

Abstract
Purpose
β-Lapachone (β-lap, ARQ 501) is a novel anticancer agent with selectivity against prostate cancer cells over-expressing the NAD(P)H:quinone oxidoreductase-1 (NQO1) enzyme. Lack of solubility and an efficient drug delivery strategy limits this compound in clinical applications. In this study, we aimed to develop β-lap-containing polymer implants (millirods) for direct implantation into prostate tumors to test the hypothesis that the combination of a tumor-specific anticancer agent with site-specific release of the agent will lead to significant antitumor efficacy.
Experimental Design
Survival assays in vitro were used to test β-lap killing effect in different prostate cancer cells. β-Lap release kinetics from millirods was determined both in vitro and in vivo. PC-3 prostate tumor xenografts in athymic nude mice were employed for antitumor efficacy studies in vivo.
Results
β-Lap killed three different prostate cancer cell lines in an NQO1-dependent manner. Upon incorporation of solid-state inclusion complexes of β-lap with hydroxypropyl-β-cyclodextrin (HPβ-CD) into poly(D,L-lactide-co-glycolide) (PLGA) millirods, β-lap release kinetics in vivo showed a burst release of ~0.5 mg within 12 h and a subsequently sustained release of the drug (~0.4 mg/kg/day) comparable to that observed in vitro. Antitumor efficacy studies demonstrated significant tumor growth inhibition by β-lap millirods compared to controls (p value <0.0001, n =10/group). Kaplan-Meier survival curves showed that tumor-bearing mice treated with β-lap millirods survived nearly two-fold longer than controls, without observable systemic toxicity.
Conclusions
Intratumoral delivery of β-lap using polymer millirods demonstrated the promising therapeutic potential for human prostate tumors.
Keywords: β-Lapachone; controlled release drug delivery; poly(D,L-lactide-co-glycolide) (PLGA); prostate cancer; intratumoral chemotherapy
Introduction
The incidence of prostate cancer is the highest among all estimated new cancer cases in American males, nearly doubling that of lung cancer. Prostate cancer was the second leading cause of cancer-related deaths in men in 2007 (1). With improvements in therapeutic technologies in early stage prostate cancer treatment, especially less invasive therapies such as brachytherapy, cryotherapy, and high-intensity focused ultrasound, the morbidity associated with prostate cancer treatment has been substantially reduced. However, concerns remain over incomplete and inadequate treatment given the high rate of recurrence after primary salvage therapy (2, 3). Moreover, most procedures have notable side-effects, including high rates of incontinence and impotence. Therefore, alternate strategies have been explored to treat the disease. The development of targeted strategies that exclusively kill prostate cancer cells in a tumor- and site-specific manner should yield increased antitumor efficacy, reduced normal tissue toxicity, and improved quality of life.
β-Lapachone (β-lap, ARQ 501) is a novel, 1,2-orthonaphthoquinone originally derived from the bark of the Lapacho tree in South America. It possesses a wide range of activities including antifungal, antiviral, antitrypanosomal, and antitumor properties (4). Recent studies have shown that the mechanism of action of β-lap is highly specific and depends on expression of NAD(P)H:quinone oxidoreductase-1 (NQO1), a two electron oxidoreductase. NQO1 is a flavoprotein found overexpressed up to 20-fold compared to normal adjacent tissue in a variety of tumors, including those of the pancreas (5), lung (6), breast (7), and prostate (8). Elevated endogenous levels of NQO1 have been noted in 70% of early and late stage human prostate cancers. NQO1 induces a futile cycling of drug that exhausts NAD(P)H from the cell, leading to a substantial amount of reactive oxygen species (ROS) that causes DNA damage (6) (Fig. 1A). A concomitant rise in endoplasmic reticulum-derived cytosolic Ca2+ results in PARP1 hyperactivation, NAD+/ATP depletion, and stimulation of a unique apoptotic response involving substrate proteolysis, DNA fragmentation, and μ-calpain-mediated cell death (9). This unique mechanism of action of β-lap sets it apart from all other traditional chemotherapeutic agents. The compound kills cells independent of caspases, p53 status, cell cycle stage and all known anti-apoptotic factors (10). In light of its heightened antitumor activity specifically in cells overexpressing NQO1, β-lap is a powerful agent capable of offering tumor-selective killing.
Fig. 1.

Mechanism of β-lap redox cycling. (A)β-Lap undergoes a futile redox cycle in an NQO1-dependent manner. The hydroquinone form is unstable and through two one-electron oxidation steps converts back to the parent β-lap form. This cycling “bioactivates” β-lap and results in the generation of Reactive Oxygen Species (ROS). (B) Schematic of complexation and incorporation of β-lap and β-lap·HPβ-CD complexes into PLGA millirods.
Despite this advantage, the low solubility of β-lap in water (0.038 mg/ml or 0.16 mM) (11) hinders its clinical translation by traditional routes, such as intravenous administration (12). Moreover, the lack of site-specificity following intravenous (i.v.) injection may result not only in low drug concentrations in the tumor, but possible toxicity in normal cells lacking NQO1 activity due to high doses and prolonged exposure (6). Although ARQ 501, a formulation of β-lap complexed with hydroxypropyl β-cyclodextrin (β-lap•HPβ-CD), is currently under phase II clinical trial as systemic treatment for cancers, there is no report for local application of this drug which may be more suitable for treating solid tumors in early stages. Recently, several biodegradable polymer depot systems were developed with the hope of achieving intratumoral, controlled release of anticancer drugs. This strategy proves advantageous, since therapeutic levels of a desired anticancer agent are maintained for prolonged periods of time while reducing systemic side effects (13). Given these potential advantages, several implantable devices were established for use in a wide variety of cancers. Gliadel® Wafer, a carmustine (BCNU)-containing polymer implant, was approved by the FDA in 1996 for treatment of glioblastoma multiforme (14–17). Qiao and coworkers developed 5-fluorouracil (5-FU)-releasing silicone implants capable of zero-order drug release for 24 weeks in vitro (18). Work by our own laboratory resulted in the fabrication of cylindrical, poly(D,L-lactide-co-glycolide) (PLGA) polymer implants (millirods) releasing carboplatin or doxorubicin for intratumoral treatment of liver cancers (19–21). Upon incorporation of the latter drug within millirods, the antitumor efficacy in vivo was noted by decreased tumor volumes four and eight days after millirod implantation in VX2 liver tumors in rabbits (22).
Based on this concept, we produced several polymer millirod formulations containing β-lap complexed with different cyclodextrins (CDs) to achieve variable release kinetics (23). Depending on the cyclodextrin utilized, as well as the presence or lack of excipient molecules in the millirod, release of drug was modulated between rapid burst and sustained release kinetics. For example, when complexed with HPβ-CD, release of β-lap showed burst kinetics (~80% release after two days), while drug alone released in a more sustained manner (~9% after 22 days). Previously, we reported that the therapeutic window of β-lap in lung (6), prostate (4) and breast (7) cancer cells was provided by a short high-dose pulse of β-lap to exploit elevated basal levels of NQO1 for cell death responses. Extended treatment with high doses of β-lap would lead to normal tissue complications due to the two one-electron reductions by the b5 and P450 oxidoreductases present in normal tissues (Fig. 1A). Therefore, we aimed to optimize a formulation that can give a burst release in a short time period to reach the highest killing efficiency in tumor while a prolonged low dose of β-lap release to kill the residual cancer cells with sparing normal cells. The ultimate goal was to examine the antitumor efficacy in vivo of β-lap-containing polymer millirods in prostate tumor-bearing mice. HPβ-CD was used to form inclusion complexes with β-lap to achieve initial burst release of drug from PLGA millirods, while free, non-complexed drug was incorporated to provide prolonged release. PLGA millirods were directly implanted inside PC-3 tumor xenografts followed by the examination of drug release, antitumor efficacy, and systemic toxicity over time. The significant antitumor response highlights the unique advantage of integrating a novel and tumor-selective therapeutic agent (β-lap) with enabling drug delivery technology (polymer millirods) to achieve tumor- and site-specific therapy of human prostate cancers.
Materials and Methods
Materials
Poly(D,L-lactide-co-glycolide) (lactide:glycolide=50/50, MW 50,000 Da, inherent viscosity 0.65 dl/g) was purchased from Birmingham Polymers, Inc. (Birmingham, AL). β-Lap was synthesized following a previously published procedure (24) and prepared as described (8). HPβ-CD was obtained from Cyclodextrin Technologies Development, Inc. (High Springs, FL) with >98% purity. Glucose anhydrous was obtained from Fisher Scientific (Pittsburgh, PA). Dicoumarol, an NQO1 inhibitor, was purchased from the Sigma Chemical Co. (St. Louis, MO).
Cell culture
DU145, PC-3, and LNCaP human prostate cancer cells were originally obtained from Dr. George Wilding (University of Wisconsin-Madison). DU145 and PC-3 cells were grown in GIBCO™ RPMI Medium 1640 (1X) (Invitrogen) with 5% fetal bovine serum (FBS) and LNCaP cells were grown in Dulbecco’s minimal essential medium (DMEM, Invitrogen) with 10% FBS. All media contained 2 mM/L L-glutamine, penicillin (100 units/ml) and streptomycin (100 mg/mL). Cells were cultured at 37 °C in a 5% CO2, 95% air humidified atmosphere and were free from mycoplasma contamination.
NQO1 expression analyses
Western blots were prepared using standard methods. Briefly, cultured cells or xenograft tissues were harvested for assessment of NQO1 expression. Cells were washed in ice-cold PBS, lysed, sonicated, and stored at −20°C for future analyses. Western blots were first incubated with PBS containing 0.2% Tween 20 and 5% milk for 1 h to prevent nonspecific binding, then developed as previously described using SuperSignal® West Pico Chemiluminescent substrate (Thermo Scientific, Rockford, IL) and exposed using Autoradiography Film (Denville Scientific Inc., Metuchen, NJ) (8). An anti-human NQO1 antibody was kindly provided by Dr. David Ross (University of Colorado Health Science Center, Denver, CO) and used at a 1:5,000 dilution overnight at 4 °C. GAPDH or tubulin levels were monitored for loading control.
NQO1 enzyme assays
Enzymatic reactions from S9 cell preparations were performed in reactions containing 77 μM cytochrome c (Sigma) and 0.14% bovine serum albumin in Tris-HCl buffer (50 mM, pH 7.5). NQO1 activity was measured using NADH (200 μM) as an immediate electron donor and menadione (10 μM) as an intermediate electron acceptor as described (8). Dicoumarol (40 μM) was used to block NQO1 activity, and each sample was measured in triplicate. Enzymatic activities were calculated as nmol cytochrome c reduced/min/mg protein, based on initial rate of change in OD (absorbance) at 550 nm.
Relative survival assays
Cell survival was examined using a DNA assay as described (8). Briefly, cells were seeded at 5 × 103 per well in a 48-well plate and allowed to attach overnight. Cells were then treated for 2 h with various β-lap concentrations, alone or with 40 μM dicoumarol. Drug-free medium was then added and cells were allowed to grow for 5–7 days until control cells reached 100% confluence. DNA content was then determined by Hoescht dye staining and fluorescence detection using a Perkin-Elmer plate reader (Boston, MA). Results were reported as means ± standard error (SE) from at least three independent experiments performed six times.
Preparation of β-lap·HPβ-CD inclusion complexes
Following a previously published procedure (11), 25 g o HPβ-CD were weighed and dissolved in 50 mL PBS (pH 7.4). After complete dissolution, approximately 1 g of β-lap was added to the HPβ-CD solution. The solution was covered and allowed to stir at room temperature for 3 days, after which the contents were filtered using a 0.45 μm nylon filter, yielding a solution of β-lap·HPβ-CD complex. The solution was then lyophilized and the resulting solid powder was grounded using a mortar and pestle.
Fabrication of polymer millirods
Polymer millirods were produced using a previously established compression-heat molding procedure (20). Millirod composition was modified to achieve the desired drug release profile. Briefly, millirod components (31%β-lap·HPβ-CD complex, 19% free β-lap, and 50% PLGA) were weighed separately, placed in a mortar, and mixed well utilizing a pestle. Control millirods were composed of 26% HPβ-CD and 74% PLGA. The contents were then placed into a Teflon tube (1.6 mm I.D.) within a stainless steel mold. The mold was then placed in an oven (Fisher Model 282A) at 90 °C for 2 h with a compression pressure of 4.6 MPa. The resulting cylindrical millirods, with a diameter of 1.6 mm, were cut into lengths of 4 mm for subsequent biology studies in vitro and in vivo (Fig. 1B).
Drug release studies in vitro
β-Lap-loaded PLGA millirods (n = 3) were placed in glass scintillation vials containing PBS (pH 7.4) at 37 °C. Sample vials were placed in an orbital shaker (C24 model, New Brunswick Scientific) with a rotating speed of 150 rpm. At various times, millirods were removed and placed into new scintillation vials containing fresh buffer at 37 °C. The concentration of released β-lap was measured using UV-Vis spectrophotometry (Perkin-Elmer Lambda 20 model) at the maximum absorption wavelength of drug (λmax = 257.2 nm).
Characterization of β-lap release in vivo from polymer millirods
Drug release studies in vivo were conducted by intratumorally implanting β-lap millirods into PC-3 xenograft-bearing athymic nude mice. All animal procedures were approved by the UT Southwestern IACUC committee. PC-3 cells were grown to 80–90% confluence, harvested, prepared at 5 × 106/50 μl cell suspensions, and inoculated on both flanks of 6–8 week-old athymic nude mice with an average weight of 25 ± 1.6 g (Charles River Labs). Tumor dimensions were measured regularly with calipers and volumes calculated: volume (mm3) = length × width × width/2. The mean tumor size used to characterize β-lap release in vivo was ~300 mm3, similar to mean tumor sizes in randomized studies used to determine antitumor efficacy. At different time points (6, 12, 24 h, day 2, 3, 4, 6, 8, 10, 13, 17, 20 and 24), mice were sacrificed and millirods retrieved. Three animals were used at each time point. Experiments were repeated at least three times. Millirods were dissolved in acetonitrile to recover and quantify the remaining drug by HPLC using a ZORBAX C-18 column (150 × 4.6 mm, 5.0 mm) with a mobile phase consisting of 70% acetonitrile and 30% 25 mM ammonium formate buffer at pH 7.0. Three millirods from three individual mice were measured for statistical analysis at each time point.
Antitumor efficacy
Subcutaneous tumors were inoculated in athymic nude mice as described above. In general, mice were randomly distributed into two groups. The mean sizes of tumors used at the start of these experiments were 325.1 ± 7.4 mm3 for the experimental group and 354.6 ± 10.1 mm3 for the control group. Statistically, these two groups were not different. β-lap loaded (2.6 mg) or control millirods measuring 4 mm in length were intratumorally implanted using a 13-gauge trochar. Tumor volumes were calculated every other day after implantation. Mice were sacrificed when tumors reached 2 cm3 or 10% total body weight. 10 mice/group were used in this study, which were repeated at lest twice. Tumor, liver, and kidney tissues were removed and sent for histological examination at the UT Southwestern Medical Center histological core. Portions of tumor tissues were also frozen in liquid nitrogen for western analyses.
Histological staining
Tumor, liver, and kidney tissues were harvested at day six, when the drug release reached the maximum, and at all the endpoints when the mice were sacrificed. Tissues were fixed in 10% formalin overnight, paraffin embedded and processed by the Department of Pathology, UT Southwestern Medical Center. Briefly, 5 μm thick sections were prepared from paraffin-embedded tissues and baked overnight at 37 °C. Hematoxylin and eosin (H&E) staining was conducted and histology images taken using a Nikon E400 microscope with a Nikon coolpix 4500 camera.
Statistical analyses
Tumor growth profiles in two tested groups were analyzed using a mixed model approach. Log-rank tests were applied to survival analyses (Kaplan-Meier curves). P values of ≤0.05 were considered significant. All statistical analyses were performed using SAS 9.1.3 Service Pack 3.
Results
NQO1-dependent cell killing by β-lap
NQO1 is considerably elevated in multiple tumor types, including human prostate cancers (4). Immunoblotting and enzymatic methods were used to confirm and quantify NQO1 expression levels in three different human prostate cancer cell lines: DU145, PC-3 and LNCaP (Fig. 2A; Table 1). DU145 cells have the highest NQO1 expression and activity (556 ± 18 nmol cytochrome c reduced/min/mg protein), with PC-3 cells having elevated but lower levels (108 ± 17 nmol cytochrome c reduced/min/mg protein). A particular clone of LNCaP cells lacks NQO1 expression, even though they have a wild-type NQO1 gene (25). We previously isolated an isogenic LNCaP clone transfected with CMV-directed NQO1 (4). LNCaP NQO1 + cells showed similar NQO1 activity (108 ± 21 nmol cytochrome c reduced/min/mg protein) as PC-3 cells.
Fig. 2.

β-Lap killed human prostate cancer cells in an NQO1-dependent mechanism. (A) NQO1 protein expression levels in various human prostate cancer cell lines. LNCaP cells are DNA mismatch repair deficient (25) and one clone was isolated with an NQO1-deficiency. Cells were transfected with NQO1, creating an isogenic NQO1+/− system (4). (B) Relative survival assays of three different cell lines exposed to either β-lap alone at the indicated doses or β-lap + concomitant dicoumarol (40 μM) for 2 h. Error bars represent standard error of means (SEM) from six replicates performed each in duplicate.
Table 1.
NQO1 enzymatic activity and LD50 of β-lap ± Dic in three different prostate cancer cell lines
| Cell Lines | NQO1 level (nmol cytochrome c reduced/min/mg protein)* | LD50 values of β-Lap (μM, 2 h) | |
|---|---|---|---|
| Dicoumarol (−) | Dicoumarol (+)§ | ||
| DU145 | 556 ± 18 | 3.0 ± 0.1 | 9.1 ± 0.6 |
| PC-3 | 107 ± 17 | 1.5 ± 0.1 | 20.0 ± 4.4 |
| LNCaP NQO1+ | 108 ± 21 | 2.7 ± 0.9 | > 20 |
| LNCaP NQO1− | 2.4 ± 0.1 | 11.8 ± 1.7 | ND |
Values are mean ± SEM; n = 6 per group.
Dicoumarol was administered at 40 μM concomitant with 2 h β-lap treatments.
We then assessed the survival of the three prostate cancer cells after transient exposure to β-lap (Fig. 2B). The LD50 values of the three cell lines following a 2 h exposure of β-lap can be found in Table 1. Dose-response data showed that after a 2 h exposure, all three cell lines were killed by β-lap at minimum concentrations of 4 μM (DU145 and PC-3 cells) and 6 μM (LNCaP NQO1+ cells), respectively. These and other data in a variety of other breast, pancreatic, non-small cell lung and colon cancer cell lines suggest that a minimum NQO1 activity of ~100 nmol cytochrome c reduced/min/mg protein was required for β-lap to irreversibly kill cells when exposed at 4–6 μM for 2 h. Dicoumarol, an NQO1 inhibitor, rescued DU145, LNCaP NQO1+ and PC-3 cells from β-lap-induced cell death, confirming the NQO1-dependent cell killing mechanism of β-lap. In contrast, LNCaP NQO1-cells were able to survive an 8 μM dose of β-lap, with increasing doses (e.g. 10 μM and 20 μM) resulting in lethality by NQO1-independent processes as described (7, 9, 10). Administration of dicoumarol failed to protect NQO1− cells at high doses of β-lap > 20 μM, and confirmed the roles of other low affinity reductases, such as cytochrome P450 and b5 oxidoreductases, in NQO1− independent β-lap cytotoxicity.
Enhanced delivery of β-lap via incorporation of free β-lap and β-lap·HPβ-CD complexes into millirods
Polymer millirods for the intratumoral delivery of β-lap to prostate tumors were produced as described in ‘Materials and Methods’, and release kinetics of the drug in vitro were examined. Since a 2 h exposure of β-lap resulted in irreversible cell death, an initial burst of the drug was desired to rapidly achieve therapeutic levels. Optimal millirod delivery would then provide a sustained release over a prolonged period of time. A burst release of ~0.5 mg occurred within 12 h (Fig. 3), due predominantly to the complexation of the drug with HPβ-CD that dramatically enhanced its solubility and drug release (23). Thereafter, release of drug decreased over time, with a release rate of ~0.05 mg/day for the next three days. By day 6, 0.6 mg of drug was released from the millirod, and the release rate for the subsequent four days was ~0.01 mg β-lap/day. By day 23, total release of drug from the millirod was ~0.7 mg, highlighting the sustained release of drug due most likely to the fraction of free drug remaining as a molecular dissolution in the PLGA polymer (23).
Fig. 3.
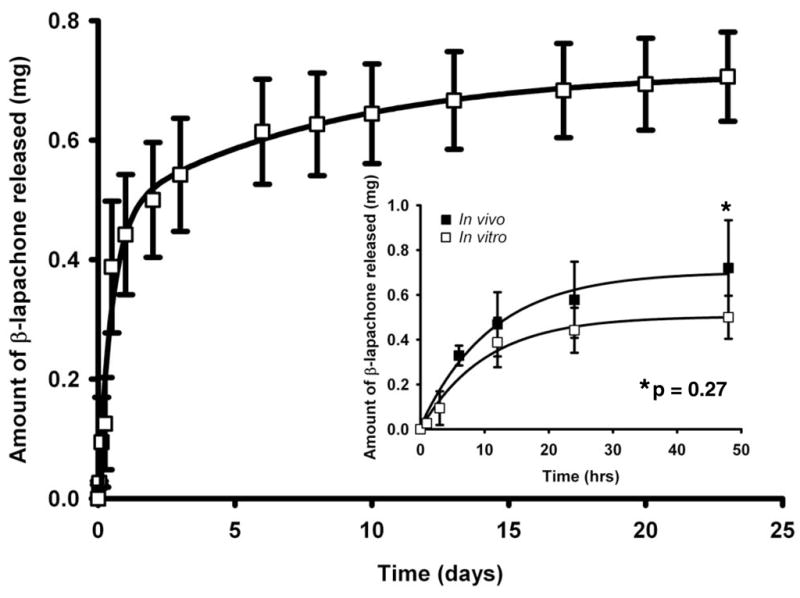
Cumulative levels of β-lap released from PLGA millirods were plotted as a function of time (h). The figure inset depicts cumulative β-lap released in vitro compared to in vivo following millirod implantation into PC-3 tumor xenografts within 48 h. Release levels were calculated as the differences between initial amount of β-lap loaded and amount remaining in millirods over time. Values are means ± SEM (n=3).
β-Lap release kinetics in vitro was similar to that in PC-3 tumor xenografts in vivo within 48 h after millirod implantation (Fig. 3, inset). After 6 h in vivo, a burst dose of 0.33 ± 0.04 mg of the drug was released from the millirod, compared to 0.21 ± 0.03 mg released in vitro. At the transition point between burst and sustained release (12 h), 0.47 ± 0.14 mg of drug was released in vivo compared to 0.39 ± 0.11 mg in vitro. After two days, 0.72 ± 0.21 mg and 0.50 ± 0.10 mg were released in vivo and in vitro, respectively. Although release of drug in vivo was slightly greater than release in vitro, the difference was not statistically significant at all times examined (p value = 0.27).
Antitumor efficacy of intratumoral delivery of β-lap-loaded millirods
To examine the antitumor efficacy of β-lap-containing millirods, implantation into subcutaneous PC-3 xenografts located on the flanks of female athymic nude mice were performed. Tumor volumes were calculated every other day and compared with tumors containing control (vehicle alone) millirods. Three days after implantation, significant tumor regression was noted in the treatment group (204 ± 12 mm3) compared to control tumors that nearly doubled in volume to an average of 551 ± 39 mm3 (Fig. 4A). Delayed tumor growth lasted for nearly two weeks before β-lap-treated tumors grew to their original volume of 371 ± 54 mm3 at day 13, while the average volume of control tumors surpassed 1000 mm3 (1130 ± 98 mm3). It took approximately 25 days for tumors in the treatment group to surpass the 1000 mm3 mark. It is important to note that after day 17, tumors treated with β-lap millirods began to grow at a similar rate as control tumors, as evidenced by comparable growth rates (51.1 mm3/day for control tumors and 55.6 mm3/day for treatment tumors). In spite of tumor regrowth, statistical analyses at each time point demonstrated significant tumor volume differences (p value ≤ 0.01) between treated and control groups. Mixed model statistical analyses showed a more pronounced difference (p value ≤ 0.0001) in total tumor growth behavior between these two groups.
Fig. 4.

Intratumoral delivery of β-lap using polymer millirods as a vehicle showed significant antitumor efficacy of β-lap in PC-3 tumor xenograft models. (A) Comparison of tumor volume after intratumoral implantation of either β-lap-loaded millirods or control millirods containing HPβ-CD alone. Values are means ± SEM (n=10). (B) Animal weight data over time (days) in both control and treatment groups. Values are means ± SEM (n=10). (C) Kaplan-Meier curves comparing the antitumor efficacies of two different types of millirods as shown in (A). Open circles represent either accidentally animal loss due to skin infection at day 27 or animal survival at the end of the experiment.
To monitor possible toxicity to normal tissue resulting from the proposed treatment regimen, animal weights were recorded over the duration of the study (Fig. 4B). Weights in both control and treated groups remained similar throughout the course of the experiment, with the average weight being 25 ± 1.6 g. No statistical difference was found between the average weights of the two groups, suggesting no apparent adverse toxicity in mice treated with β-lap millirods. While there appeared to be no difference in systemic toxicity, the two groups possessed significant differences in survival (See Kaplan-Meier survival curve, Fig. 4C). An approximate 50% loss in animals was observed after 25 days in the control group, whereas 50% loss in survival of β-lap-treated animals did not occur until after 35 days, a time when all animals in the control group were sacrificed. A fraction (2/10) of mice in the treatment group survived > 40 days. A Log-rank test of the Kaplan-Meier survival curve indicated that there was a significant delay for tumor growth in β-lap-treated mice to reach the size for sacrifice when compared to control tumors (p value = 0.0024).
Histological examination of tumor tissues following β-lap millirod treatment confirmed significant antitumor response
To determine whether or not PC-3 tumor xenografts maintained NQO1 expression, a key target for achieving cell killing specificity by β-lap-loaded PLGA millirods in vivo, xenograft tissues were harvested and western analyses conducted to monitor NQO1 levels over time (Fig. 5A). Results indicated that high NQO1 expression levels were preserved following subcutaneous implantation and growth in the flanks of mice, and that these levels were comparable to PC-3 cells harvested in vitro.
Fig. 5.

Histological examination (H&E) of PC-3 tumor xenograft sections after millirod implantation confirmed significant anti-tumor effects of β-lap in vivo. (A) Western analyses demonstrating NQO1 expression in explanted PC-3 xenografts. Cell lysate of parental LNCaP cells (NQO1−) was used as negative control. (B) A cross section of tumors treated with HPβ-CD-loaded millirod (4× magnification). Circled ‘R’ represents the millirod implantation site, while * indicates areas of patchy necrosis. (C) A cross section (4× magnification) of a tumor xenografts 6 days after implantation of β-lap-loaded millirods. (D) and (E) represent increased magnifications (40×) of (B) and (C), respectively.
Histological examination of tumor tissues explanted from mice six days after millirod implantation showed that the area immediately adjacent to control millirods contained numerous viable tumor cells in H&E stained sections, easily identifiable by clusters of dark staining nuclei (Fig. 5B). Magnification of this implant boundary region (Fig. 5D) showed tumor cells that have large nuclei, are irregular in shape and size, and appear mitotic. These observations corroborate the presence of viable tumor cells in this area. At regions farther away from the implant, a typical patchy necrotic area (region labeled *), due to fast proliferation rates in high-grade tumors was observed. In contrast, β-lap-treated tumors (Fig. 5C) showed pronounced areas of coagulative necrosis in the region surrounding the millirod implant. A sharp demarcation line between necrotic and darkly stained cells at distances away from the implant was easily discernible. A higher magnification of this zone (Fig. 5E) revealed the presence of a large number of inflammatory cells, mostly neutrophils and lymphocytes. Immediately adjacent to the inflammatory cells were viable tumor cells noted by their large nuclei, irregular shape, and mitotic state that was more and more pronounced at distances farther away from the immediate treatment radius. As in control tumors, an area of patchy necrotic tissue at distances away from the implant was observed, interspersed with viable and apoptotic tumor cells, as well as inflammatory cells. Examining the tissues from the endpoints, we were encouraged by observing typical coagulative necrosis with sharp tissue demarcation after β-lap·HPβ-CD-encoded millirod implantation on day 26, even though clear tumor regrowth was noted in surrounding areas (not shown) which will be further discussed.
To evaluate possible systemic side-effects of β-lap-loaded millirods, we performed histological examination of liver and kidney tissues one month after millirod implantation (Fig. 6). It has been reported that mice express abundant NQO1 levels in their livers and high activity of NQO1 was detected in kidneys (26). These tissues were also chosen since β-lap is likely cleared through these organs in vivo. H&E staining of these tissues did not show any pathological changes after β-lap treatment compared to those in animals receiving control millirods. The absence of tissue damage or cell abnormalities in the liver and kidney tissues strongly suggested that there were no noticeable signs of systemic toxicity brought about by millirod-delivered β-lap·HPβ-CD.
Fig. 6.

Histological examination (H&E, 40× magnification) of liver and kidney tissues further corroborated negligible systemic toxicity observed with implant-delivered β-lap. (A) and (B) represent liver tissues while (C) and (D) represent kidney tissue. (A) and (C) are labeled as blank and refer to samples from control groups, while (B) and (D) represent tissues from experiments involving β-lap-loaded millirods.
Discussion
The objective of the present study was to evaluate the antitumor efficacy of β-lap in vivo, a novel and unique anticancer drug, delivered intratumorally via a polymer depot. This drug, complexed as β-lap·HPβ-CD (ARQ 501), is currently used in clinical trials against pancreatic cancers. Currently, 10–15 year survival rates for patients with primary prostate cancer are 65% after surgery (2), and 67–87% after brachytherapy (27). Invasiveness of the surgical procedure and high local recurrence rates provide motivation for the search for efficacious alternatives and supplemental therapies. Docetaxel is a frequently used drug for chemotherapy of prostate cancer that offers moderate success, but results in high systemic toxicity (28, 29). Moreover, human prostate tumors are inherently resistant to many clinically used drugs against other tumors, given the fact that loss of androgen dependence and tumor suppressor genes (e.g., p53) are important factors in prostate cancer progression (30–32). Additionally, these tumors are commonly disregulated with regard to calcium homeostasis (33), and lack caspases that mediate apoptosis (34). To overcome the shortcomings of current treatments, a novel agent is required that can offer an effective prostate tumor-specific treatment, while sparing normal cells. The unique mechanism of action of β-lap, involving its bioactivation through the NQO1 enzyme, makes it an ideal agent for the treatment of tumors that overexpress this enzyme (Fig. 1A). Results from cell culture studies demonstrated that a short exposure of β-lap for 2 h caused irreversible cell death, contrasting tremendously with other known anticancer drugs, where prolonged drug exposures might be required to elicit significant antitumor activity.
Matrix-based drug delivery devices represent a rising trend in cancer chemotherapy, yielding the following specific advantages: 1) exposure of tumors to therapeutic levels of the drug for a prolonged time; 2) reduction of toxicity to healthy cells; and 3) potential tailoring of release kinetics for the design of most efficacious delivery regimen. Several implant strategies have been explored for intratumoral treatment of cancers. Besides Gliadel® wafers as we have mentioned before (35), Wientjes et. al., developed a doxorubicin-releasing poly(lactide-co-glycolide) (PLG) implant for the treatment of prostate tumors, demonstrating feasibility for in vivo use as well as low systemic concentrations of the drug after implantation into the prostate of beagle dogs (36). Recently, we reported β-lap-loaded millirods with tailorable release kinetics depending on inclusion of excipient molecules (e.g., glucose) or complexation with different cyclodextrin complexes (e.g., α-CD, β-CD, and γ-CD) (23). We were able to demonstrate that β-lap complexation with HPβ-CD allowed for faster release of β-lap from millirods, in part due to the increase in drug solubility from 0.038 mg/mL to 16 mg/mL. On the other hand, free drug showed the slowest release due to formation of a molecular-level mixture with the PLGA polymer and the lack of excipient molecules in the polymer matrix.
Based on the fact that a 2 h exposure of β-lap led to irreversible cell death, we designed a polymer millirod that featured a burst release of drug within a short time period to achieve an elevated, therapeutic dose of the drug (6), followed by a sustained release of the drug over a prolonged period of time. This was achieved by incorporating both complexed and free drug within the millirod, with the complexed form of β-lap providing a burst release of drug (~0.5 mg released within the first 12 h) and the free form providing a sustained release of β-lap (release rate of 0.01 mg of drug/day over 22 days). The kinetics of drug release in the tumor is concluded from the drug release study in vivo as shown in Figure 3. The burst release led to elevated drug concentrations within tumors that quickly exerted antitumor effects, while the sustained release maintained drug concentrations inside tumors for a prolonged period. Data showed that in vivo burst release kinetics of β-lap correlated very closely with release in vitro, with no statistical difference between the two conditions.
To evaluate antitumor efficacy and systemic toxicity, β-lap-loaded polymer millirods were implanted into PC-3 prostate tumor xenografts. Results from this study showed significant tumor regression and delayed growth after intratumoral implantation of the β-lap millirods. Moreover, survival of mice treated with β-lap millirods was significantly increased compared to untreated mice. Minimal, if any, systemic toxicity was observed, as evidenced by the lack of weight loss and normal appearing histology of major organs (e.g. liver, kidney). No complications were observed in the surrounding normal tissues. Taken together, these data highlight the potential of utilizing intratumoral delivery of β-lap via polymer millirods as a viable treatment option for prostate cancer.
Results from this study showed tumor recurrence as a potential limitation of the current treatment. Tumor regrowth was found two weeks after millirod implantation (Fig. 4A), which was likely due to viable cancer cells in the region adjacent to the exposed area resulted from the limited drug penetration (Fig. 5B-E). To overcome this limitation, it may be necessary to combine the current treatment with another therapeutic modality, in an adjuvant or neoadjuvant manner. For example, patients may undergo radiation therapy or a minimally invasive ablative technique, such as cryotherapy or radiofrequency ablation, followed by implantation of β-lap-containing polymer millirods to maximize therapeutic potential. Previously, our laboratory demonstrated that the combination of radiofrequency ablation of VX2 liver tumors followed by the intratumoral implantation of a doxorubicin-containing millirod resulted in increased efficacy compared to millirod implants alone (37). The selective killing feature of β-lap makes it more appealing than other drugs due to its tumor selectivity and reduced toxicity in tissues that do not express NQO1. Furthermore, prior studies have shown that β-lap functions as a radiosensitizer, with in vitro studies demonstrating that a low dose of β-lap together with a low dose of ionizing radiation (IR) led to synergistic cell death (38). Therefore, β-lap millirods may be implanted initially, followed by low-doses of IR to provide synergy between the two treatment modalities and enhance antitumor efficacy. The combination can also be achieved by applying either external beam or co-implantation of low dose irradiation materials, such as radioactive seeds during brachytherapy together with β-lap millirods. Thus, several viable combination strategies exist that can potentially maximize the therapeutic outcome of intratumoral β-lap implants.
Conclusion
β-Lap-containing polymer millirods represent an exciting therapeutic option for the targeted treatment of prostate tumors that overexpress the enzyme, NQO1. By combining a novel drug, β-lap, whose mechanism of action is enhanced by an enzyme overexpressed in prostate tumors, with a local polymer delivery device capable of controlled-release, we were able to demonstrate significantly improved antitumor efficacy. Moreover, results from this study suggested minimal systemic toxicity and prolonged animal survival. Work is currently in progress to further maximize treatment efficacy by combining the therapy with existing radiotherapy strategies.
Statement of Translational Relevance
Prostate cancer is the second leading cause of cancer-related death for American men. Enormous side-effects including incontinence and impotence after salvage therapy, have severely limited the quality of life of prostate cancer patients. β-Lapachone is a novel anticancer agent, and an effective radiosensitizer, with specific selectivity against cancer cells that over-express the two-electron oxidoreductase, NAD(P)H:quinone oxidoreductase 1 (NQO1). However, lack of solubility, inactivation of the drug in human serum, and the lack of an efficient drug delivery strategy limit the use of this compound in the clinics. Combined with a local polymer delivery device capable of controlled-release, we demonstrate significant antitumor efficacy with minimal systemic toxicity. Thus, implantation of β-lapachone-containing millirods after salvage surgery, or co-implantation of low dose irradiation materials (such as radioactive seeds during brachytherapy) together with β-lapachone millirods, should greatly increase efficacy of treating prostate cancer, as well as bring a new era of synergistic therapy for prostate cancer.
Acknowledgments
This work was supported by NIH grant RO1 CA90696 to JG, DOD grant W81XWH-04-1-0164 and NIH grant RO1 CA102792-06 to DAB. YD appreciates the support of DOD Prostate Cancer Research Program Training Award, W81XWH-08-1-0344. EB is grateful for the support of DOD predoctoral grant, W81XWH-05-1-0258. This is report CSCN016 from the Cell Stress and Cancer Nanomedicine program in the Simmons Comprehensive Cancer Center at the University of Texas Southwestern Medical Center at Dallas. We also acknowledge the histology and biostatistical cores supported by the Simmons Comprehensive Cancer Center.
References
- 1.Jemal A, Siegel R, Ward E, Murray T, Xu J, Thun MJ. Cancer statistics, 2007. CA Cancer J Clin. 2007;57:43–66. doi: 10.3322/canjclin.57.1.43. [DOI] [PubMed] [Google Scholar]
- 2.Huang WC, Lee CL, Eastham JA. Locally ablative therapies for primary radiation failures: a review and critical assessment of the efficacy. Curr Urol Rep. 2007;8:217–23. doi: 10.1007/s11934-007-0009-5. [DOI] [PubMed] [Google Scholar]
- 3.Bracarda S, de Cobelli O, Greco C, et al. Cancer of the prostate. Crit Rev Oncol Hematol. 2005;56:379–96. doi: 10.1016/j.critrevonc.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 4.Planchon SM, Pink JJ, Tagliarino C, Bornmann WG, Varnes ME, Boothman DA. beta-Lapachone-induced apoptosis in human prostate cancer cells: involvement of NQO1/xip3. Exp Cell Res. 2001;267:95–106. doi: 10.1006/excr.2001.5234. [DOI] [PubMed] [Google Scholar]
- 5.Ough M, Lewis A, Bey EA, et al. Efficacy of beta-lapachone in pancreatic cancer treatment: exploiting the novel, therapeutic target NQO1. Cancer Biol Ther. 2005;4:95–102. doi: 10.4161/cbt.4.1.1382. [DOI] [PubMed] [Google Scholar]
- 6.Bey EA, Bentle MS, Reinicke KE, et al. An NQO1− and PARP-1-mediated cell death pathway induced in non-small-cell lung cancer cells by beta-lapachone. Proc Natl Acad Sci USA. 2007;104:11832–7. doi: 10.1073/pnas.0702176104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bentle MS, Reinicke KE, Bey EA, Spitz DR, Boothman DA. Calcium-dependent modulation of poly(ADP-ribose) polymerase-1 alters cellular metabolism and DNA repair. J Biol Chem. 2006;281:33684–96. doi: 10.1074/jbc.M603678200. [DOI] [PubMed] [Google Scholar]
- 8.Pink JJPS, Tagliarino C, Varnes ME, Siegel D, Boothman DA. NAD(P)H:Quinone oxidoreductase activity is the principal determinant of β-lapachone cytotoxicity. J Biol Chem. 2000;275:5416–24. doi: 10.1074/jbc.275.8.5416. [DOI] [PubMed] [Google Scholar]
- 9.Tagliarino C, Pink JJ, Dubyak GR, Nieminen AL, Boothman DA. Calcium is a key signaling molecule in beta-lapachone-mediated cell death. J Biol Chem. 2001;276:19150–9. doi: 10.1074/jbc.M100730200. [DOI] [PubMed] [Google Scholar]
- 10.Reinicke KE, Bey EA, Bentle MS, et al. Development of beta-lapachone prodrugs for therapy against human cancer cells with elevated NAD(P)H:quinone oxidoreductase 1 levels. Clin Cancer Res. 2005;11:3055–64. doi: 10.1158/1078-0432.CCR-04-2185. [DOI] [PubMed] [Google Scholar]
- 11.Nasongkla N, Wiedmann AF, Bruening A, et al. Enhancement of solubility and bioavailability of beta-lapachone using cyclodextrin inclusion complexes. Pharm Res. 2003;20:1626–33. doi: 10.1023/a:1026143519395. [DOI] [PubMed] [Google Scholar]
- 12.Tewari A, Raman JD, Chang P, Rao S, Divine G, Menon M. Long-term survival probability in men with clinically localized prostate cancer treated either conservatively or with definitive treatment (radiotherapy or radical prostatectomy) Urology. 2006;68:1268–74. doi: 10.1016/j.urology.2006.08.1059. [DOI] [PubMed] [Google Scholar]
- 13.LaVan DA, McGuire T, Langer R. Small-scale systems for in vivo drug delivery. Nat Biotechnol. 2003;21:1184–91. doi: 10.1038/nbt876. [DOI] [PubMed] [Google Scholar]
- 14.Fleming AB, Saltzman WM. Pharmacokinetics of the carmustine implant. Clin Pharmacokinet. 2002;41:403–19. doi: 10.2165/00003088-200241060-00002. [DOI] [PubMed] [Google Scholar]
- 15.Ewend MG, Elbabaa S, Carey LA. Current treatment paradigms for the management of patients with brain metastases. Neurosurgery. 2005;57:S66–77. doi: 10.1227/01.neu.0000182739.84734.6e. discusssion S1–4. [DOI] [PubMed] [Google Scholar]
- 16.Limentani SA, Asher A, Heafner M, Kim JW, Fraser R. A phase I trial of surgery, Gliadel wafer implantation, and immediate postoperative carboplatin in combination with radiation therapy for primary anaplastic astrocytoma or glioblastoma multiforme. J Neurooncol. 2005;72:241–4. doi: 10.1007/s11060-004-2339-1. [DOI] [PubMed] [Google Scholar]
- 17.Westphal M, Ram Z, Riddle V, Hilt D, Bortey E. Gliadel wafer in initial surgery for malignant glioma: long-term follow-up of a multicenter controlled trial. Acta Neurochir (Wien) 2006;148:269–75. doi: 10.1007/s00701-005-0707-z. discussion 75. [DOI] [PubMed] [Google Scholar]
- 18.He YC, Chen JW, Cao J, Pan DY, Qiao JG. Toxicities and therapeutic effect of 5-fluorouracil controlled release implant on tumor-bearing rats. World J Gastroenterol. 2003;9:1795–8. doi: 10.3748/wjg.v9.i8.1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Qian F, Stowe N, Liu EH, Saidel GM, Gao J. Quantification of in vivo doxorubicin transport from PLGA millirods in thermoablated rat livers. J Control Release. 2003;91:157–66. doi: 10.1016/s0168-3659(03)00237-2. [DOI] [PubMed] [Google Scholar]
- 20.Qian F, Szymanski A, Gao J. Fabrication and characterization of controlled release poly(D,L-lactide-co-glycolide) millirods. J Biomed Mater Res. 2001;55:512–22. doi: 10.1002/1097-4636(20010615)55:4<512::aid-jbm1044>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 21.Szymanski-Exner A, Gallacher A, Stowe NT, Weinberg B, Haaga JR, Gao J. Local carboplatin delivery and tissue distribution in livers after radiofrequency ablation. J Biomed Mater Res A. 2003;67:510–6. doi: 10.1002/jbm.a.10038. [DOI] [PubMed] [Google Scholar]
- 22.Weinberg BD, Ai H, Blanco E, Anderson JM, Gao J. Antitumor efficacy and local distribution of doxorubicin via intratumoral delivery from polymer millirods. J Biomed Mater Res A. 2007;81:161–70. doi: 10.1002/jbm.a.30914. [DOI] [PubMed] [Google Scholar]
- 23.Wang F, Blanco E, Ai H, Boothman DA, Gao J. Modulating beta-lapachone release from polymer millirods through cyclodextrin complexation. J Pharm Sci. 2006;95:2309–19. doi: 10.1002/jps.20721. [DOI] [PubMed] [Google Scholar]
- 24.Planchon SM, Wuerzberger S, Frydman B, et al. Beta-lapachone-mediated apoptosis in human promyelocytic leukemia (HL-60) and human prostate cancer cells: a p53-independent response. Cancer Res. 1995;55:3706–11. [PMC free article] [PubMed] [Google Scholar]
- 25.Steiner M, Hillenbrand M, Borkowsi M, Seiter H, Schuff-Werner P. 609 C --> T polymorphism in NAD(P)H:quinone oxidoreductase gene in patients with prostatic adenocarcinoma or benign prostatic hyperplasia. Cancer Lett. 1999;135:67–71. doi: 10.1016/s0304-3835(98)00269-9. [DOI] [PubMed] [Google Scholar]
- 26.Radjendirane V, Joseph P, Lee YH, et al. Disruption of the DT diaphorase (NQO1) gene in mice leads to increased menadione toxicity. J Biol Chem. 1998;273:7382–9. doi: 10.1074/jbc.273.13.7382. [DOI] [PubMed] [Google Scholar]
- 27.Heysek RV. Modern brachytherapy for treatment of prostate cancer. Cancer Control. 2007;14:238–43. doi: 10.1177/107327480701400306. [DOI] [PubMed] [Google Scholar]
- 28.Beer TM, El-Geneidi M, Eilers KM. Docetaxel (taxotere) in the treatment of prostate cancer. Expert Rev Anticancer Ther. 2003;3:261–8. doi: 10.1586/14737140.3.3.261. [DOI] [PubMed] [Google Scholar]
- 29.Small EJ. Docetaxel in prostate cancer. Anticancer Drugs. 2001;12(Suppl 1):S17–20. [PubMed] [Google Scholar]
- 30.Martel CL, Gumerlock PH, Meyers FJ, Lara PN. Current strategies in the management of hormone refractory prostate cancer. Cancer Treat Rev. 2003;29:171–87. doi: 10.1016/s0305-7372(02)00090-7. [DOI] [PubMed] [Google Scholar]
- 31.Downing SR, Jackson P, Russell PJ. Mutations within the tumour suppressor gene p53 are not confined to a late event in prostate cancer progression. a review of the evidence. Urol Oncol. 2001;6:103–10. doi: 10.1016/s1078-1439(00)00119-8. [DOI] [PubMed] [Google Scholar]
- 32.Downing SR, Russell PJ, Jackson P. Alterations of p53 are common in early stage prostate cancer. Can J Urol. 2003;10:1924–33. [PubMed] [Google Scholar]
- 33.Prevarskaya N, Skryma R, Shuba Y. Ca2+ homeostasis in apoptotic resistance of prostate cancer cells. Biochem Biophys Res Commun. 2004;322:1326–35. doi: 10.1016/j.bbrc.2004.08.037. [DOI] [PubMed] [Google Scholar]
- 34.Coffey RN, Watson RW, Fitzpatrick JM. Signaling for the caspases: their role in prostate cell apoptosis. J Urol. 2001;165:5–14. doi: 10.1097/00005392-200101000-00003. [DOI] [PubMed] [Google Scholar]
- 35.Perry J, Chambers A, Spithoff K, Laperriere N. Gliadel wafers in the treatment of malignant glioma: a systematic review. Curr Oncol. 2007;14:189–94. doi: 10.3747/co.2007.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ortiz R, Au JL, Lu Z, Gan Y, Wientjes MG. Biodegradable intraprostatic doxorubicin implants. Aaps J. 2007;9:E241–50. doi: 10.1208/aapsj0902027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weinberg BD, Blanco E, Lempka SF, Anderson JM, Exner AA, Gao J. Combined radiofrequency ablation and doxorubicin-eluting polymer implants for liver cancer treatment. J Biomed Mater Res A. 2007;81:205–13. doi: 10.1002/jbm.a.30926. [DOI] [PubMed] [Google Scholar]
- 38.Miyamoto S, Huang TT, Wuerzberger-Davis S, et al. Cellular and molecular responses to topoisomerase I poisons. Exploiting synergy for improved radiotherapy. Ann N Y Acad Sci. 2000;922:274–92. doi: 10.1111/j.1749-6632.2000.tb07045.x. [DOI] [PubMed] [Google Scholar]