

Abstract
Primary myelofibrosis (PMF) belongs to the Philadelphia-negative myeloproliferative neoplasms and is a hematological disorder caused by abnormal function of the hematopoietic stem cells. The disease manifests itself with a plethora of alterations, including anemia, splenomegaly and extramedullary hematopoiesis. Its hallmarks are progressive marrow fibrosis and atypical megakaryocytic hyperplasia, two distinctive features used to clinically monitor disease progression. In an attempt to investigate the role of abnormal megakaryocytopoiesis in the pathogenesis of PMF, several transgenic mouse models have been generated. These models are based either on mutations that interfere with the extrinsic (thrombopoietin and its receptor, MPL) and intrinsic (the GATA1 transcription factor) control of normal megakaryocytopoiesis, or on known genetic lesions associated with the human disease. Here we provide an up-to-date review on the insights into the pathobiology of human PMF achieved by studying these animal models, with particular emphasis on results obtained with Gata1low mice.
Keywords: animal model, GATA1, hematopoietic stem cell, megakaryocyte, primary myelofibrosis, transcription factor
Primary myelofibrosis (PMF) is a rare hematological malignancy, with an estimated incidence of 0.3–1.5 cases per 100,000 individuals per year [1]. PMF is a member of a family of diseases named chronic myeloproliferative disorders (MPDs) in 1951 by Dameshek [2] and very recently renamed myeloproliferative neoplasms (MPNs) [3,4]. According to the WHO, chronic MPNs are classified into Philadelphia-positive (Ph+) and Philadelphia-negative (Ph-) diseases. The first group is represented by chronic myelogenous leukemia (CML). Thanks to the discovery of the Philadelphia chromosome by Nowell and Hungerford in 1960, CML was the first cancer associated with a specific genomic aberration [5]. The subsequent characterization of the Ph t(9;22)(q34;q11) translocation in 1984 [6] and of the deriving BCR—ABL fusion gene in 1985 [7] marked the beginning of a new era in understanding the pathobiology of this disease. BCR—ABL is a powerful oncogene that results from the fusion of the BCR on chromosome 22 and the human homolog of the ABL tyrosine kinase gene on chromosome 9 [7]. The resulting BCR—ABL fusion protein displays constitutive tyrosine kinase activity that triggers several intermediate cytosolic signals overcoming the normal signaling of a variety of cytokines, including SCF [8]. The identification of BCR—ABL led to the important discovery of imatinib, a small molecule that reverses the clinical manifestations of CML by inhibiting the deregulated activity of this oncoprotein [9].
Philadelphia-negative MPNs include three classical MPDs, polycytemia vera (PV), essential thrombocythemia and PMF, as well as chronic neutrophilic leukemia, chronic eosinophilic leukemia/hypereosinophilic syndrome, mast cell diseases and other nonclassifiable MPNs [3,4]. PMF is characterized by anemia, splenomegaly, leukoerythroblastosis, abnormal trafficking of CD34+ stem/progenitor cells in the peripheral blood, multiorgan extramedullary erythropoiesis, especially involving spleen and liver, and destruction of the bone marrow structure due to progressive fibrosis, neoangiogenesis and increased bone deposits (Table 1) [1,2,10–17]. The disease begins with an asymptomatic phase, also defined as prefibrotic, which proceeds with the first manifestations (usually splenomegaly and fatigue) and culminates into the fibrotic stage (Table 1). The symptomatic stage of the disease normally has poor prognosis (life span < 5 years). The principal causes of death include infections, thrombohemorragic events and leukemic transformation. In fact, 3.9–20% of PMF patients develop lethal acute myeloid leukemia [18,19]. In addition, Ph- MPNs are clonal stem cell malignancies originating from genetic anomalies that alter the physiological response of the pluripotent hematopoietic stem/progenitor cells to growth factors, thus affecting the balance between proliferation and differentiation in these cells.
Table 1.
Clinical manifestations of primary myelofibrosis.
| Prefibrotic stage | Fibrotic stage |
|---|---|
| Non-bone marrow | |
| Mild to absent splenomegaly Mild anemia Mild thrombocytosis |
Marked splenomegaly Marked anemia Pancytopenia with tear-drop poikilocytes Extramedullary hematopoiesis |
| Bone marrow | |
|---|---|
| Hypercellularity Megakaryocytic hyperplasia Mild fibrosis |
Hypocellularity Megakaryocytic hyperproliferation and abnormal differentiation Marked fibrosis Osteosclerosis Neoangiogenesis May evolve to leukemia |
Recent studies have highlighted the presence of gain-of-function mutations in the Janus kinase-2 (JAK2), protein (JAK2 V617F, JAK2 T875N and exon 12 mutant alleles) in patients with Ph- MPNs [20–26]. JAK2 is constitutively associated with the receptors for several hematopoietic growth factors, including thrombopoietin (TPO) [27,28], erythropoietin (EPO) [29–31], granulocyte colony-stimulating factor [32] and SCF [33–35], and is, therefore, the first protein to be activated when these receptors are engaged by the corresponding growth factors. The JAK2 V617F substitution replaces valine with phenylalanine in the autoinhibitory domain of the enzyme, leading to its inability to fold back to the off-modus after activation [36,37]. The JAK2 T875N activating mutation replaces a threonine with an asparagine at position 875 of the JAK2 JH1 kinase domain [26]. Both JAK2 V617F and the JAK2 T875N, result in permanent signaling that partially explains the hypersensitivity to cytokines that characterizes hematopoietic cells from MPDs [38,39]. In addition, a set of JAK2 exon 12 mutant alleles have recently been described that induced constitutive activation of JAK—STAT signaling [40]. Furthermore, evidence linking JAK2 signaling with the onset of Ph- MPNs is provided by the identification of the TEL—JAK2 oncogene not only in patients with T- and B-cell acute lymphoblastic leukemia but also in some patients with atypical CML [41–44]. The TEL—JAK2 translocation juxtaposes the oligomerization domain of the Ets-family transcription factor TEL with the catalytic domain of JAK2. The corresponding fusion protein is constitutively activated, inducing signaling abnormalities comparable to those that arise due to the BCR—ABL oncogene [44]. However, although the JAK2 V617F mutation accounts for 90% of PV patients it is found in only 50% of PMF patients [9–14]. Within a minority (between 5–10%) of JAK2 V617F-negative PMF patients, two somatic mutations have been identified in the autoinhibitory loop of the transmembrane domain of MPL (MPL W515L and MPL W515K) [25,45,46]. The W515L, and possibly W515K, substitution leads to constitutive activation of the JAK—STAT pathway and thereby confers cytokine-independence to hematopoietic progenitor cells (HPCs) [46]. MPL is not only expressed by megakaryocytic cells but also by stem cells and early HPCs. In particular, the interaction between TPO, expressed by the bone marrow stroma, and MPL, expressed by the HPCs, has been shown to play a critical role in maintaining the homeostasis of the stem cell niche [47–50]. Thus the TPO—MPL axis plays an essential role not only in platelet development, but also in the biology of the hematopoietic stem cells [50,51].
In spite of the great body of evidence linking mutations of JAK2 signaling with PMF, the mechanism by which a single mutation can give rise to all the pleiotropic traits of PMF is still poorly understood. It is clear that PMF is the result of multiple genetic alterations accumulated as disease progresses. In fact, although PMF is mainly sporadic, familiar predisposition has been observed, indicating the importance of silent mutations that may favor accumulation of JAK2 mutations [52–55]. Furthermore, diverse chromosomal aberrations, such as del(5q), del(13q), del(20q) and trisomy(1q) (Table 2) arise during the transition of the disease to its leukemic stage [56–61]. However, because of their sporadic occurrence these mutations cannot be considered as a diagnostic karyotype. Additional genetic alterations observed in PMF are represented by high levels of expression and/or mutations of c-KIT, the gene encoding the SCF receptor, found in HPCs circulating in these patients [62–67]. However, since none of these c-Kit mutations are unique to PMF, they may also represent secondary events occurring in an already destabilized stem cell compartment (Table 2).
Table 2.
Genetic abnormalities associated with primary myelofibrosis.
The similarity between the biochemical defects induced by the JAK2 mutation and those induced by BCR—ABL had raised hope that a JAK2 inhibitor with similar effects to those exerted by imatinib on BCR—ABL might represent an effective therapy for PMF [68]. The importance of finding medications to cure the disease is stressed by the fact that the only cure available to these patients at present is allogeneic bone marrow transplantation [10–16]. Unfortunately, only a minority of PMF patients are eligible for this therapy, either because of difficulties in finding allogeneic matched donors, or for risk considerations usually linked to the advanced age of these patients. Several small-molecules capable of inhibiting the JAK2 kinase (e.g. INCB018424, XL019 and TG101348) [69], and many others that have off-target kinase-inhibitory activity (e.g., MKS-0457, CEP-701 and AT9283) have now been identified (reviewed in [69,70]). The therapeutic efficacy of many of these is being tested in ongoing Phase II clinical studies. However, as predicted on the basis of the importance of JAK2 signaling in hematopoietic cells, the preliminary results of these trials have produced significant evidence showing dose-dependent cytopenia as a major side effect of such drugs. The difficulty of finding a chemical approach to cure PMF highlights the importance of studies in animal models, not only for preclinical evaluation of efficacy and toxicity of JAK2 inhibitors, but also to better understand the pathobiological pathway of PMF in order to identify additional targets for therapy.
Megakaryocyte abnormalities as a ‘cellular signature’ of PMF
Megakaryocyte (MK) development is a tightly regulated process characterized by specific ultrastructural changes that are very similar between mice and men. The transition from promegakaryoblast to MK is associated with a progressive formation of platelet secretory granules (α-granules) and the production of an elaborate system of cisternae and tubules, known as demarcation membrane system (DMS), that spread through the cytoplasm of the cells. The DMS serves both to identify fields, or territories, from which platelets will be released via fragmentation and to increase the membrane surface necessary for the elaboration of proplatelets [71]. The composition of the α-granules is conserved between MKs and platelets. As discovered by Italiano et al., the α-granules of MKs and platelets belongs to two subtypes (Figure 1) [72]. One subtype includes proangiogenic factors, including VEGF and angiopoietin 1 (ANG1). The other subtype stores anti-angiogenic molecules, such as endostatin and thrombospondin-1 (TSP-1). Anti-angiogenic α-granules also contain von Willebrand factor (VWF), which is thought to act as a receptor to regulate the release of the α-granules’ content in response to thrombin. The corresponding proangiogenic α-granule-specific receptor has not yet been identified. However, the observation that the leukocyte receptor P-selectin and VWF are localized on different α-granules [ZINGARIELLO M ET AL., MANUSCRIPT SUBMITTED], suggests that it may be represented by P-selectin.
Figure 1. Role of MKs and platelets in the control of tissue repair in normal individuals (left) and in a patient with primary myelofibrosis (PMF; right).

Under normal conditions, mature MKs extend their protrusions through the endothelial cells of the blood vessels of the bone marrow, shedding platelets in the blood flux. MKs and platelets both contain the same two types of α-granules: those that contain proangiogenic factors (and an adhesion receptor still to be identified, probably represented by P-selectin) and those containing anti-angiogenic factors (and VWF as adhesion receptor). Upon injury, platelets are recruited to the site of damage and activated, flip-flopping their membranes and exposing the adhesion receptors on the α-granules. VWF stored in the anti-angiogenic α-granules then becomes capable of binding thrombin, triggering the dismissal of anti-angiogenic factors in the microenvironment. Later on, probably triggered by the interaction between P-selectin and neutrophils recruited in the mean time at the site of injury, proangiogenic α-granules also release their content, stimulating proliferation of multiple types of cell. Thus, sequential and concerted releases of anti- and pro-angiogenic growth factors at the inflammation site is first responsible for stilling the hemorrhage and then promotes tissue repair. Probably due to their reduced GATA1 content, MKs from PMF patients remain immature shedding reduced numbers of platelets in the bloodstream. These cells accumulate in the marrow, and, lacking VWF expression, respond poorly to stimuli that would normally induce the release of anti-angiogenic factors. Moreover, abnormal localization of P-selectin on the DMS results in the pathological process of emperipolesis, with neutrophils increasing the release of the content of stimulatory α-granules in the bone marrow microenvironment. This release stimulates stromal cells of all types (fibroblasts, endothelial cells and osteoblasts), inducing fibrosis, neo-angiogenesis and osteosclerosis, respectively.
ANG1: Angiopoietin 1; DMS: Demarcation membrane system; MK: Megakaryocyte; OPG: Osteoprogesterin; VWF: Von Willebrand factor.
Platelets are not only involved in clotting but also in inflammation and wound healing, as they represent efficient long-distance delivery units for several biologically active substances all around the body. The mechanism that mediates the release of the content of the platelet α-granules in the microenvironment in response to injury or inflammation is starting to emerge (Figure 1). It is conceivable that platelets are first trapped to the site of damage by bridges established through the platelet receptor (GPIbα), with plasma VWF and collagen exposed by the vessel injury. Tighter platelet contacts established by direct interaction between collagen and glycoprotein VI allow the platelets to be activated through locally produced thrombin. As part of this activation process, platelets flip-flop the α-granule membranes exposing the molecules present in the inner layer, including VWF, to external agents. In the early phase of the response to injury, α-granule-bound VWF binds thrombin, inducing the release of anti-angiogenic factors. At a later stage, neutrophils recruited to the inflammation sites, by interacting with P-selectin, may induce release of the content of proangiogenic α-granules (FGF, TGF-β, TGF- α osteoprotegerin [OPG] and bone morphogenic protein 4 [BMP4]) (Figure 1) [73–81] inducing tissue repair.
One of the diagnostic features of PMF is the presence of MKs abnormal in both number and morphology in bone marrow biopsies. These alterations include an increased proliferation with delayed maturation, a normal expression level but abnormal localization of P-selectin on the DMS and a reduced expression of VWF (Figure 1). In addition, in spite of the fact that the GATA1 gene is normal, MKs from PMF patients express reduced levels of GATA1 [82], a transcription factor that controls the expression of many MK-specific genes, hence the reduced levels of VWF expression [83]. The abnormal P-selectin localization on the DMS of the MKs triggers a pathological form of neutrophil emperipol esis within these cells [84,85]. Emperipolesis is defined as the passive passage of a cell through the cytoplasm of another cell that, under normal circumstances, does not alter the biological functions of either of the two cells involved. As a result of their size and of the tubular structure of the cytoplasm, emperipolesis of cells of many types occurs within MKs with a frequency of 1–2% [84]. In PMF patients, however, due to the high amount of P-selectin on the DMS, more than 30% of the MKs are engaged in emperipolesis with as many as three to five neutrophils embedded in each one of them. In addition, the process actively changes the properties of the two cells types that fuse their membranes, triggering the release of proteolytic enzymes by neutrophils and proangiogenic factors by α-granules, first within the cytoplasm of the MKs and then in the microenvironment [85]. These proteolytic enzymes and proangiogenic factors are then responsible for the development of the other traits of the disease, such as fibrosis (TGF-β, FGF), neoangiogenesis (VEGF, ANG1) and osteosclerosis (OPG, BMP4) (Figure 1) [74,76–86].
Mouse models of myelofibrosis
The crucial role played by MK abnormalities in promoting PMF is confirmed by the observation that the phenotype of mice carrying all the genetic mutations in the extrinsic and intrinsic control of MK differentiation, which induce MK alterations similar to those found in PMF, includes the development of myelofibrosis (Figure 2 & Table 3).
Figure 2. Unifying pathobiological pathways leading to the abnormal MK development in mouse models of myelofibrosis and in primary myelofibrosis (PMF) patients.

TPO exerts the major extrinsic control on megakaryocytopoiesis. By interfering with TPO signaling it is possible to reproduce in mice many of the MK alterations observed in PMF patients, including reduced levels of GATA1 expression. In addition to the MK abnormalities, mice either overexpressed TPO or exhibited hyperactive TPO signals that led to myelofibrosis. Hyperactive TPO signals can be represented by activating mutations of MPL or of its downstream effector JAK2, or even genetic deletion of the gene (Lnk) encoding the protein that inhibits MPL transport to the cell surface. On the other hand, downregulation of GATA1 and BACH1 expression, two genes encoding transcription factors exerting an intrinsic control on MK development, induced MK abnormalities in mice similar to those observed in PMF and led to the development of myelofibrosis. The major consequence of the blockade in MK development induced by all these mutations is that the P-selectin is delocalized on the demarcation membrane system, which triggers an abnormal process of neutrophil emperipolesis that increases the proteolytic and growth factor milieu of the microenvironment.
MK: Megakaryocyte; MPL: Monophosphoryl lipid; TPO: Thrombopoeitin;
VWF: von Willebrand factor.
Table 3.
Available mouse models for primary myelofibrosis.
| Mouse model | Mutated in primary myelofibrosis patients |
Mouse phenotype/application | Ref. |
|---|---|---|---|
|
TPOhigh Systemic administration Transplantation Transgenic expression |
No | Decrease in hematocrit, pancytopenia, extramedullary hematopoiesis, osteosclerosis, fibrosis/genetic complementation studies |
[76,108,109] [111–117] [118,119] |
| MPL W515L | Yes | Splenomegaly, extramedullary hematopoiesis, thrombocytosis, megakaryocytic hyplasia, bone marrow fibrosis/ preclinical testing of JAK2 inhibitors |
[46] |
| JAK2 V617F Transplantation Transgenic expression |
Yes | Polycythemia vera-like disease, erythroid, megakaryocytic, and granulocytic hyperplasia in the bone marrow and spleen, splenomegaly and reduced level of erythropoietin and thrombopoietin in the plasma/preclinical testing of JAK2 inhibitors |
[88,89] |
| Lnk-/- | No | Splenomegaly, leukocytosis, thrombocytosis, MK hyperplasia, extramedullary hematopoiesis, increased response of HPCs to cytokines |
[91–96] |
| Osteoprotegerin | No | Osteopetrosis, splenomegaly and extramedullary hematopoiesis | [75,119] |
| TEL—JAK2 | No (ALL, CML) |
Myelo- and lympho-proliferative disorder. Myelofibrosis | [42,43,127] |
| TEL—PDGFβR | No | Hyperproliferation of neutrophils, abnormal MKs, splenomegaly, extramedullary hematopoiesis |
[128–130] |
| GATA1 low | No | Severe anemia and thrombocytopenia, extramedullary hematopoiesis, MK hyperproliferation and abnormal differentiation, bone marrow fibrosis/pathobiology of HPC trafficking and extramedullary hematopoiesis |
[97,146,149,150] |
| GATA1 0.5 | No | Erythroblastic leukemia, no myelofibrosis | [190] |
| BACH1 | No | Aberrant MKs, thrombocytopenia, myelofibrosis | [92] |
| Trisomy 21 | Yes | Accumulation of abnormal MKs in bone marrow and spleen, profound thrombocytosis and myelofibrosis. Abnormal HPC compartment |
[194] |
| RARγ-/- | No | Age-dependent myeloproliferation, extramedullary hematopoiesis, splenomegaly, bone loss |
[99] |
| Mx-Cre Rbfl/fl | No | Myeloproliferation, HSCs mobilization, extramedullary hematopoiesis | [100] |
| Op/opFlt1TK-/- | No | Bone marrow fibrosis due to osteoclast deficiency, extramedullary hematopoiesis |
[199] |
ALL: Acute lymphoblastic leukemia; CML: Chronic myeloid leukemia; HPC: Hematopoietic progenitor cell; HSC: Hematopoietic stem cell; MK: Megakaryocyte; MPL: Monophosphoryl lipid.
The first models established to study the pathobiological mechanism of myelofibrosis were represented by mice with altered TPO/MPL expression (TPOhigh mice) (Table 3). Upregulation of TPO expression in mice induced MK abnormalities similar to those observed in PMF patients, including reduced levels of GATA1 expression (Figure 3). The mechanism that links high levels of TPO with reduced Gata1 expression is unclear. The observation that TPO treatment of TPO-responsive cell lines [87] does not alter Gata1 mRNA expression, while reducing in a time- and concentration-dependent fashion the levels of GATA1 protein (ARM) (Figure 3) [MIGLIACCIO AR, UNPUBLISHED DATA], suggests that TPO may reduce the expression of GATA1 in the MKs from TPOhigh mice via a post-transcriptional mechanism. It also suggests that constitutively activated elements downstream of TPO signaling, such as MPL and JAK2 mutations, might reduce GATA1 expression in MKs from PMF patients with a similar post-transcriptional mechanism [82]. Additional animal models may be considered as TPOhigh mice because they mimic reinforced cytokine signal and are represented by mice bearing the mutated JAK2 V617F [88,89] or MPL W515L [46,90] gene, or lacking the Lnk gene (Lnk-/- mice; Lnk encodes a protein that retains MPL in the cell cytoplasm [91–96]). All these animal models develop a disease similar in several ways to that developed by TPOhigh mice (see later).
Figure 3. Megakaryocytes (MKs) from TPOhigh and Gata1low mice express similar reduced levels of GATA1.

(A) Spleen sections from TPOhigh , wild-type and GATA1low mice stained with an anti-GATA1 antibody. Similar numbers of clusters of MKs negative for GATA1 staining are present in sections from TPOhigh and Gata1low mice. By contrast, the isolated MKs observed in the section of wild-type mice are positive for GATA1. (B) Effect of TPO stimulation on the levels of GATA1 protein expressed by a TPO-dependent MK cell line (32DTPO) [83]. 32D cells were stimulated with different amounts of TPO for the indicated time periods. Untreated GM979 cells were used as control. Protein lysates were electrophoretically separated on SDS PAGE followed by western blotting and immunostained with antibodies against either GATA1 or FOG1 (as control). Equal amount of protein loading was monitored by β-tubulin immunostaining. As shown, the observed decrease in GATA1 signal upon TPO stimulation is dose dependent and probably due to post-transcriptional events, since comparable levels of GATA1 mRNA were detected in all the experimental points investigated (ARM, results not shown).
(C) MKs from wild-type and Gata1low mice were isolated by sorting and the expression of GATA1 mRNA measured by quantitative reverse transcriptase PCR. As expected, the presence of the hypomorphic Gata1low mutation significantly reduced the GATA1 mRNA levels in MKs.
TPO: Thrombopoietin.
Modified with permission from [76].
A second model of myelofibrosis was generated by deleting the regulatory sequences of the Gata1 gene that guides its expression in MKs (Gata1low mice) (Figure 2) (Table 3) [97]. Since the levels of GATA1 in MKs of TPOhigh and Gata1low mice are comparably low (Figure 3), it is not surprising that the two animal models develop similar MK abnormalities and myelofibrosis. The most important phenotypic difference between the two models is represented by the time required for the disease to manifest itself. TPOhigh mice rapidly develop a complete myelofibrosis trait, while Gata1low mice develop the disease in an orderly sequence of events that span over more than 2 years [97]. For this reason, the two systems are suited to investigating different aspects of the pathobiology of myelofibrosis in mice. The rapidity with which TPOhigh mice develop the disease makes this model particularly suited for genetic complementation studies such as those that have proved the essential role played by TGF-β and OPG in the disruption of the bone marrow structure. Alternately, the progressive development of myelofibrosis in Gata1low mice makes this model specifically suited for studying the pathobiological mechanisms that determine increased stem/progenitor cell trafficking and extramedullary hematopoiesis. Additional genetic mutations that, like the Gata1low mutation, target genes that regulate gene expression in MKs (BACH1) also induce myelofibrosis in mice [98].
The third most recent mouse model of myelofibrosis has been generated by manipulating genes that control the ability of the marrow microenvironment to support hematopoiesis [99,100]. In this model, development of myelofibrosis is not due to a stem cell defect but rather to a microenvironmental defect. As such, the MKs of these mice are normal. However the microenvironment is not normal and presents many of the alterations characteristic of the microenvironment of PMF patients. This last model highlights the fact that PMF is a disease of abnormal interactions between the stem cells and their niches.
TPO high mouse models
Thrombopoietin is the major cytokine controlling growth, develop ment and turnover of MKs, and is thus responsible for the regulation of platelet homeostasis in the blood [101–107]. This soluble factor is mainly produced by the liver and kidney [103,104] and is strongly induced by drops in platelet numbers or by increases in IL-6 during acute inflammatory stress [105,106]. The first observation that high TPO doses may lead to the development of myelofibrosis was the occasional result of an animal study to monitor the side effects of the long-term administration of recombinant TPO, utilized as a drug to rescue platelet deficiency in individuals with hemorrhagic issues [108]. To date, several approaches have been used to generate TPOhigh mouse models of myelofibrosis: repeated systemic administration, bone marrow reconstitution with stem cells virally transduced to overexpress the human TPO gene and transgenic mice overexpressing the human TPO gene in liver cells (Table 3).
Systemic administration
Increasing amounts of a truncated and polyethylene glycol-linked version of the TPO protein (PEG-rHuMGDF) have been injected on a daily basis into BALB/c mice. After 2 weeks all the treated animals developed anemia, splenomegaly and extramedullary hematopoiesis, with more drastic effects at higher TPO concentrations. Reversible bone marrow fibrosis was observed only at the highest TPO doses [108]. Transient increases in marrow MKs and platelet counts were also observed in normal rats injected with PEG-rHuMGDF at suprapharmacologic doses for 5 days. Emerging myelofibrosis normalized by day 20 and was paralleled by increased TGF-β levels in extracellular fluids of bone marrow, and in plasma and platelets [109].
Transplantation approach
Different mouse lines have been generated using bone marrow transplantation. Wild-type HSCs from lethally irradiated recipient mice were replaced with donor HSCs previously transduced to overexpress the TPO murine gene, by the use of either adenoviral [110–114] or retroviral vectors [115,116]. In spite of some strain-variability in results observed with adenoviral vectors [110–114], two retrovirally-based TPOhigh models developed myelofibrosis, the severity of which was positively correlated with the levels of TPO expressed in the blood of the animals (30 ng/ml vs 1000 ng/ml). At high plasma levels, mice presented a two-step disease, which strongly resembled the course of the human disorder [115]. The first phase occurred 7–9 weeks after bone marrow transplantation and was characterized by decreased hematocrit and high numbers of platelets and leukocytes in peripheral blood. The bone marrow presented hypoplastic MKs, erythroblasts and granulocytes. The second phase took place 10 weeks after bone marrow transplantation. In this stage the level of blood cells dropped to pancytopenia. Extramedullary hematopoiesis, severe fibrosis in bone marrow and spleen and increased bone deposition were also observed. Finally, all animals died within 7 months after bone marrow replacement [117].
Genetic approach
To date, two transgenic animal models have been generated showing contradictory phenotypes. In one case, expression of the human TPO gene was driven by the liver apolipoprotein E enhancer/promoter [117]. These animals, despite increased platelet and tissue MK counts, do not develop bone marrow fibrosis [117]. In the second system, high levels of murine TPO expression were induced by placing the full length TPO gene under the control of the mouse IgH-chain promoter [118]. These mice showed a more drastic but still mild phenotype, with anemia, leukocytosis, slightly high TPO levels (increased by 2.3-fold) in plasma, bone marrow fibrosis and osteosclerosis 9 months after birth [118].
The TPOhigh mouse model has been instrumental in formally demonstrating the link between marrow fibrosis and overproduction of TGF-β. In fact, wild-type mice transplanted with TGF-βnull stem cells that had previously been transduced with a retrovirus containing the TPO gene, did not develop myelofibrosis. By contrast, animals transplanted with TGF-β+/+ HPCs expressing similar high levels of TPO manifested the disease (TPOhigh model of PMF, see later) (Table 3) [44]. These complementation studies have also clarified the role of OPG overproduction, a recently identified biomarker of PMF patients [119], in the development of myelofibrosis. In these experiments, marrow cells from Opg-/- or wild type (Opg+/+) littermates were infected with a retrovirus encoding TPO and engrafted into either Opg-/- or wild-type mice. The phenotype of the four combinations of graft/host mice (Opg+/+/Opg+/+, Opg-/-/Opg-/-, Opg-/-/Opg+/+, Opg+/+/Opg-/-) was then analyzed [75]. All four experimental groups showed similarly elevated TPO and TGF-β levels in plasma and developed severe myelofibrosis. In addition, wild-type hosts engrafted with either Opg+/+ or Opg-/- hematopoietic cells developed osteosclerosis associated with increased OPG levels in plasma and decreased osteoclastogenesis. By contrast, Opg-/- hosts did not exhibit osteosclerosis even if transplanted with TPOhigh wild-type stem cells [75]. These experiments highlighted the importance of a reactive microenvironment for development of the entire myelofibrotic trait. On the other hand, Evrard et al. showed with a similar transplantation approach that HPCs from mice lacking the Tsp gene (Tsp-/- mice) expressing high levels of TPO induce myelofibrosis in wild-type mice, indicating that the high levels of TSP similar to those found in PMF patients have no detectable pathological consequences in mice [120].
Biological effects similar to those promoted by TPO overexpression are mimicked in mice by any mutation that activates signaling pathways downstream of this growth factor. Also, ectopic expression of MPL W515L, an active form of the TPO receptor, in a murine bone marrow transplant assay induced a fully penetrant myeloproliferative disease, associated with splenomegaly, extramedullary hematopoiesis, marked thrombocytosis, bone marrow fibrosis and megakaryocytic proliferation (Table 3) [46]. Hematopoietic cells that express the MPL W515L mutation are hypersensitive to stimulation with TPO and present a proliferative advantage in bone marrow and spleen, as assessed by colony-forming assay.
The role of the TPO/MPL/JAK2 signaling in PMF is further supported by the observation that linker of T-cell receptor pathway (Lnk-/-) mice, in addition to defective B lymphopoiesis, display many of the manifestations of myelofibrosis, including splenomegaly, hyperplasia of the MK lineage and thrombocytosis due to hyperactivation of TPO signaling, increased number of erythroid cells and hematopoietic progenitors in the spleen [94–95]. Lnk, recently designated as SH2B3, is a Src-homology 2 (SH2) domain-containing adaptor protein [91,92]. Lnk is highly expressed on HPCs [94,95]. The SH2 domain of LNK negatively regulates translocation to the plasma membrane of several cytokine receptors, including MPL [87,96,121] and the EPO [122] and SCF [123,124] receptor. As a consequence, Lnk-/- cells express higher levels of MPL on their surface and respond more readily to TPO [27,119–124]. Given its role in downregulating MPL, LNK may represent a promising target for therapeutic intervention in those MPDs associated with the MPL W515L mutation [125].
The JAK2 mouse models
JAK2 is an important element of the TPO/MPL signaling pathway and it is, therefore, not surprising that the introduction of two activating mutations in this gene (JAK2 V617F, exon 12 mutations and TEL—JAK2), produce similar phenotypes to TPOhigh and MPL W515L mutations in mice (Table 3). In a mouse transplantation model, HPCs overexpressing either JAK2 V617F [88,89] or JAK2K539L (exon 12 mutation) induced a MPD that strongly resembled PV. Recently, two transgenic mouse lines expressing JAK2 V617F in the hematopoietic system under the control of the tissue-specific vav promoter have been generated [40]. The line expressing the lower JAK2 V617F level showed a moderate increase in blood cell counts, whereas the line with the higher level of expression displayed marked elevation in blood cellularity and developed a phenotype resembling essential thrombocytopenia and PV. These mice also developed myelofibrosis as they aged with erythroid, megakaryocytic and granulocytic hyperplasia in bone marrow and spleen, as well as splenomegaly and reduced levels of EPO and TPO in the plasma [126].
Since in all of the JAK2 transgenic mice developed so far the fibrotic phase was preceded by a polycythemic or thrombocythemic phase, these animals more closely represent a model of secondary myelofibrosis rather than PMF. Nevertheless these mice are of great value as preclinical models for screening the in vivo efficacy of newly developed JAK2 inhibitors. Notably, the small-molecule JAK inhibitor 1 (Calbiochem, San Diego, CA) also blocks proliferation of MPL W515L transformed 32D cells, suggesting that drugs targeting the JAK—STAT pathway may represent an effective therapy to cure both JAK- and MPL-dependent diseases [46].
Other models of myelofibrosis have been induced in mice by genetic mutations encoding forms of activated kinases found in other human leukemias. In fact, retrovirally transduced murine bone marrow overexpressing the oncogenic TEL—JAK2 fusion gene promotes a mixed myelo- and lympho-proliferative disorder when transplanted in irradiated syngenic hosts [42,43]. Furthermore, CD34+ cells obtained from the human umbilical cord and expressing TEL—JAK2 rapidly induce myelofibrosis in vivo in a nonobese diabetic/severe combined immunodeficiency xenotransplantation model [127]. However, since myelofibrosis is not observed in patients harboring this translocation, the genomic rearrangement is unlikely to be responsible for the human disease. These results may instead reflect the biological functions acquired by TEL when fused with a tyrosin kinase in mice. In agreement with this hypothesis, also the TEL—PDGF-βR (the β chain of the PDGF receptor [PDGF-βR] has tyrosine kinase activity) fusion gene, the result of the t(5;12)(q33;p13) translocation associated with chronic myelomonocytic leukemia [128], introduced into whole bone marrow by retroviral transduction also caused a rapidly fatal MPD in mice. TEL—PDGF-βR-transplanted mice developed leuko cytosis, splenomegaly, extramedullary hematopoiesis and bone marrow fibrosis (Table 3) [129,130].
Gata1 low mouse models
GATA1 is a gene localized both in mice and in humans on the X chromosome that encodes a transcription factor necessary for the development of hematopoietic cells of many lineages, including erythroid, megakaryocytic, mast, eosinophilic and denditric cells [131–135]. Gata1 was first identified as one of the factors up regulated in response to erythroid differentiation induced in murine erythroleukemic cells by dimethylsulfoxide by differential display [135]. GATA1 binds the promoter region of many erythroid-specific genes, such as EPO and GATA1 itself [136,137], and its crucial role for erythroid maturation was first demonstrated by the observation that the germinal knockout of this gene in mice results in fatal anemia before birth [138]. GATA1 belongs to a family of zinc finger transcription factors that share a well conserved common architecture. This similarity ensures redundancy, with different proteins being interchangeable in regulating the same function [139]. It is conceivable that accurate control of gene expression, rather than the functional structure of individual proteins, drives the biological specificity of the different family members [137,139–142]. The lineage specificity of GATA1 expression is ensured by the complex promoter region through a specific activation of the chromatin configuration of the locus. The Gata1 locus, in fact, contains two untranslated first exons: the proximal exon, mostly used by erythroid cells and MKs, named IE exon, and the distal exon, named the IT exon, which was first identified because it is used as a transcription start site by Sertoli cells of the testis [141,142]. The hypomorphic Gata1low (or Gata1neoδHS) mutation is a targeted deletion that replaces approximately 8 kb of the upstream region, which includes the IT promoter and the DNaseI hypersensitivity site (HS1), with a neomycin-resistance cassette. This construct was used to generate mutant embryonic stem cells, which were then injected into C57BL/6 blastocysts. Gata1low pups are severely anemic and thrombocytopenic and more than 95% of them die around birth [143], highlighting the importance of HS1 for appropriate Gata1 expression in MKs and erythroid cells. Anemia was due to a high apoptotic rate of the erythroblasts, linked to low GATA1 expression (∼20% of wild-type expression levels) [143]. Thrombocytopenia was also caused by reduced levels of GATA1 protein in MKs, which, therefore, remained immature and released abnormally large platelets [144,145]. Surprisingly when the mutation was transferred in the CD1 background, perinatal mortality was lost [146]. CD1 mutant mice were still born anemic but recovered from anemia within the first month by establishing extramedullary erythropoiesis in the spleen and developing splenomegaly. Severe thrombocytopenia, however, remained a life-long trait, with platelet counts at 30% of the physiological value. Thrombocytopenia was associated with MK hyperproliferation and retarded maturation in the spleen and bone marrow. By 18 months Gata1low mice showed marked myelofibrosis with collagen deposition and new bone formation, reduction of bone marrow cavity and extramedullary hematopoiesis in the liver (Figure 4) [97,146].
Figure 4. Gata1low mice (CD1 background) develop MF, a disease that mimicks the progression of human primary MF.

At birth Gata1low mice are anemic and thrombocytopenic. In the pre-MF stage (1–6 months of age) mice recover from the anemia by developing extramedullary hematopoiesis in the spleen and splenomegaly. In this phase increased osteogenesis is also observed. In the early-MF phase (8–12 months) mice develop bone marrow fibrosis and neovascularization of the bone marrow stroma. By 15 months of age, animals show an increased mobilization of hematopoietic progenitor cells and extramedullary hematopoiesis in the liver. Nevertheless, the life expectancy of Gata1low mice remains normal [97].
MF: Myelofibrosis.
By contrast, in the DBA/2 background, the mutation induced a milder myelofibrosis picture, while a similar mutation in BALB/C mice induced thrombocytopenia and osteopetrosis, but not myelofibrosis, and the mice died of megakaryocytic leukemia at an old age (Figure 5) [METCALF D, PERS. COMM.]. The different penetrance of the myelofibrotic trait induced by the hypomorphic Gata1low mutation in different mouse strains suggests the existence of gene modifiers that cooperate with the hypomorphic mutation in determining the mouse phenotype. The observation that the severity of the phenotype is positively correlated with the ability of the strains to recruit the spleen as an erythropoietic site to recover from phenylhydrazine-induced hemolytic anemia [146] suggests that at least one of these gene modifiers is represented by sf-Stk. The activation of spleen erythropoiesis in response to stress is controlled by a truncated form of the Stk receptor, sf-Stk, transcribed from an alternative GATA1-responsive promoter located inside the Fv2 locus. The presence of sf-Stk also confirms the susceptibility to infection by the Friend leukemia virus (FLV) [147,148]. Strains that are susceptible to FLV infection, such as BALB/C, express a truncated version of sf-Stk, while C57B/6 mice, which harbor a point mutation destroying the GATA1 binding site, are unable to express sf-Stk and are resistant to FLV infection (Figure 5) [147,148]. Alternatively, CD1 and DBA/2 mice that express levels of sf-Stk intermediate between those expressed by C57BL/6 (null) and BALB/C (maximal) mice, present a consequent variegation in the extent of FLV susceptibility (Figure 5) [147].
Figure 5. Correlation between Sf-Stk expression and background-dependent variegation of the phenotype induced by the Gata1low mutation.
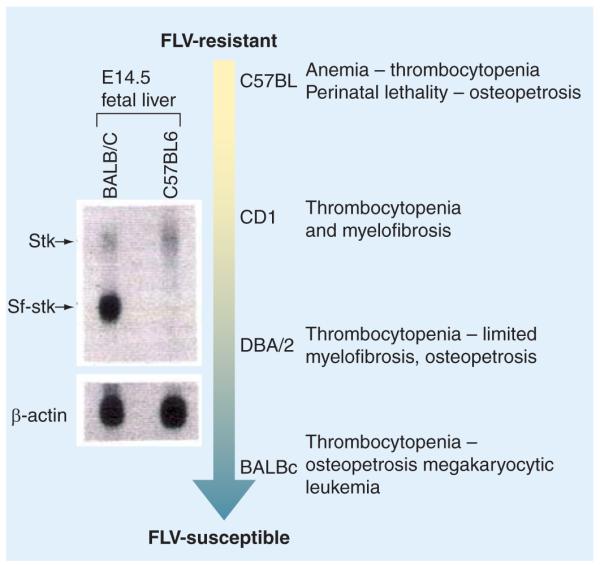
The left panel shows the difference in expressions of Sf-Stk mRNA in the fetal liver of BALB/C and C57BL6 mice by northern blotting [147]. The Sf-Stk mRNA is transcribed from an alternative promoter within the Stk gene under the control of a GATA1 binding site mutated in C57BL6 mice. The erythroid cells from these mice are, therefore, incapable of expressing Sf-Stk and are resistant to FLV infection. By contrast, CD1, DBA/2 and BALB/c all contain the GATA1 binding site and their livers express Sf-Stk. The lowest levels of expression are observed in CD1 mice, while maximal levels of expression are observed in BALB/c mice (as indicated by the shade of the arrow). These levels of expression correlate with the susceptibility of the different strains to FLV infection. On the right, phenotypes of the different Gata1low strains are indicated. The severity of the phenotype of these mutants correlates with the intrinsic susceptibility to FLV infection of the single strain (the higher the susceptibility, the lower the lethality) [146].
FLV: Friend leukemia virus.
Modified with permission from [147].
MKs of Gata1low mice present all the abnormalities of the Mks observed in PMF patients
As in PMF patients, MKs from GATA1low mice are hyperplastic and immature, showing altered structure of β-granules, low or un detectable expression of VWF, abnormal P-selectin distribution associated with increased and abnormal emperipolesis of neutrophils, and augmented levels of para-apoptosis (Figure 6) [146,149,150]. Moreover, thrombocytopenia is observed in all of the strains examined, while anemia is greatly dependent on the genetic background. Therefore, the hypomorphic mutation is linked directly to the MK abnormalities observed in PMF patients with the reduced GATA1 protein in these cells (Figure 3) [146,150]. In spite of the fact that the GATA1 locus in PMF patients is normal, a large number of MKs in the bone marrow of these patients are GATA1-negative (Figure 5) [82]. It is interesting that, in contrast to the results observed in normal mice, TPO treatment increased the levels of GATA1 expression in MKs of Gata1low mice and protected these animals from developing myelofibrosis [76], indicating that the levels of GATA1 expressed by the MKs may represent a druggable target for treatment of the human disease.
Figure 6. Ultrastructural analyses of megakaryocytes (MKs) from the spleen of wild-type (A,B) and Gata1low (C,D) mice.

(A) The arrow indicates an isolated MK in the semi-thin section from a wild-type spleen that was then further analyzed by transmission electron microscopy (TEM) in (B). The ultrastructural morphology (cytoplasm rich in α-granules and platelets territories) characterizes this cell as a stage III MK. (C) The arrow and arrowhead indicates a cluster of MKs in a semi-thin spleen section from Gata1low mice further analyzed by TEM in (D). Two populations of mutant MK were identified by TEM, one with normal (arrow) and the other with high (arrowhead) electron density of the nucleus and cytoplasm. High electron-dense MKs present signs of cytoplasmic damage (swollen mitochondria with disrupted crests, numerous empty vacuoles and occasional remnants of demarcation membrane system). Their extensive heterochromatic nuclear organization and the shrunk and degraded appearance of their cytoplasm are indications of para-apoptosis, a terminal deoxynucleotide transferase-mediated dUTP nick-end labeling-negative process of cell death.
Magnification 100x in (A) and (C), and 3000x in (B) and (D).
Genetic models that target additional transcription factors that regulate MK development are also indirectly linked to the Gata1low model of PMF. Broad complex—tramtrack—bric-a-brac domain and cap‘n’collar (CNC) homolog 1 (BACH1) are transcriptional repressors closely related to the Drosophila nuclear regulatory protein CNC [151,152]. It is located on chromosome 21q22.1 within the Down syndrome-associated gene region [153,154] and is expressed in MKs as well as in erythroblasts [155]. BACH1 heterodimerizes and other members of the CNC family (e.g., the p45 subunit of the nuclear factor—erythroid 2 [NF—E2]) and musculoaponeurotic fibrosarcoma (Maf) proteins to bind Maf-recognition element (MARE) [151,152,156], present themselves in the promoter region of several erythroid and megakaryocytic genes; including the globin genes and genes encoding enzymes involved in the biosynthesis of heme [157–160]. The functions of CNC proteins in MKs have been well documented in animal studies. Homozygous disruption of the mouse p45 subunit results in defective megakaryopoiesis and profound thrombocytopenia, leading to postnatal death [161]. In addition, BACH1 overexpression in erythroid cells and MKs phenocopies p45-mutant animals [92]. Transgenic mice bearing human BACH1 under the control of GATA1 regulatory sequences present significant reductions in the numbers of circulating platelets due to arrest in MK maturation. The MK abnormalities observed in these mutants are similar to those observed in Gata1low mice with aberrant morphology, characterized by polyploidy, disorganized DMS, reduced granules and increased neuthrophil emperipolesis (Table 3). BACH1 transgenic animals develop myelofibrosis with reticulin fibers, which are detectable as early as 5 weeks of age [92].
Occurrence of osteosclerosis in Gata1low mice
New bone formation is the result of a balanced positive and negative effect exerted by osteoblasts and osteoclasts, respectively [162]. The accelerated bone deposition or impaired bone resorption both lead to osteosclerosis, a pathological increase of bone density [163]. All the Gata1low mice developed osteosclerosis by 1 month of age, although the degree of osteogenesis is greatly dependent upon the genetic background (Figure 2) [146,164]. In these mice, new bone formation is mainly due to increased osteoblast proliferation while the number of osteoclasts remains normal. Increased osteoblast proliferation is probably promoted by the altered MKs through the establishment of direct cell—cell contact and the release of bioactive molecules, such as OPG and BMP4 [146,164].
Gata1low mutation promotes neo-angiogenesis
Similar to all tumors, PMF is characterized by abnormal angiogenesis [17]. A myelofibrotic bone marrow displays marked new vessel formations with increased pericyte coverage [17,165]. Mice with low GATA1 expression present the same abnormal vessel formation observed in humans, with increased mean vessel perimeter, raised microvascular density and pathological branching [166,167]. The higher level of VEGF, which is a major pericyte-attractant, found in the extracellular fluid of these mice suggests that deregulation of pericyte recruitment may contribute to the abnormal angiogenic process induced by the hypomorphic Gata1low mutation (Figure 2) [167].
Gata1low mice present increased stem/progenitor cell trafficking & extramedullary hematopoiesis in liver
A tuned HSC/HPC trafficking is responsible for establishing and maintaining normal hematopoiesis. During development, HPCs migrate via the blood from the fetal liver to the newly formed cavities in the bone marrow, by a process that involves reprogramming of the expression of the receptors that ensure the contact with the microenvironment. Although limited HPC trafficking via the blood among different niches persists throughout adult life, HPC trafficking is thought to be regulated by the interaction between the chemokine stromal cell-derived factor (SDF)-1, expressed by stromal cells of many types, with its receptor CXCR4, expressed on the surface of the HPC [168,169]. SDF-1 is not only produced by the liver but also by several stromal cells, including endothelial cells, in the bone marrow [170,171]. In addition MKs and platelets express CXCR4, therefore, these cells sequester SDF-1 from the microenvironment and store this factor in their cytoplasm to release it upon appropriate stimulation [172,173]. The importance of the interaction between SDF-1 and CXCR4 for appropriate HPC trapping within their niches [168,169] was proved by the observation that deletion of either the SDF-1 or CXCR4 gene in mice resulted in dramatic decreases in the numbers of HPCs contained in the bone marrow with a corresponding increase in the number of circulating HPCs [168,174]. Also, experimental increases of SDF-1 in the blood and the decrease of this factor in bone marrow both result in increased HPC mobilization [175,176]. Similarly, downregulation of CXCR4 expression in the HPCs results in the increased mobilization of these cells [177].
Primary myelofibrosis is accompanied by increased (up to 200-fold) levels of HSCs/HPCs, as well as endothelial progenitor cells, in blood [1,178–180]. It is conceivable that these displaced stem cells will then leave the blood to colonize niches in the spleen and the liver where normal HPCs would not survive. Evidence for this hypothesis is provided by the observation that fibroblasts isolated from the spleen of PMF patients sustains proliferation of autologous CD34+ cells but not that of CD34+ cells from normal donors [181,182]. A similar increase in HPC trafficking and development of extramedullary hematopoiesis has been described in Gata1low mice (Figure 4) [146].
Given the central role of the SDF-1—CXCR4 axis in stem/progenitor cell trafficking, several investigators have analyzed whether increased HPC trafficking and extramedullary hemato poiesis observed in Gata1low mice and PMF patients might be related to abnormalities in SDF-1—CXCR4 expression. Not surprisingly, very similar SDF-1—CXCR4 levels were detected in the two systems. Both PMF patients and Gata1low mice had modestly increased levels of SDF-1 in blood, highly increased SDF-1 immunostaining in bone marrow and reduced levels of CXCR4 expression in stem/progenitor cells [179,183,184].
It is unlikely that increased levels of SDF-1 in the blood may play a major role in an increased stem/progenitor cell mobilization in PMF and myelofibrosis because of their modest nature (twofold) and of the fact that similar increases are detectable in MPD patients, such as those with PV, who do not experience increased HPC mobilization. On the other hand, dramatic increases in SDF-1 immunostaining were evident in bone marrow sections from Gata1low mice and in marrow biopsy of PMF patients (Figure 7). In both cases a strong reactivity toward the SDF-1 antibody was associated with both MKs and endothelial cells (Figure 7) [183]. Augmented levels of SDF-1 in bone marrow should, in theory, promote a stronger interaction between the HPCs and their niche and inhibit egression of the cells from the bone marrow. However, because of the pathological process of MKs emperipolesis observed in both the Gata1low mice and PMF patients, both bone marrow microenvironments are rich in neutrophil proteases that might cleave and inactivate SDF-1. Therefore, in spite of high SDF-1 staining, the microenvironment of the mutant mice, as well as that of the PMF patients, might be deprived of active SDF-1. Since it is not possible to discriminate between bioactive and proteolytically degraded SDF-1 by immuno staining, it is not possible to assess whether reduced levels of active SDF-1 in the bone marrow are responsible for increased stem cell trafficking in PMF patients and Gata1low mice [183].
Figure 7. Increased stromal cell-derived factor (SDF)-1 immunostaining of marrow sections from Gata1low mice with age and the marrow biopsies from PMF patients.

(A) SDF-1 immunostaining of representative sections from the marrow of the young (7 months) and old (10–12 months) wild-type (top panels) and Gata1low (bottom panels) littermates are presented as indicated. Slides were counterstained with hematoxylin—eosin. (B) SDF-1 immunohistochemistry of marrow biopsies from a normal donor (upper panel) and a PMF patient (bottom panel) are shown. Bone marrow biopsy sections were stained with a monoclonal anti-SDF-1 antibody and then counterstained with Light Green (200x magnification).
PMF: Primary myelofibrosis.
Reproduced with permission from [183].
Alternatively, HPCs from both PMF patients and Gata1low mice express reduced levels of CXCR4 protein and mRNA [179,183,184]. In the case of PMF patients it has been possible to demonstrate that reduced levels of CXCR4 mRNA are associated with hyper-methylation of the promoter of the CXCR4 gene [184], linking abnormal CXCR4 expression directly to an intrinsic property of the PMF HPC clone [179,185]. It is possible that the decreased CXCR4 expression may impair the HPC affinity for the marrow niche, determining, at least in part, the increased HPC trafficking observed in PMF.
Another feature of myelofibrosis common both to PMF patients and mice, that may contribute to the increased HPC trafficking, is represented by the augmented number of HPCs associated with this disease [179,180]. In the case of Gata1low mice, the augmented number of HPCs may be a direct result of the reduced GATA1 expression in these cells. In fact, the fate of hematopoietic progenitor cells is regulated by a balance between the levels of GATA2 and GATA1 expression. When the GATA2 levels are higher than those of GATA1, the cells proliferate, while high GATA1 levels favor maturation [186,187]. Since GATA1, in association with Friend of GATA1 (FOG-1) suppresses GATA2 expression by binding to regulatory sequences on the GATA2 promoter [188,189], Gata1low HPCs are impaired in their ability to suppress GATA2 expression and so remain in a proliferative state for a longer time [91,146].
To summarize, extramedullary hematopoiesis, which is associated with PMF in humans and myelofibrosis in mice, may partly be the result of increased HPC trafficking, due to reduced CXCR4 expression in the HPCs and the destruction of a supportive bone marrow microenvironment caused by fibrosis, and the increased numbers of progenitor cells.
Leukemic transformation in the Gata1low mouse model
In humans, leukemic transformation is frequently associated with PMF progression; by contrast, Gata1low mice do not develop leukemia. Exceptions were represented by the Gata1low BALB/C mice, which die of megakaryoblastic leukemia at old age [METCALF D, PERS. COMM.], and by the most stringent Gata10.5 mouse model. In this last model, replacement of the IE exon promoter by a neomycin resistance gene induces drops in Gata1 expression down to 5% of the levels observed in wild-type cells [190]. These animals do not have the time to develop myelofibrosis because they die of erythroblastic leukemia a few months after birth (Figure 5) (Table 3) [190].
Alternatively, an amino-terminal truncated form of GATA1 (sGATA1), a hypomorphic GATA1 protein which is tenfold less active than the full length one, is detectable in megakaryocytic leukemia associated with Down syndrome in humans [191]. Down syndrome is caused by trisomy of the human chromosome 21, the affected children display a 500-fold increased risk of developing megakaryocytic leukemia associated with myelofibrosis [192]. In order to clarify the respective roles of sGATA1 and trisomy 21 in the development of leukemia in these patients the phenotypes of the mouse models carrying the two genetic abnormalities have been analyzed. sGATA1 mice develop a transient hyperproliferation of a unique fetal-specific progenitor cell, highlighting the possible tumor-suppressor role of GATA1 during fetal development in mice [193]. Unexpectedly, overexpression of the chromosome 21 mouse ortholog (Ts65Dn strain) induced a phenotype that strongly resembled MPD (Table 3). All of the Ts65Dn mice, in fact, presented morphological alteration and accumulation of MKs in bone marrow and the spleen, a profound thrombocytosis and a developed myelofibrosis [194].
Microenvironment-defective mouse models
Hematopoiesis depends on an appropriate response of progenitor/stem cells to extrinsic signals provided by the bone marrow microenvironment. Walkley et al. generated two mouse models that demonstrate that the myeloproliferation may result from perturbed interaction between HSCs and their niche. Indeed, germline deletion of retinoic acid receptor subtype-γ (RARγ) gene [99] or the conditional knockout of the retinoblastoma (Rb) protein in the murine hematopoietic compartment [100] resulted in a myeloproliferative syndrome. RARs are nuclear hormone receptors that act as transcriptional repressors in the free form and as transcriptional activators when bound to their ligand [195]. RARγ-/- mice develop age dependent myeloproliferation, associated with extramedullary hematopoiesis, splenomegaly and loss of trabecular bone [99]. The syndrome partially involves elevated levels of TNF-α expression, since the hematologic defects are mostly rescued when knockout mice are transplanted with TNF-α-/- hematopoietic cells [99]. On the other hand, Rb is a negative cell cycle regulator, which has been extensively studied for its relevance as a tumor suppressor [196,197]. Inactivation of Rb in hematopoietic cells by crossing the interferon-inducible Mx—Cre transgenes and Rbfl/fl animals resulted in profound myeloproliferation, with loss of HSCs from the bone marrow due to the migration to extramedullary sites [100]. Neither the RARγ-/- nor the Mx—Cre+ Rbfl/fl phenotypes are rescued by the transplantion with wild-type HSCs or induced by transplantation of the mutated HSC into wild-type mice, indicating that the myeloproliferation is the result of an aberrant niche [99,100].
Expert commentary
Hematopoiesis is strictly dependent on the establishment of appropriate interactions between stem cells and their niches in the marrow. The quantitative stem cell assays and the numerous genetic tools developed in recent years have elucidated the nature of these interactions in mice. Much work remains to be done, however, to validate these discoveries in humans. PMF is a disease in which the interactions between the stem cells and their niches in the marrow are disrupted. Therefore, the comparison of the biological properties of the stem cells and of the marrow microenvironment between PMF patients and normal donors provides the necessary tool to understand how the hematopoietic process is regulated in humans. It is anticipated that in the next few years many investigators will study PMF as a disease model to understand the relationship between stem cells and their niches in humans.
Five-year view
In spite of the numerous important discoveries made in recent years on the pathobiology of PMF, the primary molecular defect leading to development of this disease is still obscure and allogeneic stem cell transplantation remains the only curative therapy. Given that not all patients are eligible for this procedure and that the transplant-related mortality is relatively high, development of alternative and safer therapeutic approaches for this disease remains a priority. In the search for the ultimate druggable target to cure PMF, several animal models of the disease have been generated. These models provide invaluable tools to better understand the pathobiology of such a complex disease. Although mutations of the GATA1 gene have not been discovered in PMF patients yet, the MKs from these patients are characterized by reduced GATA1 content. Therefore, these cells phenocopy MK alterations induced by the presence of the hypomorphic Gata1low mutation in mice. Owing to the similarities between the MK abnormalities observed in PMF patients and those induced by the Gata1low mutation, Gata1low mice are of inestimable value for dissecting the mechanisms that underlie the development of myelofibrosis, in particular those responsible for traits such as abnormal stem cell trafficking and development of extramedullary hematopoiesis, which are very difficult to study in humans. In addition, the affinities between the MK abnormalities found in Gata1low and TPOhigh (another animal model of myelofibrosis) mice and those observed in PMF patients allow us to predict that PMF may be the result of multiple gene lesions occurring on a single pathobiological pathway that regulates the interaction between the stem cells and their niches. In summary, the crucial link between MK abnormalities, alterations of the microenvironment and PMF developments discovered thanks to the murine models of the disease suggests that agents that restore MK development in PMF patients might also be able to cure the disease.
Key issues.
Primary myelofibrosis (PMF) is a chronic myeloproliferative disease caused by a clonal stem cell defect and is characterized by progressive bone marrow fibrosis, splenomegaly, extramedullary hematopoiesis, osteosclerosis and pancytopenia. The most frequent (50% of cases) genetic mutation associated with PMF is the JAK2 V617F mutation. Nevertheless, the role of the biochemical abnormalities associated with the presence of this mutation in the etiology of PMF has not yet been fully characterized.
In spite of the fact that the genetic cause of PMF is still unclear, PMF is characterized by distinctive abnormalities in megakaryocyte (MK) development. These abnormalities are represented by reduced levels of expression of the transcription factor GATA1 and of those MK-specific genes, such as von Willebrand factor, whose expression is regulated by these transcription factors. In addition, P-selectin, although expressed at normal levels in MKs from PMF patients, is delocalized on the DMS region of these cells. Such delocalization induces a pathological process of neutrophil emperipolesis, with consequent para-apoptosis of the MKs and the release of growth factors stored in these cells into the bone marrow microenvironment.
Any mutation in mice that induces abnormalities in MK development similar to those observed in PMF also induces myelofibrosis, a syndrome similar to PMF. These mutations define a pathobiological pathway that links the extrinsic (thrombopoietin [TPO]) to the intrinsic (GATA1) control of MK development. Only some of these mutations have been identified in PMF patients or in other hematological malignancies so far.
A number of different animal models are presently available to study the pathobiological pathway of myelofibrosis. Each of these models has appropriate applications. Models that generate the disease within a few months, such as TPOhigh mice, are useful for gene complementation studies with the aim to analyze factors downstream of the primary lesion that contribute to the establishment of the myelofibrotic trait. Those generated by introducing mutations in mice found in PMF patients, such as JAK2 V617F mice, are more suited for preclinical studies aimed at identifying drugs that may cure PMF by inhibiting the biological activity of JAK2 V617F. Interestingly, mice such as Gata1low mice that develop the disease according to well-defined sequential phases are more suited to dissect the hierarchy of events occurring during disease progression and to define stage-specific strategies to cure the PMF.
The most intriguing of all the abnormalities observed in PMF is increased stem/progenitor cell trafficking and the development of extramedullary hematopoiesis in multiple organs, including the spleen and the liver. A vis-à-vis comparison of the abnormalities observed in PMF patients and in Gata1low mice highlights the fact that this complex trait is likely a consequence of both a stem cell defect (reduced expression of the adhesion receptor CXCR4) and of a microenvironment defect (increased growth factor milieu induced by MK abnormalities). This concept paves the way to identify strategies to cure PMF based on a better understanding of the interactions between the stem cells and their niches and how this interaction is regulated by MKs.
Acknowledgments
Financial & competing interests disclosure This study was supported by Ministero per la Ricerca Scientifica and Alleanza sul Cancro, Italy and the National Cancer Institute, grant number P01-CA108671, NIH, USA. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Writing assistance was utilized in the production of this manuscript. The editorial help of Thomas Hayes is gratefully acknowledged.
Contributor Information
Lilian Varricchio, Department of Medicine, Division of Hematology/Oncology, Mount Sinai School of Medicine, One Gustave L. Levy Place, Box 1079, New York, NY 10029, USA Tel.: +1 212 241 6974 Fax: +1 212 241 4096 lilian.varricchio@mssm.edu.
Annalisa Mancini, Department of Medicine, Division of Hematology/Oncology, Mount Sinai School of Medicine, One Gustave L. Levy Place, Box 1079, New York, NY 10029, USA Tel.: +1 212 241 6974 Fax: +1 212 241 4096 annalisa.mancini@mssm.edu.
Anna Rita Migliaccio, Department of Medicine, Division of Hematology/Oncology, Mount Sinai School of Medicine, One Gustave L. Levy Place, Box 1079, New York, NY 10029, USA Tel.: +1 212 241 6974 Fax: +1 212 241 4096 annarita.migliaccio@mssm.edu.
References
Papers of special note have been highlighted as:
• of interest
•• of considerable interest
- 1.Ahmed A, Chang CC. Chronic idiopathic myelofibrosis: clinicopathologic features, pathogenesis, and prognosis. Arch. Pathol. Lab. Med. 2006;130(8):1133–1143. doi: 10.5858/2006-130-1133-CIM. [DOI] [PubMed] [Google Scholar]
- 2.Dameshek W. Some speculations on the myeloproliferative syndromes. Blood. 1951;6(4):372–375. [PubMed] [Google Scholar]
- 3.Tefferi A, Vardiman JW. Classification and diagnosis of myeloproliferative neoplasms: the 2008 World Health Organization criteria and point-of-care diagnostic algorithms. Leukemia. 2008;22(1):14–22. doi: 10.1038/sj.leu.2404955. [DOI] [PubMed] [Google Scholar]
- 4.Orazi A, Germing U. The myelodysplastic/myeloproliferative neoplasms: myeloproliferative diseases with dysplastic features. Leukemia. 2008;22(7):1308–1319. doi: 10.1038/leu.2008.119. [DOI] [PubMed] [Google Scholar]
- 5.Nowell PC, Hungerford DA. Chromosome studies on normal and leukemic human leukocytes. J. Natl Cancer Inst. 1960;25:85–109. [PubMed] [Google Scholar]
- 6.Groffen J, Stephenson JR, Heisterkamp N, et al. Philadelphia chromosomal breakpoints are clustered within a limited region, bcr, on chromosome 22. Cell. 1984;36(1):93–99. doi: 10.1016/0092-8674(84)90077-1. [DOI] [PubMed] [Google Scholar]
- 7.Shtivelman E, Lifshitz B, Gale RP, Canaani E. Fused transcript of abl and bcr genes in chronic myelogenous leukaemia. Nature. 1985;315(6020):550–554. doi: 10.1038/315550a0. [DOI] [PubMed] [Google Scholar]
- 8.Hallek M, Danhauser-Riedl S, Herbst R, et al. Interaction of the receptor tyrosine kinase p145c-kit with the p210bcr/abl kinase in myeloid cells. Br. J. Haematol. 1996;94(1):5–16. doi: 10.1046/j.1365-2141.1996.6102053.x. [DOI] [PubMed] [Google Scholar]
- 9 ••.Druker BJ, Tamura S, Buchdunger E, et al. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr—Abl positive cells. Nat. Med. 1996;2(5):561–566. doi: 10.1038/nm0596-561. Describes the first therapeutic drug identified on the basis of its activity to inhibit an oncogene.
- 10.Tefferi A, Thiele J, Orazi A, et al. Proposals and rationale for revision of the World Health Organization diagnostic criteria for polycythemia vera, essential thrombocythemia, and primary myelofibrosis: recommendations from an ad hoc international expert panel. Blood. 2007;110(4):1092–1097. doi: 10.1182/blood-2007-04-083501. [DOI] [PubMed] [Google Scholar]
- 11.Abdel-Wahab OI, Levine RL. Primary myelofibrosis: update on definition, pathogenesis, and treatment. Annu. Rev. Med. 2008 doi: 10.1146/annurev.med.60.041707.160528. [DOI] [PubMed] [Google Scholar]
- 12.Barosi G, Hoffman R. Idiopathic myelofibrosis. Semin. Hematol. 2005;42(4):248–258. doi: 10.1053/j.seminhematol.2005.05.018. [DOI] [PubMed] [Google Scholar]
- 13.Reilly JT. Idiopathic myelofibrosis: pathogenesis to treatment. Hematol. Oncol. 2006;24(2):56–63. doi: 10.1002/hon.771. [DOI] [PubMed] [Google Scholar]
- 14.Thiele J, Kvasnicka HM. Myelofibrosis in chronic myeloproliferative disorders — dynamics and clinical impact. Histol. Histopathol. 2006;21(12):1367–1378. doi: 10.14670/HH-21.1367. [DOI] [PubMed] [Google Scholar]
- 15.Cervantes F. Myelofibrosis: biology and treatment options. Eur. J. Haematol. Suppl. 2007;68:13–17. doi: 10.1111/j.1600-0609.2007.00937.x. [DOI] [PubMed] [Google Scholar]
- 16.Tefferi A. Primary myelofibrosis. Cancer Treat. Res. 2008;142:29–49. [PubMed] [Google Scholar]
- 17.Lundberg LG, Lerner R, Sundelin P, et al. Bone marrow in polycythemia vera, chronic myelocytic leukemia, and myelofibrosis has an increased vascularity. Am. J. Pathol. 2000;157(1):15–19. doi: 10.1016/S0002-9440(10)64511-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Visani G, Finelli C, Castelli U, et al. Myelofibrosis with myeloid metaplasia: clinical and haematological parameters predicting survival in a series of 133 patients. Br. J. Haematol. 1990;75(1):4–9. doi: 10.1111/j.1365-2141.1990.tb02609.x. [DOI] [PubMed] [Google Scholar]
- 19.Mesa RA, Silverstein MN, Jacobsen SJ, Wollan PC, Tefferi A. Population-based incidence and survival figures in essential thrombocythemia and agnogenic myeloid metaplasia: an Olmsted County Study, 1976–1995. Am. J. Hematol. 1999;61(1):10–15. doi: 10.1002/(sici)1096-8652(199905)61:1<10::aid-ajh3>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 20.Zhao R, Xing S, Li Z, et al. Identification of an acquired JAK2 mutation in polycythemia vera. J. Biol. Chem. 2005;280(24):22788–22792. doi: 10.1074/jbc.C500138200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kralovics R, Passamonti F, Buser AS, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N. Engl. J. Med. 2005;352(17):1779–1790. doi: 10.1056/NEJMoa051113. [DOI] [PubMed] [Google Scholar]
- 22 ••.Levine RL, Wadleigh M, Cools J, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7(4):387–397. doi: 10.1016/j.ccr.2005.03.023. Identification of a genetic defect associated with primary myelofibrosis (PMF).
- 23 ••.James C, Ugo V, Le Couedic JP, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434(7037):1144–1148. doi: 10.1038/nature03546. Identification of a genetic defect associated with PMF.
- 24 ••.Baxter EJ, Scott LM, Campbell PJ, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365(9464):1054–1061. doi: 10.1016/S0140-6736(05)71142-9. Identification of a genetic defect associated with PMF.
- 25.Kilpivaara O, Levine RL. JAK2 and MPL mutations in myeloproliferative neoplasms: discovery and science. Leukemia. 2008;22(10):1813–1817. doi: 10.1038/leu.2008.229. [DOI] [PubMed] [Google Scholar]
- 26.Mercher T, Wernig G, Moore SA, et al. JAK2T875N is a novel activating mutation that results in myeloproliferative disease with features of megakaryoblastic leukemia in a murine bone marrow transplantation model. Blood. 2006;108(8):2770–2779. doi: 10.1182/blood-2006-04-014712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bersenev A, Wu C, Balcerek J, Tong W. Lnk controls mouse hematopoietic stem cell self-renewal and quiescence through direct interactions with JAK2. J. Clin. Invest. 2008;118(8):2832–2844. doi: 10.1172/JCI35808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Royer Y, Staerk J, Costuleanu M, Courtoy PJ, Constantinescu SN. Janus kinases affect thrombopoietin receptor cell surface localization and stability. J. Biol. Chem. 2005;280(29):27251–27261. doi: 10.1074/jbc.M501376200. [DOI] [PubMed] [Google Scholar]
- 29.Grebien F, Kerenyi MA, Kovacic B, et al. Stat5 activation enables erythropoiesis in the absence of EpoR and Jak2. Blood. 2008;111(9):4511–4522. doi: 10.1182/blood-2007-07-102848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Funakoshi-Tago M, Pelletier S, Moritake H, Parganas E, Ihle JN. Jak2 FERM domain interaction with the erythropoietin receptor regulates Jak2 kinase activity. Mol. Cell. Biol. 2008;28(5):1792–1801. doi: 10.1128/MCB.01447-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Funakoshi-Tago M, Tago K, Kasahara T, Parganas E, Ihle JN. Negative regulation of Jak2 by its auto-phosphorylation at tyrosine 913 via the Epo signaling pathway. Cell. Signal. 2008;20(11):1995–2001. doi: 10.1016/j.cellsig.2008.07.008. [DOI] [PubMed] [Google Scholar]
- 32.Wang L, Xue J, Zadorozny EV, Robinson LJ. G-CSF stimulates Jak2-dependent Gab2 phosphorylation leading to Erk1/2 activation and cell proliferation. Cell. Signal. 2008;20(10):1890–1899. doi: 10.1016/j.cellsig.2008.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Radosevic N, Winterstein D, Keller JR, et al. JAK2 contributes to the intrinsic capacity of primary hematopoietic cells to respond to stem cell factor. Exp. Hematol. 2004;32(2):149–156. doi: 10.1016/j.exphem.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 34.Drayer AL, Boer AK, Los EL, Esselink MT, Vellenga E. Stem cell factor synergistically enhances thrombopoietin-induced STAT5 signaling in megakaryocyte progenitors through JAK2 and Src kinase. Stem Cells. 2005;23(2):240–251. doi: 10.1634/stemcells.2004-0153. [DOI] [PubMed] [Google Scholar]
- 35.Arcasoy MO, Jiang X. Co-operative signalling mechanisms required for erythroid precursor expansion in response to erythropoietin and stem cell factor. Br. J. Haematol. 2005;130(1):121–129. doi: 10.1111/j.1365-2141.2005.05580.x. [DOI] [PubMed] [Google Scholar]
- 36 ••.Tefferi A. JAK and MPL mutations in myeloid malignancies. Leuk. Lymphoma. 2008;49(3):388–397. doi: 10.1080/10428190801895360. Up-to-date review on the role of JAK2 and MPL mutations in the pathogenesis of hematological disorders.
- 37.Lu X, Huang LJ, Lodish HF. Dimerization by a cytokine receptor is necessary for constitutive activation of JAK2 V617F. J. Biol. Chem. 2008;283(9):5258–5266. doi: 10.1074/jbc.M707125200. [DOI] [PubMed] [Google Scholar]
- 38.Tefferi A, Gilliland DG. JAK2 in myeloproliferative disorders is not just another kinase. Cell Cycle. 2005;4(8):1053–1056. [PubMed] [Google Scholar]
- 39.Zhao ZJ, Vainchenker W, Krantz SB, Casadevall N, Constantinescu SN. Role of tyrosine kinases and phosphatases in polycythemia vera. Semin. Hematol. 2005;42(4):221–229. doi: 10.1053/j.seminhematol.2005.05.019. [DOI] [PubMed] [Google Scholar]
- 40.Scott LM, Tong W, Levine RL, et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N. Engl. J. Med. 2007;356(5):459–468. doi: 10.1056/NEJMoa065202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cramer K, Nieborowska-Skorska M, Koptyra M, et al. BCR/ABL and other kinases from chronic myeloproliferative disorders stimulate single-strand annealing, an unfaithful DNA double-strand break repair. Cancer Res. 2008;68(17):6884–6888. doi: 10.1158/0008-5472.CAN-08-1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tomasson MH, Williams IR, Li S, et al. Induction of myeloproliferative disease in mice by tyrosine kinase fusion oncogenes does not require granulocyte—macrophage colony-stimulating factor or interleukin-3. Blood. 2001;97(5):1435–1441. doi: 10.1182/blood.v97.5.1435. [DOI] [PubMed] [Google Scholar]
- 43.Schwaller J, Frantsve J, Aster J, et al. Transformation of hematopoietic cell lines to growth-factor independence and induction of a fatal myelo- and lymphoproliferative disease in mice by retrovirally transduced TEL/JAK2 fusion genes. EMBO J. 1998;17(18):5321–5333. doi: 10.1093/emboj/17.18.5321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lacronique V, Boureux A, Valle VD, et al. A TEL—JAK2 fusion protein with constitutive kinase activity in human leukemia. Science. 1997;278(5341):1309–1312. doi: 10.1126/science.278.5341.1309. [DOI] [PubMed] [Google Scholar]
- 45.Pardanani AD, Levine RL, Lasho T, et al. MPL515 mutations in myeloproliferative and other myeloid disorders: a study of 1182 patients. Blood. 2006;108(10):3472–3476. doi: 10.1182/blood-2006-04-018879. [DOI] [PubMed] [Google Scholar]
- 46.Pikman Y, Lee BH, Mercher T, et al. MPL W515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006;3(7):e270. doi: 10.1371/journal.pmed.0030270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yoshihara H, Arai F, Hosokawa K, et al. Thrombopoietin/MPL signaling regulates hematopoietic stem cell quiescence and interaction with the osteoblastic niche. Cell Stem Cell. 2007;1(6):685–697. doi: 10.1016/j.stem.2007.10.020. [DOI] [PubMed] [Google Scholar]
- 48.Adams GB, Scadden DT. The hematopoietic stem cell in its place. Nat. Immunol. 2006;7(4):333–337. doi: 10.1038/ni1331. [DOI] [PubMed] [Google Scholar]
- 49.Moore KA, Lemischka IR. Stem cells and their niches. Science. 2006;311(5769):1880–1885. doi: 10.1126/science.1110542. [DOI] [PubMed] [Google Scholar]
- 50.Migliaccio AR, Rana RA, Vannucchi AM, Manzoli FA. Role of thrombopoietin in mast cell differentiation. Ann. NY Acad. Sci. 2007;1106:152–174. doi: 10.1196/annals.1392.024. [DOI] [PubMed] [Google Scholar]
- 51.Chang Y, Bluteau D, Debili N, Vainchenker W. From hematopoietic stem cells to platelets. J. Thromb. Haemost. 2007;5(Suppl 1):318–327. doi: 10.1111/j.1538-7836.2007.02472.x. [DOI] [PubMed] [Google Scholar]
- 52.Rumi E. Familial chronic myeloproliferative disorders: the state of the art. Hematol. Oncol. 2008;26(3):131–138. doi: 10.1002/hon.863. [DOI] [PubMed] [Google Scholar]
- 53.Rossbach HC. Familial infantile myelofibrosis as an autosomal recessive disorder: preponderance among children from Saudi Arabia. Pediatr. Hematol. Oncol. 2006;23(5):453–454. doi: 10.1080/08880010600623240. [DOI] [PubMed] [Google Scholar]
- 54.Sheikha A. Fatal familial infantile myelofibrosis. J. Pediatr. Hematol. Oncol. 2004;26(3):164–168. doi: 10.1097/00043426-200403000-00005. [DOI] [PubMed] [Google Scholar]
- 55.Bonduel M, Sciuccati G, Torres AF, Pierini A, Gallo G. Familial idiopathic myelofibrosis and multiple hemangiomas. Am. J. Hematol. 1998;59(2):175–177. doi: 10.1002/(sici)1096-8652(199810)59:2<175::aid-ajh13>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 56.Pastore C, Nomdedeu J, Volpe G, et al. Genetic analysis of chromosome 13 deletions in BCR/ABL negative chronic myeloproliferative disorders. Genes Chromosomes Cancer. 1995;14(2):106–111. doi: 10.1002/gcc.2870140204. [DOI] [PubMed] [Google Scholar]
- 57.Reilly JT, Snowden JA, Spearing RL, et al. Cytogenetic abnormalities and their prognostic significance in idiopathic myelofibrosis: a study of 106 cases. Br. J. Haematol. 1997;98(1):96–102. doi: 10.1046/j.1365-2141.1997.1722990.x. [DOI] [PubMed] [Google Scholar]
- 58.Juneau AL, Kaehler M, Christensen ER, et al. Detection of RB1 deletions by fluorescence in situ hybridization in malignant hematologic disorders. Cancer Genet. Cytogenet. 1998;103(2):117–123. doi: 10.1016/s0165-4608(97)00408-1. [DOI] [PubMed] [Google Scholar]
- 59.Wurster-Hill DH, Cornwell GG, 3rd, McIntyre OR. Chromosomal aberrations and neoplasm — a family study. Cancer. 1974;33(1):72–81. doi: 10.1002/1097-0142(197401)33:1<72::aid-cncr2820330114>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 60.Ohyashiki K, Kodama A, Ohyashiki JH. Recurrent der(9;18) in essential thrombocythemia with JAK2 V617F is highly linked to myelofibrosis development. Cancer Genet. Cytogenet. 2008;186(1):6–11. doi: 10.1016/j.cancergencyto.2008.04.021. [DOI] [PubMed] [Google Scholar]
- 61.Santana-Davila R, Tefferi A, Holtan SG, et al. Primary myelofibrosis is the most frequent myeloproliferative neoplasm associated with del(5q): clinicopathologic comparison of del(5q)-positive and -negative cases. Leuk. Res. 2008;32(12):1927–1930. doi: 10.1016/j.leukres.2008.04.022. [DOI] [PubMed] [Google Scholar]
- 62.Nakata Y, Kimura A, Katoh O, Kawaishi K, Hyodo H. c-kit point mutation of extracellular domain in patients with myeloproliferative disorders. Br. J. Hematol. 1995;91(3):661–663. doi: 10.1111/j.1365-2141.1995.tb05364.x. [DOI] [PubMed] [Google Scholar]
- 63.Kimura A, Nakata Y, Katoh O, Hyodo H. c-kit Point mutation in patients with myeloproliferative disorders. Leuk. Lymphoma. 1997;25(3–4):281–287. doi: 10.3109/10428199709114167. [DOI] [PubMed] [Google Scholar]
- 64.Longley BJ, Jr., Metcalfe DD, Tharp M, et al. Activating and dominant inactivating c-KIT catalytic domain mutations in distinct clinical forms of human mastocytosis. Proc. Natl Acad. Sci. USA. 1999;96(4):1609–1614. doi: 10.1073/pnas.96.4.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Longley BJ, Tyrrell L, Lu SZ, et al. Somatic c-KIT activating mutation in urticaria pigmentosa and aggressive mastocytosis: establishment of clonality in a human mast cell neoplasm. Nat. Genet. 1996;12(3):312–314. doi: 10.1038/ng0396-312. [DOI] [PubMed] [Google Scholar]
- 66.Munugalavadla V, Sims EC, Chan RJ, Lenz SD, Kapur R. Requirement for p85α regulatory subunit of class IA PI3K in myeloproliferative disease driven by an activation loop mutant of KIT. Exp. Hematol. 2008;36(3):301–308. doi: 10.1016/j.exphem.2007.11.008. [DOI] [PubMed] [Google Scholar]
- 67.Vidovic A, Jankovic G, Colovic M, et al. The proto-oncogene expression varies over the course of chronic myeloid leukemia. Hematology. 2008;13(1):34–40. doi: 10.1179/102453308X315807. [DOI] [PubMed] [Google Scholar]
- 68.Kaushansky K. On the molecular origins of the chronic myeloproliferative disorders: it all makes sense. Hematology Am. Soc. Hematol. Educ. Program. 2005;2005:533–537. doi: 10.1182/asheducation-2005.1.533. [DOI] [PubMed] [Google Scholar]
- 69.Geron I, Abrahamsson AE, Barroga CF, et al. Selective inhibition of JAK2-driven erythroid differentiation of polycythemia vera progenitors. Cancer Cell. 2008;13(4):321–330. doi: 10.1016/j.ccr.2008.02.017. [DOI] [PubMed] [Google Scholar]
- 70.Pardanani A. JAK2 inhibitor therapy in myeloproliferative disorders: rationale, preclinical studies and ongoing clinical trials. Leukemia. 2008;22(1):23–30. doi: 10.1038/sj.leu.2404948. [DOI] [PubMed] [Google Scholar]
- 71.Schulze H, Korpal M, Hurov J, et al. Characterization of the megakaryocyte demarcation membrane system and its role in thrombopoiesis. Blood. 2006;107(10):3868–3875. doi: 10.1182/blood-2005-07-2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Italiano JE, Jr, Richardson JL, Patel-Hett S, et al. Angiogenesis is regulated by a novel mechanism: pro- and antiangiogenic proteins are organized into separate platelet alpha granules and differentially released. Blood. 2008;111(3):1227–1233. doi: 10.1182/blood-2007-09-113837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Le Bousse-Kerdiles MC, Martyre MC. Dual implication of fibrogenic cytokines in the pathogenesis of fibrosis and myeloproliferation in myeloid metaplasia with myelofibrosis. Ann. Hematol. 1999;78(10):437–444. doi: 10.1007/s002770050595. [DOI] [PubMed] [Google Scholar]
- 74 ••.Chagraoui H, Komura E, Tulliez M, et al. Prominent role of TGF-β 1 in thrombopoietin-induced myelofibrosis in mice. Blood. 2002;100(10):3495–3503. doi: 10.1182/blood-2002-04-1133. Gene complementation study proving the prominent role of high levels of TGF-β expression in the pathogenesis of myelofibrosis in the thrombopoietin (TPO)high mouse model.
- 75.Chagraoui H, Tulliez M, Smayra T, et al. Stimulation of osteoprotegerin production is responsible for osteosclerosis in mice overexpressing TPO. Blood. 2003;101(8):2983–2989. doi: 10.1182/blood-2002-09-2839. [DOI] [PubMed] [Google Scholar]
- 76 ••.Vannucchi AM, Bianchi L, Paoletti F, et al. A pathobiologic pathway linking thrombopoietin, GATA-1, and TGF-β1 in the development of myelofibrosis. Blood. 2005;105(9):3493–3501. doi: 10.1182/blood-2004-04-1320. Comparison of the TPOhigh and Gata1low models of myelofibrosis and describe the paradoxic curative effect of TPO in Gata1low mice
- 77.Border WA, Noble NA. Transforming growth factor β in tissue fibrosis. N. Engl. J. Med. 1994;331(19):1286–1292. doi: 10.1056/NEJM199411103311907. [DOI] [PubMed] [Google Scholar]
- 78.Kimura A, Katoh O, Hyodo H, Kuramoto A. Transforming growth factor-β regulates growth as well as collagen and fibronectin synthesis of human marrow fibroblasts. Br. J. Haematol. 1989;72(4):486–491. doi: 10.1111/j.1365-2141.1989.tb04310.x. [DOI] [PubMed] [Google Scholar]
- 79.Roberts AB, Sporn MB, Assoian RK, et al. Transforming growth factor type β: rapid induction of fibrosis and angiogenesis in vivo and stimulation of collagen formation in vitro. Proc. Natl Acad. Sci. USA. 1986;83(12):4167–4171. doi: 10.1073/pnas.83.12.4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nachman RL, Rafii S. Platelets, petechiae, and preservation of the vascular wall. N. Engl. J. Med. 2008;359(12):1261–1270. doi: 10.1056/NEJMra0800887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mohle R, Green D, Moore MA, Nachman RL, Rafii S. Constitutive production and thrombin-induced release of vascular endothelial growth factor by human megakaryocytes and platelets. Proc. Natl Acad. Sci. USA. 1997;94(2):663–668. doi: 10.1073/pnas.94.2.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Vannucchi AM, Pancrazzi A, Guglielmelli P, et al. Abnormalities of GATA-1 in megakaryocytes from patients with idiopathic myelofibrosis. Am. J. Pathol. 2005;167(3):849–858. doi: 10.1016/S0002-9440(10)62056-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Orkin SH, Shivdasani RA, Fujiwara Y, McDevitt MA. Transcription factor GATA-1 in megakaryocyte development. Stem Cells. 1998;16(Suppl 2):79–83. doi: 10.1002/stem.5530160710. [DOI] [PubMed] [Google Scholar]
- 84.Cashell AW, Buss DH. The frequency and significance of megakaryocytic emperipolesis in myeloproliferative and reactive states. Ann. Hematol. 1992;64(6):273–276. doi: 10.1007/BF01695470. [DOI] [PubMed] [Google Scholar]
- 85 ••.Schmitt A, Jouault H, Guichard J, et al. Pathologic interaction between megakaryocytes and polymorphonuclear leukocytes in myelofibrosis. Blood. 2000;96(4):1342–1347. The first characterization of the specific megakacyotic abnormalities associated with PMF.
- 86.Rameshwar P, Narayanan R, Qian J, et al. NF-κ B as a central mediator in the induction of TGF-β in monocytes from patients with idiopathic myelofibrosis: an inflammatory response beyond the realm of homeostasis. J. Immunol. 2000;165(4):2271–2277. doi: 10.4049/jimmunol.165.4.2271. [DOI] [PubMed] [Google Scholar]
- 87.Amabile G, Di Noia A, Alfani E, et al. Isolation of TPO-dependent subclones from the multipotent 32D cell line. Blood Cells Mol. Dis. 2005;35(2):241–252. doi: 10.1016/j.bcmd.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 88.Wernig G, Mercher T, Okabe R, et al. Expression of Jak2V617F causes a polycythemia vera-like disease with associated myelofibrosis in a murine bone marrow transplant model. Blood. 2006;107(11):4274–4281. doi: 10.1182/blood-2005-12-4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lacout C, Pisani DF, Tulliez M, et al. JAK2 V617F expression in murine hematopoietic cells leads to MPD mimicking human PV with secondary myelofibrosis. Blood. 2006;108(5):1652–1660. doi: 10.1182/blood-2006-02-002030. [DOI] [PubMed] [Google Scholar]
- 90.Chaligne R, James C, Tonetti C, et al. Evidence for MPL W515L/K mutations in hematopoietic stem cells in primitive myelofibrosis. Blood. 2007;110(10):3735–3743. doi: 10.1182/blood-2007-05-089003. [DOI] [PubMed] [Google Scholar]
- 91.Takaki S, Watts JD, Forbush KA, et al. Characterization of Lnk. An adaptor protein expressed in lymphocytes. J. Biol. Chem. 1997;272(23):14562–14570. doi: 10.1074/jbc.272.23.14562. [DOI] [PubMed] [Google Scholar]
- 92.Huang X, Li Y, Tanaka K, Moore KG, Hayashi JI. Cloning and characterization of Lnk, a signal transduction protein that links T-cell receptor activation signal to phospholipase C γ 1, Grb2, and phosphatidylinositol 3-kinase. Proc. Natl Acad. Sci. USA. 1995;92(25):11618–11622. doi: 10.1073/pnas.92.25.11618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Velazquez L, Cheng AM, Fleming HE, et al. Cytokine signaling and hematopoietic homeostasis are disrupted in Lnk-deficient mice. J. Exp. Med. 2002;195(12):1599–1611. doi: 10.1084/jem.20011883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ema H, Sudo K, Seita J, et al. Quantification of self-renewal capacity in single hematopoietic stem cells from normal and Lnk-deficient mice. Dev. Cell. 2005;8(6):907–914. doi: 10.1016/j.devcel.2005.03.019. [DOI] [PubMed] [Google Scholar]
- 95.Takaki S, Morita H, Tezuka Y, Takatsu K. Enhanced hematopoiesis by hematopoietic progenitor cells lacking intracellular adaptor protein, Lnk. J. Exp. Med. 2002;195(2):151–160. doi: 10.1084/jem.20011170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Takizawa H, Eto K, Yoshikawa A, et al. Growth and maturation of megakaryocytes is regulated by Lnk/Sh2b3 adaptor protein through crosstalk between cytokine- and integrin-mediated signals. Exp. Hematol. 2008;36(7):897–906. doi: 10.1016/j.exphem.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 97 ••.Vannucchi AM, Bianchi L, Cellai C, et al. Development of myelofibrosis in mice genetically impaired for GATA-1 expression (GATA-1low mice) Blood. 2002;100(4):1123–1132. doi: 10.1182/blood-2002-06-1913. First report on the Gata1low mouse model of myelofibrosis.
- 98.Toki T, Katsuoka F, Kanezaki R, et al. Transgenic expression of BACH1 transcription factor results in megakaryocytic impairment. Blood. 2005;105(8):3100–3108. doi: 10.1182/blood-2004-07-2826. [DOI] [PubMed] [Google Scholar]
- 99 ••.Walkley CR, Olsen GH, Dworkin S, et al. A microenvironment-induced myeloproliferative syndrome caused by retinoic acid receptor γ deficiency. Cell. 2007;129(6):1097–1110. doi: 10.1016/j.cell.2007.05.014. Identification of a genetic defect associated with PMF.
- 100 ••.Walkley CR, Shea JM, Sims NA, Purton LE, Orkin SH. Rb regulates interactions between hematopoietic stem cells and their bone marrow microenvironment. Cell. 2007;129(6):1081–1095. doi: 10.1016/j.cell.2007.03.055. First description of a microenvironment mediated mouse model of myelofibrosis.
- 101.de Sauvage FJ, Carver-Moore K, Luoh SM, et al. Physiological regulation of early and late stages of megakaryocytopoiesis by thrombopoietin. J. Exp. Med. 1996;183(2):651–656. doi: 10.1084/jem.183.2.651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kaushansky K. Thrombopoietin: the primary regulator of platelet production. Blood. 1995;86(2):419–431. [PubMed] [Google Scholar]
- 103.Burmester H, Wolber EM, Freitag P, Fandrey J, Jelkmann W. Thrombopoietin production in wild-type and interleukin-6 knockout mice with acute inflammation. J. Interferon Cytokine Res. 2005;25(7):407–413. doi: 10.1089/jir.2005.25.407. [DOI] [PubMed] [Google Scholar]
- 104.Linthorst GE, Folman CC, van Olden RW, von dem Borne AE. Plasma thrombopoietin levels in patients with chronic renal failure. Hematol. J. 2002;3(1):38–42. doi: 10.1038/sj.thj.6200153. [DOI] [PubMed] [Google Scholar]
- 105 ••.Kaushansky K. The molecular mechanisms that control thrombopoiesis. J. Clin. Invest. 2005;115(12):3339–3347. doi: 10.1172/JCI26674. Extensive review on the role of TPO in megakaryocytopoiesis.
- 106.Kaushansky K. Thrombopoietin: a tool for understanding thrombopoiesis. J. Thromb. Haemost. 2003;1(7):1587–1592. doi: 10.1046/j.1538-7836.2003.00273.x. [DOI] [PubMed] [Google Scholar]
- 107.Kuter DJ. Thrombopoietin: biology and clinical applications. Oncologist. 1996;1(1–2):98–106. [PubMed] [Google Scholar]
- 108 ••.Ulich TR, del Castillo J, Senaldi G, et al. Systemic hematologic effects of PEG-rHuMGDF-induced megakaryocyte hyperplasia in mice. Blood. 1996;87(12):5006–5015. First report that systemic TPO administration induces myelofibrosis in mice.
- 109.Yanagida M, Ide Y, Imai A, et al. The role of transforming growth factor-β in PEG-rHuMGDF-induced reversible myelofibrosis in rats. Br. J. Haematol. 1997;99(4):739–745. doi: 10.1046/j.1365-2141.1997.4843288.x. [DOI] [PubMed] [Google Scholar]
- 110.Frey BM, Rafii S, Teterson M, et al. Adenovector-mediated expression of human thrombopoietin cDNA in immune-compromised mice: insights into the pathophysiology of osteomyelofibrosis. J. Immunol. 1998;160(2):691–699. [PubMed] [Google Scholar]
- 111.Abina MA, Tulliez M, Duffour MT, et al. Thrombopoietin (TPO) knockout phenotype induced by cross-reactive antibodies against TPO following injection of mice with recombinant adenovirus encoding human TPO. J. Immunol. 1998;160(9):4481–4489. [PubMed] [Google Scholar]
- 112.Cannizzo SJ, Frey BM, Raffi S, et al. Augmentation of blood platelet levels by intratracheal administration of an adenovirus vector encoding human thrombopoietin cDNA. Nat. Biotechnol. 1997;15(6):570–573. doi: 10.1038/nbt0697-570. [DOI] [PubMed] [Google Scholar]
- 113.Ohwada A, Rafii S, Moore MA, Crystal RG. In vivo adenovirus vector-mediated transfer of the human thrombopoietin cDNA maintains platelet levels during radiation-and chemotherapy-induced bone marrow suppression. Blood. 1996;88(3):778–784. [PubMed] [Google Scholar]
- 114.Wagner-Ballon O, Chagraoui H, Prina E, et al. Monocyte/macrophage dysfunctions do not impair the promotion of myelofibrosis by high levels of thrombopoietin. J. Immunol. 2006;176(11):6425–6433. doi: 10.4049/jimmunol.176.11.6425. [DOI] [PubMed] [Google Scholar]
- 115 ••.Villeval JL, Cohen-Solal K, Tulliez M, et al. High thrombopoietin production by hematopoietic cells induces a fatal myeloproliferative syndrome in mice. Blood. 1997;90(11):4369–4383. Describes a retrovirally induced TPOhigh model of myelofibrosis
- 116 ••.Yan XQ, Lacey D, Fletcher F, et al. Chronic exposure to retroviral vector encoded MGDF (mpl-ligand) induces lineage-specific growth and differentiation of megakaryocytes in mice. Blood. 1995;86(11):4025–4033. Describes a retrovirally-induced TPOhigh model of myelofibrosis
- 117.Zhou W, Toombs CF, Zou T, Guo J, Robinson MO. Transgenic mice overexpressing human c-mpl ligand exhibit chronic thrombocytosis and display enhanced recovery from 5-fluorouracil or antiplatelet serum treatment. Blood. 1997;89(5):1551–1559. [PubMed] [Google Scholar]
- 118.Kakumitsu H, Kamezaki K, Shimoda K, et al. Transgenic mice overexpressing murine thrombopoietin develop myelofibrosis and osteosclerosis. Leuk. Res. 2005;29(7):761–769. doi: 10.1016/j.leukres.2004.12.009. [DOI] [PubMed] [Google Scholar]
- 119.Bock O, Loch G, Schade U, et al. Osteosclerosis in advanced chronic idiopathic myelofibrosis is associated with endothelial overexpression of osteoprotegerin. Br. J. Haematol. 2005;130(1):76–82. doi: 10.1111/j.1365-2141.2005.05573.x. [DOI] [PubMed] [Google Scholar]
- 120.Evrard S, Tulliez M, Zetterberg E, et al. Increased myelofibrosis in thrombospondin-1 null mice is associated with enhanced TGF-β1-mediated response by bone marrow fibroblasts. American Society of Hematology 50th Annual Meeting; San Fransisco, CA, USA. 6–9 December (2008). [Google Scholar]
- 121.Tong W, Lodish HF. Lnk inhibits Tpo—mpl signaling and Tpo-mediated megakaryocytopoiesis. J. Exp. Med. 2004;200(5):569–580. doi: 10.1084/jem.20040762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Tong W, Zhang J, Lodish HF. Lnk inhibits erythropoiesis and Epo-dependent JAK2 activation and downstream signaling pathways. Blood. 2005;105(12):4604–4612. doi: 10.1182/blood-2004-10-4093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Gueller S, Gery S, Nowak V, et al. Adaptor protein Lnk associates with Tyr(568) in c-Kit. Biochem. J. 2008;415(2):241–245. doi: 10.1042/BJ20080102. [DOI] [PubMed] [Google Scholar]
- 124.Simon C, Dondi E, Chaix A, et al. Lnk adaptor protein down-regulates specific Kit-induced signaling pathways in primary mast cells. Blood. 2008;112(10):4039–4047. doi: 10.1182/blood-2008-05-154849. [DOI] [PubMed] [Google Scholar]
- 125.Gery S, Gueller S, Chumakova K, et al. Adaptor protein Lnk negatively regulates the mutant MPL, MPL W515L associated with myeloproliferative disorders. Blood. 2007;110(9):3360–3364. doi: 10.1182/blood-2007-05-089326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Xing S, Wanting TH, Zhao W, et al. Transgenic expression of JAK2 V617F causes myeloproliferative disorders in mice. Blood. 2008;111(10):5109–5117. doi: 10.1182/blood-2007-05-091579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kennedy JA, Barabe F, Patterson BJ, et al. Expression of TEL—JAK2 in primary human hematopoietic cells drives erythropoietin-independent erythropoiesis and induces myelofibrosis in vivo. Proc. Natl Acad. Sci. USA. 2006;103(45):16930–16935. doi: 10.1073/pnas.0604902103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Sternberg DW, Tomasson MH, Carroll M, et al. The TEL/PDGFβR fusion in chronic myelomonocytic leukemia signals through STAT5-dependent and STAT5-independent pathways. Blood. 2001;98(12):3390–3397. doi: 10.1182/blood.v98.12.3390. [DOI] [PubMed] [Google Scholar]
- 129.Ritchie KA, Aprikyan AA, Bowen-Pope DF, et al. The Tel-PDGFRβ fusion gene produces a chronic myeloproliferative syndrome in transgenic mice. Leukemia. 1999;13(11):1790–1803. doi: 10.1038/sj.leu.2401494. [DOI] [PubMed] [Google Scholar]
- 130.Tomasson MH, Sternberg DW, Williams IR, et al. Fatal myeloproliferation, induced in mice by TEL/PDGFβR expression, depends on PDGFβR tyrosines 579/581. J. Clin. Invest. 2000;105(4):423–432. doi: 10.1172/JCI8902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Yu C, Cantor AB, Yang H, et al. Targeted deletion of a high-affinity GATA-binding site in the GATA-1 promoter leads to selective loss of the eosinophil lineage in vivo. J. Exp. Med. 2002;195(11):1387–1395. doi: 10.1084/jem.20020656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Migliaccio AR, Rana RA, Vannucchi AM, Manzoli FA. Role of GATA-1 in normal and neoplastic hemopoiesis. Ann. NY Acad. Sci. 2005;1044:142–158. doi: 10.1196/annals.1349.019. [DOI] [PubMed] [Google Scholar]
- 133.Migliaccio AR, Rana RA, Sanchez M, et al. GATA-1 as a regulator of mast cell differentiation revealed by the phenotype of the GATA-1low mouse mutant. J. Exp. Med. 2003;197(3):281–296. doi: 10.1084/jem.20021149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Romeo PH, Prandini MH, Joulin V, et al. Megakaryocytic and erythrocytic lineages share specific transcription factors. Nature. 1990;344(6265):447–449. doi: 10.1038/344447a0. [DOI] [PubMed] [Google Scholar]
- 135.Tsai SF, Martin DI, Zon LI, et al. Cloning of cDNA for the major DNA-binding protein of the erythroid lineage through expression in mammalian cells. Nature. 1989;339(6224):446–451. doi: 10.1038/339446a0. [DOI] [PubMed] [Google Scholar]
- 136.Whyatt DJ, Karis A, Harkes IC, et al. The level of the tissue-specific factor GATA-1 affects the cell-cycle machinery. Genes Funct. 1997;1(1):11–24. doi: 10.1046/j.1365-4624.1997.00003.x. [DOI] [PubMed] [Google Scholar]
- 137.Cantor AB, Orkin SH. Hematopoietic development: a balancing act. Curr. Opin. Genet. Dev. 2001;11(5):513–519. doi: 10.1016/s0959-437x(00)00226-4. [DOI] [PubMed] [Google Scholar]
- 138.Pevny L, Simon MC, Robertson E, et al. Erythroid differentiation in chimaeric mice blocked by a targeted mutation in the gene for transcription factor GATA-1. Nature. 1991;349(6306):257–260. doi: 10.1038/349257a0. [DOI] [PubMed] [Google Scholar]
- 139.Patient RK, McGhee JD. The GATA family (vertebrates and invertebrates) Curr. Opin. Genet. Dev. 2002;12(4):416–422. doi: 10.1016/s0959-437x(02)00319-2. [DOI] [PubMed] [Google Scholar]
- 140.Cantor AB, Orkin SH. Transcriptional regulation of erythropoiesis: an affair involving multiple partners. Oncogene. 2002;21(21):3368–3376. doi: 10.1038/sj.onc.1205326. [DOI] [PubMed] [Google Scholar]
- 141.Fujiwara Y, Chang AN, Williams AM, Orkin SH. Functional overlap of GATA-1 and GATA-2 in primitive hematopoietic development. Blood. 2004;103(2):583–585. doi: 10.1182/blood-2003-08-2870. [DOI] [PubMed] [Google Scholar]
- 142.Kobayashi M, Yamamoto M. Regulation of GATA1 gene expression. J. Biochem. 2007;142(1):1–10. doi: 10.1093/jb/mvm122. [DOI] [PubMed] [Google Scholar]
- 143 •.McDevitt MA, Shivdasani RA, Fujiwara Y, Yang H, Orkin SH. A “knockdown” mutation created by cis-element gene targeting reveals the dependence of erythroid cell maturation on the level of transcription factor GATA-1. Proc. Natl Acad. Sci. USA. 1997;94(13):6781–6785. doi: 10.1073/pnas.94.13.6781. Historical paper describing the generation of the hypomorphic Gata1low mutation in mice and the effects of this mutation on erythroid development.
- 144 ••.Shivdasani RA, Fujiwara Y, McDevitt MA, Orkin SH. A lineage-selective knockout establishes the critical role of transcription factor GATA-1 in megakaryocyte growth and platelet development. EMBO J. 1997;16(13):3965–3973. doi: 10.1093/emboj/16.13.3965. First paper to describe the megakaryocytic defects induced by the hypomorphic Gata1low mutation.
- 145.Vyas P, Ault K, Jackson CW, Orkin SH, Shivdasani RA. Consequences of GATA-1 deficiency in megakaryocytes and platelets. Blood. 1999;93(9):2867–2875. [PubMed] [Google Scholar]
- 146.Martelli F, Ghinassi B, Panetta B, et al. Variegation of the phenotype induced by the Gata1low mutation in mice of different genetic backgrounds. Blood. 2005;106(13):4102–4113. doi: 10.1182/blood-2005-03-1060. [DOI] [PubMed] [Google Scholar]
- 147.Persons DA, Paulson RF, Loyd MR, et al. Fv2 encodes a truncated form of the Stk receptor tyrosine kinase. Nat. Genet. 1999;23(2):159–165. doi: 10.1038/13787. [DOI] [PubMed] [Google Scholar]
- 148.Lilly F. Fv-2: identification and location of a second gene governing the spleen focus response to Friend leukemia virus in mice. J. Natl Cancer Inst. 1970;45(1):163–169. [PubMed] [Google Scholar]
- 149 ••.Ghinassi B, Sanchez M, Martelli F, et al. The hypomorphic Gata1low mutation alters the proliferation/differentiation potential of the common megakaryocytic-erythroid progenitor. Blood. 2007;109(4):1460–1471. doi: 10.1182/blood-2006-07-030726. First report of the tumor suppressing activity of GATA1 in hematopoietic stem/progenitor cells.
- 150.Centurione L, Di Baldassarre A, Zingariello M, et al. Increased and pathologic emperipolesis of neutrophils within megakaryocytes associated with marrow fibrosis in GATA-1(low) mice. Blood. 2004;104(12):3573–3580. doi: 10.1182/blood-2004-01-0193. [DOI] [PubMed] [Google Scholar]
- 151.Kobayashi A, Ito E, Toki T, et al. Molecular cloning and functional characterization of a new Cap‘n’ collar family transcription factor Nrf3. J. Biol. Chem. 1999;274(10):6443–6452. doi: 10.1074/jbc.274.10.6443. [DOI] [PubMed] [Google Scholar]
- 152.Toki T, Itoh J, Kitazawa J, et al. Human small Maf proteins form heterodimers with CNC family transcription factors and recognize the NF—E2 motif. Oncogene. 1997;14(16):1901–1910. doi: 10.1038/sj.onc.1201024. [DOI] [PubMed] [Google Scholar]
- 153.Ohira M, Seki N, Nagase T, et al. Characterization of a human homolog (BACH1) of the mouse Bach1 gene encoding a BTB-basic leucine zipper transcription factor and its mapping to chromosome 21q22.1. Genomics. 1998;47(2):300–306. doi: 10.1006/geno.1997.5080. [DOI] [PubMed] [Google Scholar]
- 154.Blouin JL, Sail G Duriaux, Guipponi M, et al. Isolation of the human BACH1 transcription regulator gene, which maps to chromosome 21q22.1. Hum. Genet. 1998;102(3):282–288. doi: 10.1007/s004390050692. [DOI] [PubMed] [Google Scholar]
- 155.Terui K, Takahashi Y, Kitazawa J, et al. Expression of transcription factors during megakaryocytic differentiation of CD34+ cells from human cord blood induced by thrombopoietin. Tohoku J. Exp. Med. 2000;192(4):259–273. doi: 10.1620/tjem.192.259. [DOI] [PubMed] [Google Scholar]
- 156.Oyake T, Itoh K, Motohashi H, et al. Bach proteins belong to a novel family of BTB-basic leucine zipper transcription factors that interact with MafK and regulate transcription through the NF—E2 site. Mol. Cell. Biol. 1996;16(11):6083–6095. doi: 10.1128/mcb.16.11.6083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Talbot D, Philipsen S, Fraser P, Grosveld F. Detailed analysis of the site 3 region of the human β-globin dominant control region. Embo. J. 1990;9(7):2169–2177. doi: 10.1002/j.1460-2075.1990.tb07386.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Talbot D, Grosveld F. The 5′HS2 of the globin locus control region enhances transcription through the interaction of a multimeric complex binding at two functionally distinct NF—E2 binding sites. EMBO J. 1991;10(6):1391–1398. doi: 10.1002/j.1460-2075.1991.tb07659.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Ney PA, Sorrentino BP, McDonagh KT, Nienhuis AW. Tandem AP-1-binding sites within the human β-globin dominant control region function as an inducible enhancer in erythroid cells. Genes Dev. 1990;4(6):993–1006. doi: 10.1101/gad.4.6.993. [DOI] [PubMed] [Google Scholar]
- 160.Ney PA, Sorrentino BP, Lowrey CH, Nienhuis AW. Inducibility of the HS II enhancer depends on binding of an erythroid specific nuclear protein. Nucleic Acids Res. 1990;18(20):6011–6017. doi: 10.1093/nar/18.20.6011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Shivdasani RA, Rosenblatt MF, Zucker-Franklin D, et al. Transcription factor NF—E2 is required for platelet formation independent of the actions of thrombopoietin/MGDF in megakaryocyte development. Cell. 1995;81(5):695–704. doi: 10.1016/0092-8674(95)90531-6. [DOI] [PubMed] [Google Scholar]
- 162.Clarke B. Normal bone anatomy and physiology. Clin. J. Am. Soc. Nephrol. 2008;3(Suppl 3):S131–S139. doi: 10.2215/CJN.04151206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Phan TC, Xu J, Zheng MH. Interaction between osteoblast and osteoclast: impact in bone disease. Histol. Histopathol. 2004;19(4):1325–1344. doi: 10.14670/HH-19.1325. [DOI] [PubMed] [Google Scholar]
- 164.Kacena MA, Shivdasani RA, Wilson K, et al. Megakaryocyte—osteoblast interaction revealed in mice deficient in transcription factors GATA-1 and NF—E2. J. Bone Miner. Res. 2004;19(4):652–660. doi: 10.1359/JBMR.0301254. [DOI] [PubMed] [Google Scholar]
- 165.Beerepoot LV, Mehra N, Vermaat JS, et al. Increased levels of viable circulating endothelial cells are an indicator of progressive disease in cancer patients. Ann. Oncol. 2004;15(1):139–145. doi: 10.1093/annonc/mdh017. [DOI] [PubMed] [Google Scholar]
- 166.Zetterberg E, Lundberg LG, Palmblad J. Characterization of blood vessels in bone marrow from patients with chronic myeloid leukemia and polycythemia vera. Scand. J. Clin. Lab. Invest. 2004;64(7):641–647. doi: 10.1080/00365510410002968. [DOI] [PubMed] [Google Scholar]
- 167.Zetterberg E, Vannucchi AM, Migliaccio AR, et al. Pericyte coverage of abnormal blood vessels in myelofibrotic bone marrows. Haematologica. 2007;92(5):597–604. doi: 10.3324/haematol.11013. [DOI] [PubMed] [Google Scholar]
- 168.Sugiyama T, Kohara H, Noda M, Nagasawa T. Maintenance of the hematopoietic stem cell pool by CXCL12—CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity. 2006;25(6):977–988. doi: 10.1016/j.immuni.2006.10.016. [DOI] [PubMed] [Google Scholar]
- 169.Ara T, Tokoyoda K, Sugiyama T, et al. Long-term hematopoietic stem cells require stromal cell-derived factor-1 for colonizing bone marrow during ontogeny. Immunity. 2003;19(2):257–267. doi: 10.1016/s1074-7613(03)00201-2. [DOI] [PubMed] [Google Scholar]
- 170.McGrath KE, Koniski AD, Maltby KM, McGann JK, Palis J. Embryonic expression and function of the chemokine SDF-1 and its receptor, CXCR4. Dev. Biol. 1999;213(2):442–456. doi: 10.1006/dbio.1999.9405. [DOI] [PubMed] [Google Scholar]
- 171.Kim CH, Broxmeyer HE. In vitro behavior of hematopoietic progenitor cells under the influence of chemoattractants: stromal cell-derived factor-1, steel factor, and the bone marrow environment. Blood. 1998;91(1):100–110. [PubMed] [Google Scholar]
- 172.Dar A, Goichberg P, Shinder V, et al. Chemokine receptor CXCR4-dependent internalization and resecretion of functional chemokine SDF-1 by bone marrow endothelial and stromal cells. Nat. Immunol. 2005;6(10):1038–1046. doi: 10.1038/ni1251. [DOI] [PubMed] [Google Scholar]
- 173.Lane WJ, Dias S, Hattori K, et al. Stromal-derived factor 1-induced megakaryocyte migration and platelet production is dependent on matrix metalloproteinases. Blood. 2000;96(13):4152–4159. [PubMed] [Google Scholar]
- 174.Ma Q, Jones D, Borghesani PR, et al. Impaired B-lymphopoiesis, myelopoiesis, and derailed cerebellar neuron migration in CXCR4- and SDF-1-deficient mice. Proc. Natl Acad. Sci. USA. 1998;95(16):9448–9453. doi: 10.1073/pnas.95.16.9448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Sweeney EA, Lortat-Jacob H, Priestley GV, Nakamoto B, Papayannopoulou T. Sulfated polysaccharides increase plasma levels of SDF-1 in monkeys and mice: involvement in mobilization of stem/progenitor cells. Blood. 2002;99(1):44–51. doi: 10.1182/blood.v99.1.44. [DOI] [PubMed] [Google Scholar]
- 176.Pelus LM, Bian H, Fukuda S, et al. The CXCR4 agonist peptide, CTCE-0021, rapidly mobilizes polymorphonuclear neutrophils and hematopoietic progenitor cells into peripheral blood and synergizes with granulocyte colony-stimulating factor. Exp. Hematol. 2005;33(3):295–307. doi: 10.1016/j.exphem.2004.11.008. [DOI] [PubMed] [Google Scholar]
- 177.Petit I, Szyper-Kravitz M, Nagler A, et al. G-CSF induces stem cell mobilization by decreasing bone marrow SDF-1 and up-regulating CXCR4. Nat. Immunol. 2002;3(7):687–694. doi: 10.1038/ni813. [DOI] [PubMed] [Google Scholar]
- 178.Massa M, Rosti V, Ramajoli I, et al. Circulating CD34+, CD133+, and vascular endothelial growth factor receptor 2-positive endothelial progenitor cells in myelofibrosis with myeloid metaplasia. J. Clin. Oncol. 2005;23(24):5688–5695. doi: 10.1200/JCO.2005.09.021. [DOI] [PubMed] [Google Scholar]
- 179.Xu M, Bruno E, Chao J, et al. Constitutive mobilization of CD34+ cells into the peripheral blood in idiopathic myelofibrosis may be due to the action of a number of proteases. Blood. 2005;105(11):4508–4515. doi: 10.1182/blood-2004-08-3238. [DOI] [PubMed] [Google Scholar]
- 180.Passamonti F, Vanelli L, Malabarba L, et al. Clinical utility of the absolute number of circulating CD34-positive cells in patients with chronic myeloproliferative disorders. Haematologica. 2003;88(10):1123–1129. [PubMed] [Google Scholar]
- 181.Brouty-Boye D, Briard D, Azzarone B, et al. Effects of human fibroblasts from myelometaplasic and non-myelometaplasic hematopoietic tissues on CD34+ stem cells. Int. J. Cancer. 2001;92(4):484–488. doi: 10.1002/ijc.1222. [DOI] [PubMed] [Google Scholar]
- 182.Briard D, Brouty-Boye D, Giron-Michel J, et al. Impaired NK cell differentiation of blood-derived CD34+ progenitors from patients with myeloid metaplasia with myelofibrosis. Clin. Immunol. 2003;106(3):201–212. doi: 10.1016/s1521-6616(02)00046-3. [DOI] [PubMed] [Google Scholar]
- 183.Migliaccio AR, Martelli F, Verrucci M, et al. Altered SDF-1/CXCR4 axis in patients with primary myelofibrosis and in the Gata1low mouse model of the disease. Exp. Hematol. 2008;36(2):158–171. doi: 10.1016/j.exphem.2007.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184 ••.Rosti V, Massa M, Vannucchi AM, et al. The expression of CXCR4 is down-regulated on the CD34+ cells of patients with myelofibrosis with myeloid metaplasia. Blood Cells Mol. Dis. 2007;38(3):280–286. doi: 10.1016/j.bcmd.2007.01.003. This paper reports the reduced expression of CXCR4 on the stem/progenitor cells from PMF patients.
- 185.Ciurea SO, Merchant D, Mahmud N, et al. Pivotal contributions of megakaryocytes to the biology of idiopathic myelofibrosis. Blood. 2007;110(3):986–993. doi: 10.1182/blood-2006-12-064626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Grass JA, Boyer ME, Pal S, et al. GATA-1-dependent transcriptional repression of GATA-2 via disruption of positive autoregulation and domain-wide chromatin remodeling. Proc. Natl Acad. Sci. USA. 2003;100(15):8811–8816. doi: 10.1073/pnas.1432147100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Terskikh AV, Miyamoto T, Chang C, Diatchenko L, Weissman IL. Gene expression analysis of purified hematopoietic stem cells and committed progenitors. Blood. 2003;102(1):94–101. doi: 10.1182/blood-2002-08-2509. [DOI] [PubMed] [Google Scholar]
- 188.Migliaccio AR, Jiang Y, Migliaccio G, et al. Transcriptional and posttranscriptional regulation of the expression of the erythropoietin receptor gene in human erythropoietin-responsive cell lines. Blood. 1993;82(12):3760–3769. [PubMed] [Google Scholar]
- 189.Pal S, Cantor AB, Johnson KD, et al. Coregulator-dependent facilitation of chromatin occupancy by GATA-1. Proc. Natl Acad. Sci. USA. 2004;101(4):980–985. doi: 10.1073/pnas.0307612100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Takahashi S, Onodera K, Motohashi H, et al. Arrest in primitive erythroid cell development caused by promoter-specific disruption of the GATA-1 gene. J. Biol. Chem. 1997;272(19):12611–12615. doi: 10.1074/jbc.272.19.12611. [DOI] [PubMed] [Google Scholar]
- 191.Vyas P, Crispino JD. Molecular insights into Down syndrome-associated leukemia. Curr. Opin. Pediatr. 2007;19(1):9–14. doi: 10.1097/MOP.0b013e328013e7b2. [DOI] [PubMed] [Google Scholar]
- 192.Lange B. The management of neoplastic disorders of haematopoiesis in children with Down’s syndrome. Br. J. Haematol. 2000;110(3):512–524. doi: 10.1046/j.1365-2141.2000.02027.x. [DOI] [PubMed] [Google Scholar]
- 193.Li Z, Godinho FJ, Klusmann JH, et al. Developmental stage-selective effect of somatically mutated leukemogenic transcription factor GATA1. Nat. Genet. 2005;37(6):613–619. doi: 10.1038/ng1566. [DOI] [PubMed] [Google Scholar]
- 194.Kirsammer G, Jilani S, Liu H, et al. Highly penetrant myeloproliferative disease in the Ts65Dn mouse model of Down syndrome. Blood. 2008;111(2):767–775. doi: 10.1182/blood-2007-04-085670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Minucci S, Pelicci PG. Retinoid receptors in health and disease: co-regulators and the chromatin connection. Semin. Cell Dev. Biol. 1999;10(2):215–225. doi: 10.1006/scdb.1999.0303. [DOI] [PubMed] [Google Scholar]
- 196.Weinberg RA. The retinoblastoma gene and gene product. Cancer Surv. 1992;12:43–57. [PubMed] [Google Scholar]
- 197.Weinberg RA. The retinoblastoma gene and cell growth control. Trends Biochem. Sci. 1990;15(5):199–202. doi: 10.1016/0968-0004(90)90162-5. [DOI] [PubMed] [Google Scholar]
- 198.Delhommeau F, Dupont S, James C, et al. TET2 is a novel tumor suppressor gene inactivated in myeloproliferative neoplasms: identification of a Pre-JAK2 V617F event. (ASH annual meeting abstracts) Blood. 2008;112 (Abstract 3) [Google Scholar]
- 199.Niida S, Kondo T, Hiratsuka S, et al. VEGF receptor 1 signaling essential for osteoclast development and bone marrow formation in colony-stimulating factor 1-deficient mice. Proc. Natl Acad. Sci. USA. 2005;102:14016–14021. doi: 10.1073/pnas.0503544102. [DOI] [PMC free article] [PubMed] [Google Scholar]