DOI] [PubMed] [Google Scholar]
- 11.Xu D, Makkinje A, Kyriakis JM. Gene 33 is an endogenous inhibitor of epidermal growth factor (EGF) receptor signaling and mediates dexamethasone-induced suppression of EGF function. J Biol Chem. 2005;280:2924–2933. doi: 10.1074/jbc.M408907200. [DOI] [PubMed] [Google Scholar]
- 12.Xu D, Patten RD, Force T, Kyriakis JM. Gene 33/RALT is induced by hypoxia in cardiac myocytes, where it promotes cell death by suppressing phosphatidylinositol 3-kinase and extracellular signal-regulated kinase survival signaling. Mol Cell Biol. 2006;26:5043–5054. doi: 10.1128/MCB.02387-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li HL, She ZG, Li TB, Wang AB, Yang Q, Wei YS, Wang YG, Liu DP. Overexpression of myofibrillogenesis regulator-1 aggravates cardiac hypertrophy induced by angiotensin II in mice. Hypertension. 2007;49:1399–1408. doi: 10.1161/HYPERTENSIONAHA.106.085399. [DOI] [PubMed] [Google Scholar]
- 14.Dubey RK, Gillespie DG, Jackson EK. Adenosine inhibits collagen and protein synthesis in cardiac fibroblasts: role of A2B receptors. Hypertension. 1998;31:943–948. doi: 10.1161/01.hyp.31.4.943. [DOI] [PubMed] [Google Scholar]
- 15.Li HL, Huang Y, Zhang CN, Liu G, Wei YS, Wang AB, Liu YQ, Hui RT, Wei C, Williams GM, Liu DP, Liang CC. Epigallocathechin-3 gallate inhibits cardiac hypertrophy through blocking reactive oxidative species-dependent and -independent signal pathways. Free Radical Biol Med. 2006;40:1756–1775. doi: 10.1016/j.freeradbiomed.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 16.Shah BH, Catt KJ. Matrix metalloproteinase-dependent EGF receptor activation in hypertension and left ventricular hypertrophy. Trends Endocrinol Metab. 2004;15:241–243. doi: 10.1016/j.tem.2004.06.011. [DOI] [PubMed] [Google Scholar]
- 17.Shah BH, Catt KJ. A central role of EGF receptor transactivation in angiotensin II–induced cardiac hypertrophy. Trends Pharmacol Sci. 2003;24:239–244. doi: 10.1016/S0165-6147(03)00079-8. [DOI] [PubMed] [Google Scholar]
- 18.Piiper A, Zeuzem S. Receptor tyrosine kinases are signaling intermediates of G-protein–coupled receptors. Curr Pharm Des. 2004;10:3539–3545. doi: 10.2174/1381612043382936. [DOI] [PubMed] [Google Scholar]
- 19.Takashima S. Pharmacological role of HB-EGF shedding by angiotensin II in cardiac myocytes. Nippon Yakurigaku Zasshi. 2004;124:69–75. doi: 10.1254/fpj.124.69. [DOI] [PubMed] [Google Scholar]
- 20.Fiorentino L, Pertica C, Fiorini M, Talora C, Crescenzi M, Castellani L, Alemà S, Benedetti P, Segatto O. Inhibition of ErbB-2 mitogenic and transforming activity by RALT, a mitogen-induced signal transducer which binds to the ErbB-2 kinase domain. Mol Cell Biol. 2000;20:7735–7750. doi: 10.1128/mcb.20.20.7735-7750.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fiorini M, Ballarò C, Sala G, Falcone G, Alemà S, Segatto O. Expression of RALT, a feedback inhibitor of ErbB receptors, is subjected to an integrated transcriptional and post-translational control. Oncogene. 2002;21:6530–6539. doi: 10.1038/sj.onc.1205823. [DOI] [PubMed] [Google Scholar]
- 22.Iwamoto R, Yamazaki S, Asakura M, Takashima S, Hasuwa H, Miyado K, Adachi S, Kitakaze M, Hashimoto K, Raab G, Nanba D, Higashiyama S, Hori M, Klagsbrun M, Mekada E. Heparin-binding EGF-like growth factor and ErbB signaling is essential for heart function. Proc Natl Acad Sci U S A. 2003;100:3221–3226. doi: 10.1073/pnas.0537588100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen B, Bronson RT, Klaman LD, Hampton TG, Wang JF, Green PJ, Magnuson T, Douglas PS, Morgan JP, Neel BG. Mice mutant for Egfr and Shp2 have defective cardiac semilunar valvulogenesis. Nat Genet. 2000;24:296–299. doi: 10.1038/73528. [DOI] [PubMed] [Google Scholar]
- 24.Chen CH, Cheng TH, Lin H, Shih NL, Chen YL, Chen YS, Cheng CF, Lian WS, Meng TC, Chiu WT, Chen JJ. Reactive oxygen species generation is involved in epidermal growth factor receptor transactivation through the transient oxidization of Src homology 2-containing tyrosine phosphatase in endothelin-1 signaling pathway in rat cardiac fibroblasts. Mol Pharmacol. 2006;69:1347–1355. doi: 10.1124/mol.105.017558. [DOI] [PubMed] [Google Scholar]
- 25.Hardie WD, Davidson C, Ikegami M, Leikauf GD, Le Cras TD, Prestridge A, Whitsett JA, Korfhagen TR. EGF receptor tyrosine kinase inhibitors diminish transforming growth factor-alpha-induced pulmonary fibrosis. Am J Physiol. 2008;294:L1217–L1225. doi: 10.1152/ajplung.00020.2008. [DOI] [PubMed] [Google Scholar]
- 26.Li HL, Zhuo ML, Wang D, Wang AB, Cai H, Sun LH, Yang Q, Huang Y, Wei YS, Liu PP, Liu DP, Liang CC. Targeted cardiac overexpression of A20 improves left ventricular performance and reduces compensatory hypertrophy after myocardial infarction. Circulation. 2007;115:1885–1894. doi: 10.1161/CIRCULATIONAHA.106.656835. [DOI] [PubMed] [Google Scholar]
- 27.Muslin AJ. MAPK signaling in cardiovascular health and disease: molecular mechanisms and therapeutic targets. Clin Sci. 2008;115:203–218. doi: 10.1042/CS20070430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li HH, Willis MS, Lockyer P, Miller N, McDonough H, Glass DJ, Patterson C. Atrogin-1 inhibits Akt-dependent cardiac hypertrophy in mice via ubiquitin-dependent coactivation of Forkhead proteins. J Clin Invest. 2007;117:3211–3223. doi: 10.1172/JCI31757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sugden PH. Signaling pathways in cardiac myocyte hypertrophy. Ann Med. 2001;33:611–622. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. The effects of RALT on cardiac hypertrophy in vivo (A) Gross hearts and H&E staining of 4 weeks of Ang II-infused mice. (B) Gross hearts and H&E staining of 4 weeks of ISO-infused mice.
Supplemental Figure 2. The effect of RALT on EGFR signaling pathway (A) The effect of RALT on the autophosphorylation of EGFR in Thr 1068 and Thr 1173 sites in response to chronic Ang II or ISO stimulation in TG and WT mice (n=5). Upper, Statistical result; bottom, representative Western blots. *P<0.01 vs WT/saline. §P<0.01 vs WT after Ang II or ISO infusion. (B) The effect of RALT on the autophosphorylation of EGFR in Tyr 1086, Tyr 1148 and Tyr 992 sites in response to chronic Ang II or ISO stimulation in TG and WT mice (n=5). (C) The effect of RALT on p38 and JNK phosphorylation in response to chronic Ang II or ISO infusion in TG and WT mice (n=5). (D) The effect of RALT on GSK3β, mTOR and FOXO1 as well as ELK, p90RSK (n=5). The results were reproducible in three separate experiments. (E) The statistical results for GSK3β, mTOR and FOXO1 as well as ELK, p90RSK phosphorylation. The results were reproducible in three separate experiments as mean±SEM. *P<0.01 vs WT/saline. §P<0.01 vs WT after Ang II or ISO infusion.
Supplemental Figure 3. The effects of RALT on inflammation (A) TNF-α, IL-1β and MCP-1 protein expression in response to chronic infusion of Ang II and ISO (n=4). Upper, Statistical result; bottom, representative Western blots. *P<0.01 vs WT/saline. §P<0.01 vs WT after Ang II or ISO infusion. (B) The effects of AG1478 on TNF-α and IL-1β protein levels in cultured cardiac myocytes from indicated groups (n=4). The results were reproducible in three separate experiments. *P<0.01 vs control/Ang II+PBS.
Supplemental Figure 4. The effects of RALT on apoptosis (A) Representative images of TUNEL staining from indicated groups. (B) TUNEL positive cells from histological sections were quantified (n=5). *P<0.01 vs WT/saline. (C) Western blot analysis of cleaved caspase-3 and caspase-8, Bcl-2 and Bax protein expression in response to chronic infusion of Ang II and ISO (n=5).