

Abstract
By evoking changes in climbing fiber activity, movement errors are thought to modify synapses from parallel fibers onto Purkinje cells (pf*Pkj) so as to improve subsequent motor performance. Theoretical arguments suggest there is an intrinsic tradeoff, however, between motor adaptation and long-term storage. Assuming a baseline rate of motor errors is always present, then repeated performance of any learned movement will generate a series of climbing fiber-mediated corrections. By reshuffling the synaptic weights responsible for any given movement, such corrections will degrade the memories for other learned movements stored in overlapping sets of synapses. The present paper shows that long-term storage can be accomplished by a second site of plasticity at synapses from parallel fibers onto stellate/basket interneurons (pf*St/Bk). Plasticity at pf*St/Bk synapses can be insulated from ongoing fluctuations in climbing fiber activity by assuming that changes in pf*St/Bk synapses occur only after changes in pf*Pkj synapses have built up to a threshold level. Although climbing fiber-dependent plasticity at pf*Pkj synapses allows for the exploration of novel motor strategies in response to changing environmental conditions, plasticity at pf*St/Bk synapses transfers successful strategies to stable long-term storage. To quantify this hypothesis, both sites of plasticity are incorporated into a dynamical model of the cerebellar cortex and its interactions with the inferior olive. When used to simulate idealized motor conditioning trials, the model predicts that plasticity develops first at pf*Pkj synapses, but with additional training is transferred to pf*St/Bk synapses for long-term storage.
Animal studies indicate that the cerebellum contributes to a variety of learned motor behaviors (1–7), a conclusion supported in humans by both behavioral experiments (8, 9) and imaging studies (10, 11). Starting with the original models of Marr (12) and Albus (13), a number of theoretical analyses have postulated that climbing fiber-dependent plasticity at synapses from parallel fibers onto Purkinje cells (pf*Pkj) in the cerebellar cortex contributes to motor adaptation (14–23). The anatomical assumptions underlying Marr/Albus-based models of cerebellar involvement in motor learning are supported by evidence of a strong topographical organization in the reciprocal interactions between the cerebellum and the inferior olive (24, 25). In addition, electrophysiological studies report changes in cerebellar activity after climbing fiber stimulation (26–28) and during motor adaptation (2, 4, 7, 29) that are consistent with the postulated role of climbing fiber-dependent plasticity at pf*Pkj synapses.
Despite evidence supporting their postulated role in motor learning, theoretical arguments suggest that memories stored at pf*Pkj synapses would be susceptible to long-term degradation as a result of ongoing motor adaptation. First, motor memories are likely to be distributed across overlapping sets of pf*Pkj synapses. If motor memories did not overlap, the storage capacity of pf*Pkj synapses would be greatly reduced (12, 17). Second, it is reasonable to assume that the pattern of synaptic weights necessary to effect any given movement is non-unique. If the activity of a Purkinje cell depends only on the total sum of the synaptic input at any given moment, then there will be a virtual infinity of synaptic weight combinations capable of producing the same Purkinje cell output. Third, it can be further assumed that repeated execution of any learned movement in the face of constantly fluctuating internal and external environmental conditions will generate a constant stream of error signals. Given these three assumptions, it follows that motor adaptation is an inherently noisy process, and that random corrections to any given movement inevitably will damage other existing motor memories. In particular, ongoing corrections to any given movement will tend to reshuffle the pattern of synaptic weights responsible for that movement, and thus destroy any information relating to other learned motor behaviors stored at the same synapses.
Anatomical arguments suggest that a second site of plasticity at parallel fiber to stellate/basket (pf*St/Bk) synapses could provide an alternative locus of stable long-term storage in a manner consistent with climbing fiber-dependent plasticity at pf*Pkj synapses. First, learned pauses in Purkinje cell activity, which in the above models results from the induction of long-term depression (LTD) at pf*Pkj synapses, also could be produced by appropriately timed increases in the inhibitory synaptic input from stellate/basket cells. Second, stellate and basket cells greatly outnumber Purkinje cells (30) and receive similar patterns of parallel fiber input from granule cells, thus pf*St/Bk synapses provide a potentially large reservoir of additional storage capacity. Third, the projection patterns of stellate/basket cells respect the parasagital motor organization of the cerebellar cortex (30, 31), suggesting that such projections may play a direct role in the execution of movements. Fourth, recent studies report that rats raised in environments requiring increased motor learning exhibit a significant expansion in the number of stellate dendrites (32). Finally, plasticity has been reported at synapses onto analogous interneurons in other brain areas (33).
In this paper, we use a mathematical model of the cerebellar cortex and its reciprocal interactions with the inferior olive to investigate how the two proposed sites of plasticity could interact in complementary manner. Although plasticity at synapses onto interneurons in the cerebellar cortex has been studied in previous models (13, 16), these analyses did not address the issue of how plasticity at pf*St/Bk synapses might be specialized for stable long-term storage. As a tractable example of motor learning, the present model is applied to the analysis of idealized Pavlovian conditioning trials. Our principal finding is that when plasticity at pf*St/Bk synapses is sensitive to changes in Purkinje cell activity, motor memories automatically are transferred from pf*Pkj to pf*St/Bk synapses during continued reinforcement training. These results suggest that pf*St/Bk synapses could provide a site of stable long-term memory storage, whereas climbing fiber-dependent plasticity pf*Pkj synapses provides a means of exploring novel motor strategies in an intrinsically noisy environment without damaging previously learned behaviors.
THEORY
The Anatomy of the Cerebellar-Olivary System.
The anatomy of the cerebellar-olivary system relevant to the present model is illustrated in Fig. 1 (34). Input to the cerebellar cortex is via both mossy fibers and climbing fibers. Each mossy fiber fans out to contact numerous granule cells, which, in turn, project to Purkinje cells and also to stellate and basket cells. Purkinje cells receive input from approximately 100,000 granule cells, but from only a single climbing fiber. Stellate/basket cells, which are approximately 10–20 times more numerous than Purkinje cells (30), make inhibitory synapses onto Purkinje cells. Despite the known differences between stellate and basket cells, the present model treats these cells as a single cell type.
Figure 1.
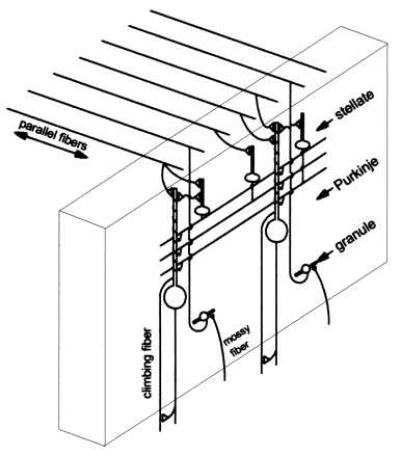
Schematic of a single cerebellar microzone. Within a microzone, the cells are arrayed in a parasagital strip aligned transversely to the long axis of the folia. Parallel fibers, running along the axis of the folia, transect each microzone at a right angle, making synapses with stellate/basket cells and Purkinje cells. Stellate/basket cells send their axons along a transverse axis, linking Purkinje cells within the same microzone. Climbing fiber input from a localized region of the inferior olive is restricted to a single microzone and is subject to feedback from this same region.
The sole output of the cerebellar cortex is via Purkinje cells. The influence of Purkinje cells on climbing fibers is via a disynaptic pathway consisting of two inhibitory synapses in series; Purkinje cells make inhibitory synapses in the cerebellar nuclei, and the cerebellar nuclei send a prominent inhibitory projection to the inferior olive (35, 36), which is the sole source of climbing fibers. Because both climbing fibers and cerebellar nucleus neurons are tonically active (4, 37), it should be possible to model the two inhibitory synapses between Purkinje cells and climbing fibers as a single excitatory connection, as shown in Fig. 1.
At the highest level of organization, the present model assumes that the cerebellar cortex is divided into functionally distinct microzones, each of which controls a specific muscle group (24, 25). As illustrated in Fig. 1, each microzone consists of a parasagital strip that runs transversely to the long axis of the folia. The dendritic planes of the individual Purkinje cells are oriented along the axis of their corresponding microzones, whereas parallel fibers run in a perpendicular direction, transecting multiple microzones as they course along the long axis of the folia. The axons of stellate and basket cells run transversely to the folia so as to potentially link different Purkinje cells within the same microzone.
Because of regular patterns of connectivity within a single cerebellar microzone, we assume that all cells of a given type within a specific microzone exhibit identical behavior. With this assumption, it becomes necessary to model only a single representative stellate/basket cell, its target Purkinje cell, and a single representative climbing fiber input.
Simplifying Assumptions.
We seek to construct the simplest mathematical model that is still able to capture important features of the synaptic dynamics affecting the cerebellar cortex. To this end, it is simplest to assume that the input/output properties of the neural elements are linear. This assumption is supported by the finding that the neurons in the present study all exhibit finite levels of background activity, thus minimizing the importance of a nonlinear firing threshold. Furthermore, because our goal is to model the slow cumulative changes caused by synaptic plasticity, rather than the real time behavior of the network, we may simplify the dynamics by using a discrete time model in which the activities of the individual neural elements, as well as the external inputs to the system, change discontinuously on each time step. Specifically, we assume that the activity of each neural element jumps immediately to the value defined by the synaptic input present on that time step. Finally, to avoid the introduction of large number of arbitrary constants, the parameters of the model are chosen so that the activities of all elements remain between zero and one.
Purkinje Cells.
Let the activity of a representative Purkinje cell be denoted by Pp. Likewise, let the activity of a representative stellate/basket cell be denoted by Ps. To develop an expression for Purkinje cell activity, let the elements {wi} denote the individual pf*Pkj synaptic weights, and the elements {Pi} denote the activity levels of the individual parallel fibers. The activity of the Purkinje cell then can be approximated by the linear sum of its synaptic input from parallel fibers minus the inhibition from stellate/basket cells:
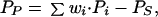 |
1 |
where wi⋅Pi represents the synaptic input to the Purkinje cell because of the ith granule cell and the sum is over all parallel fibers.
Stellate/Basket Cells.
As shown in Fig. 1, granule cells also make direct connections onto stellate/basket cells. Letting the elements {w′i} denote the individual pf*St/Bk synaptic weights, we may express the activity of a representative stellate/basket cell as
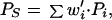 |
2 |
where w′i⋅Pi represents the ith synaptic input to a representative stellate/basket cell and the sum is over all parallel fibers.
Climbing Fibers.
Evidence suggests that climbing fibers can be activated by external stimuli that signal a need for motor adaptation (2, 7, 38). An expression for the activity of a representative climbing fiber, denoted by PC, then may be written
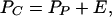 |
3 |
where E represents a source of transient external excitation that causes climbing fiber activity to increase when motor adaptation is required. For simplicity, we will assume that E = 0, except during motor conditioning trials. Eq. 3 also expresses mathematically the condition that net influence of Purkinje cells on climbing fibers is excitatory, as shown in Fig. 1. For purposes of the present model, the ability of a source of external excitation to activate the correct set of climbing fibers for promoting an adaptive movement will be assumed.
LTD/Potentiation at pf*Pkj Synapses.
Numerous electrophysiological studies have contributed to our understanding of plasticity at pf*Pkj synapses (26–28, 39–41). In terms of the combination of inputs necessary to induce plasticity, these studies suggest that pf*Pkj synapses display a bidirectional form of climbing fiber-dependent LTD/potentiation: the weight of a pf*Pkj synapse decreases when active during elevated climbing fiber input and increases when active during reduced climbing fiber input. We may express this rule mathematically as follows:
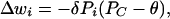 |
4 |
where δ denotes the magnitude of the changes in pf*Pkj synaptic weights produced by climbing fiber-dependent LTD/potentiation and θ represents the level of climbing fiber activity at which pf*Pkj synaptic weights remain constant. Previous analysis has shown that with a plasticity rule of this form, climbing fiber activity always is driven to this equilibrium level (20). Because of the coupling between Purkinje cells and climbing fibers, as expressed by Eq. 3, θ denotes the equilibrium level of Purkinje cell activity as well.
Plasticity at pf*St/Bk Synapses.
To our knowledge, plasticity at pf*St/Bk synapses has not been investigated, and the rules potentially governing such plasticity are completely unknown. We require, however, that any plasticity rule at pf*St/Bk synapses not only contribute to motor learning in a manner consistent with climbing fiber-dependent plasticity at pf*Pkj synapses, but provide additional functionality as well. One such rule that satisfies these requirements can be expressed mathematically as follows:
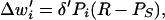 |
5 |
where δ′ characterizes the magnitude of the changes in pf*St synaptic weights produced by changes in Purkinje cell and/or stellate/basket activity and R is defined as θ − Pp. We assume that pauses in Purkinje cell activity generate increased cerebellar output by releasing projection neurons in the cerebellar nuclei from tonic inhibition, and thus may directly elicit movements. The quantity R therefore provides a rough measure of the movement amplitude generated by a given decrease in Purkinje cell activity. Qualitatively, Eq. 5 states that when a pf*St/Bk synapse is active (Pi ≈ 1) during a movement-related pause in Purkinje cell activity (Pp ≈ 0, R > 0), the synaptic weight increases by an amount proportional to δ′ as long as the activity of the stellate/basket cell remains near equilibrium (Ps ≈ 0). To account for the extinction of conditioned motor responses, Eq. 5 further specifies that increased stellate/basket activity in the absence of a pause in Purkinje cell activity promotes the induction of LTD at pf*St/Bk synapses. Although Eq. 5 implies that at equilibrium, Ps = 0, this equation may be easily modified by adding a constant value to Ps to specify a finite equilibrium level of stellate/basket activity.
The above equations can be solved exactly by using a standard linear systems analysis. The predicted consequences of the proposed plasticity rules are described below.
RESULTS
To explore cerebellar mediated motor learning, we use an idealized Pavlovian conditioning protocol, in which an initially neutral conditioned stimulus (CS) is paired repeatedly with an unconditioned stimulus (US). After a large number of paired, or reinforced, training trials, the CS comes to elicit a conditioned motor response. In eyelid conditioning, the CS typically is represented by a tone, and the US by an air puff directed at the cornea. Eventually, the subject learns to generate an anticipatory eyeblink in response to the tone. In the present model, the CS is represented by a particular pattern of parallel fiber input that is distinct from the time-averaged background activity, and the US is represented by an increase in the external excitatory drive to a representative climbing fiber, modeled by setting E = 1/2 in Eq. 3. Extinction is modeled by resetting E to zero once a stationary state has been reached. The amplitude of a conditioned response is represented by the quantity R, which measures the reduction in Purkinje cell activity during presentations of the CS. In each stimulated training trial, the CS and the US are presented simultaneously for a single time step, and the system is allowed to fully equilibrate between trials. No attempt is made to account for the temporal aspects of conditioning. The results of the model when applied to an idealized Pavlovian conditional protocol are show in Fig. 2.
Figure 2.
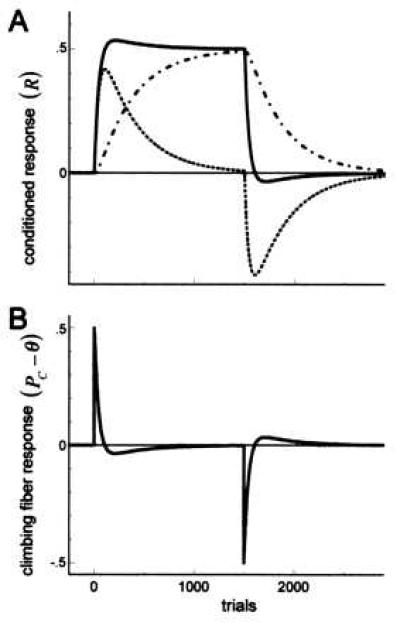
The transfer of plasticity from pf*Pkj to pf*St/Bk synapses during the acquisition and extinction of a conditioned motor response. (A) Simulated acquisition and extinction training. The solid line plots the quantity R = θ − Pp, here used as a measure of response amplitude, as a function of training trials. The parameters are chosen such that acquisition occurs in approximately 100 training trials. The dotted line shows the contribution to the conditioned response made by plasticity at pf*Pkj synapses (R − Ps). Although such plasticity accounts for much of the initial acquisition of the response, the contribution from pf*Pkj synapses eventually reverses and decays to zero with continued training. In contrast, the dotted-dashed line plots the contribution to the conditioned movement from plasticity at pf*St/Bk synapses (Ps), which grows with continued training until such plasticity becomes entirely responsible for the response after about 1,000 training trials. Analogous phenomena occur during extinction. At the behavioral level, extinction is mediated entirely by plasticity at pf*Pkj synapses, requiring approximately 100 training trials, whereas nearly 1,000 extinction trials are necessary to fully extinguish the memory stored at pf*St/Bk synapses. (B) Climbing fiber responses during acquisition and extinction. Climbing fiber responses are most prominent during the initial phases of training, during which times they mediate rapid motor adaptation at pf*Pkj synapses. As conditioned responses are acquired, climbing fiber responses become suppressed by the increased inhibition from the cerebellar nuclei. The small deviations in the level of climbing fiber activity from equilibrium after asymptotic response levels have been reached contribute to the transfer of plasticity from pf*Pkj to pf*St/Bk synapses. Analogous events occur during extinction. Plasticity at pf*Pkj synapses was chosen to be a factor of 10 times stronger than plasticity at pf*St/Bk synapses (δ = 10⋅δ′).
The solid line in Fig. 2A plots a measure of the conditioned response, R, as a function of both training and subsequent extinction trials. The learning curve defined by R is qualitatively similar to those obtained behaviorally (42). For the parameters chosen, responses approach asymptotic levels in approximately 100 training trials, with a similar number of trials required to extinguish responses.
The rather conventional nature of the learning curve defined by R masks a more complex underlying process, in which the conditioned response is mediated initially by plasticity at pf*Pkj synapses, but this plasticity subsequently is transferred to pf*St/Bk synapses with further training. To show this process, we break the response into two components. The first component, R′ = Ps measures the contribution to the total response because of plasticity at pf*St/Bk synapses (Fig. 2A, dash-dotted line). The second component, R′′ = R − R′, measures the remaining contribution because of plasticity at pf*Pkj synapses (Fig. 2A, dotted line). Early in training, conditioned responses are entirely mediated by plasticity at pf*Pkj synapses. With further training, this plasticity is transferred to pf*St/Bk synapses. Eventually, asymptotic response levels are mediated entirely by plasticity at pf*St/Bk synapses, and the weights of pf*Pkj synapses will have returned to their initial values. A very similar process occurs during extinction. Initially, the relatively rapid extinction of responses is mediated by plasticity at pf*Pkj synapses, which become sufficiently large to compensate for the increased inhibition from stellate/basket cells. There then follows a prolonged period during which the synaptic weights at both sites of plasticity return to their original values.
The model predicts that the discharge activity of cerebellar neurons will change in a characteristic fashion as the plasticity responsible for the conditioned response is transferred from pf*Pkj to pf*St/Bk synapses. Early in training, before asymptotic response levels have been attained, the CS-evoked discharge activity of Purkinje cells decreases relatively rapidly as a function of training, whereas the CS-evoked discharge activity of stellate/basket cells changes very little during this period. After asymptotic response levels have been attained, however, the situation is predicted to reverse. The CS-evoked discharge activity of Purkinje cells remains approximately constant, whereas the CS-evoked stellate/basket discharge activity slowly increases with further training. The extinction of conditioned responses exhibits a similar phenomenology. The CS-evoked Purkinje cell activity increases rapidly during the initial phase of extinction training, whereas CS-evoked stellate/basket activity decreases, but more slowly, during the final phase of extinction training.
The model predicts that the US-evoked discharge activity of climbing fibers also changes in a characteristic fashion during training. Fig. 2B shows that early in training, climbing fibers discharge robustly in response to the US. As the response amplitude approaches asymptote, however, the external excitation of climbing fibers is balanced by increased inhibition from cerebellar nucleus neurons. The asymptotic response amplitude corresponds to the point at which the increased discharge activity of cerebellar nucleus neurons evoked by the CS exactly cancels the excitatory influence of the US, and thus climbing fiber activity remains constant during the training trial. This automatic down-regulation of climbing fiber responses to CS+US trials has been modeled previously (21), and is consistent with general theories of associative learning (43) and with physiological recordings from climbing fibers during several forms of motor adaptation (2, 7, 38). A related process also is predicted to occur during extinction. Early in extinction training, climbing fiber discharge activity drops below normal background levels during presentations of the unreinforced CS. This decrease occurs because the response-related inhibition of climbing fibers by cerebellar nucleus neurons is no longer countered by the excitatory influence of the US. As conditioned responses are extinguished, however, inhibition from the nucleus is reduced, and climbing fiber discharge activity returns to normal background levels.
The theoretical learning curve plotted in Fig. 2A predicts a small overshoot in the learning curve during which the response amplitude is predicted to rise slightly above its asymptotic level, and then slowly fall back. This overshoot occurs because even though the asymptotic response amplitude has been attained, the response-related pause in Purkinje cell activity still promotes the induction of LTP in pf*St/Bk synapses, thus increasing the response amplitude still further. This overshoot, which corresponds to an slightly higher level of discharge activity in cerebellar nucleus neurons above their equilibrium activity level, drives climbing fiber activity below equilibrium, thereby promoting the induction of LTP at pf*Pkj synapses. With continued reinforcement training, both pf*Pkj and pf*St/Bk synapses get stronger until the response is entirely mediated by an increase in stellate/basket activity, and pf*Pkj synaptic weights have been driven back up to their original values. A analogous undershoot also is predicted to occur during extinction. By driving cerebellar output below zero, climbing fiber discharge activity becomes slightly elevated, thereby driving synaptic weights at both sites of plasticity back down to their original values. The proposed plasticity rules thus ensure that motor conditioning is entirely reversible.
DISCUSSION
The present study describes how the synaptic plasticity postulated to be responsible for the generation of conditioned motor responses could be smoothly transferred from pf*Pkj to pf*St/Bk synapses during continued reinforcement training. The principal assumption made by the present model is that movement-related decreases in Purkinje cell firing promote LTP at coactive pf*St/Bk synapses, whereas increases in stellate/basket activity during baseline Purkinje cell activity produce LTD. Whether or not plasticity at pf*St/Bk synapses actually is governed by such a rule can be determined only by direct experimental investigation. Unfortunately, no published study of plasticity at pf*St/Bk synapses is presently available.
In addition to the indirect lines of evidence cited previously, the strongest support for the hypothesis that plasticity at pf*St/Bk synapses could play an essential role in motor learning may derive from theoretical arguments that postulate a critical functional role for such plasticity. The present model is anchored in the assumption that movement errors activate the appropriate climbing fibers for adapting, or correcting, the corresponding movement. Despite ongoing motor adaptation, however, errors still will occur at a finite rate regardless of how much the movement has been rehearsed. No biological system can ever perform perfectly. Not only do external environmental conditions fluctuate in an unpredictable fashion, but movements themselves are generated by biological components that are inherently noisy and subject to failure. It follows that once the rate of movement errors has been driven to a minimum, it no longer will be possible to achieve additional improvements in motor performance. There are likely to be a virtual infinity of synaptic weight combinations that can produce any given movement, however. Thus, the fluctuations in climbing fiber activity produced by errors in the execution of any given movement will cause the pattern of synaptic weights responsible for that movement to be constantly reshuffled. Because storage capacity arguments suggest that motor memories are distributed across overlapping sets of synapses, it follows that motor adaptation always will involve some degradation of other previously learned movements. Over time, as movements are continuously adapted, the memories for previously learned movements, unless explicitly reinforced with additional training, will be destroyed.
Fluctuations in climbing fiber activity do not just reshuffle synaptic weights, however. The changes in pf*Pkj synaptic weights produced by such fluctuations are adaptive, and indeed necessary, for the maintenance of motor performance. The problem becomes how to explore new motor strategies that might reduce the overall rate of motor errors without destroying the memories for previously learned movements. The present results suggest that one possible solution is to use two sites of plasticity. The first site, located at pf*Pkj synapses, responds to each fluctuation in climbing fiber input, even though some of these fluctuations occur in random, or unlearnable, contexts. As argued above, such random fluctuations eventually would destroy any long-term information regarding any learned movement that is not continuously reinforced with additional training. Occasionally, however, a given pattern of climbing fiber fluctuations will be predicted by a reproducible pattern of parallel fiber input. In this case, the changes in pf*Pkj synaptic weights can accumulate until an adaptive movement has been acquired. The resulting changes in Purkinje cell activity then will induce a transfer of the plasticity responsible for the adaptive movement to pf*St/Bk synapses. If it is further assumed that the transfer of plasticity is initiated only after plasticity at pf*Pkj synapses has built up above a given threshold level, then plasticity at pf*St/Bk synapses can be effectively insulated from the ongoing random fluctuations in climbing fiber input. Thus, the proposed mechanism allows sufficiently large motor adaptations to be stored indefinitely without preventing novel motor strategies from being explored.
The role of the cerebellum in motor learning is presently the subject of much disagreement. A variety of studies have been interpreted by some investigators as evidence that the cerebellum is not a site of motor learning at all, but rather provides an essential input to a site of plasticity located elsewhere in the nervous system (44–48). Although the present model is clearly incompatible with such theories, the mechanism proposed here for storing long-term information at pf*St/Bk synapses is consistent with a variety of related cerebellar models designed to account for temporal aspects of motor learning (15, 16, 18, 19, 22, 23), as well as with models of cerebellar function based on arrays of adjustable pattern generators (49) or on temporal difference models (50). Other authors have conjectured that whereas climbing fiber-dependent plasticity underlies cerebellar involvement in motor leaning, synaptic plasticity in the deep cerebellar nuclei also contributes to the storage of long-term motor memories (14, 51–53). When plasticity in the cerebellar nuclei (or equivalently in the vestibular nuclei) is regulated by input from Purkinje cells (54), it has been argued that motor memories would be transferred from the cerebellar cortex to the cerebellar nuclei as a function of continued training (55). Because the transfer of plasticity from the cerebellar cortex to the cerebellar nuclei parallels the transfer of plasticity between pf*Pkj and pf*St/Bk synapses, it follows that much of the present discussion regarding the function of plasticity at pf*St/Bk synapses could apply to the storage of motor memories in the cerebellar nuclei as well. The two sites of long-term storage then might be specialized for different aspects of learned motor behavior, as suggested by experiments showing that the cerebellar cortex may play a primary role in the proper timing of learned movements (4, 51).
Although anatomical evidence supporting the present model was cited earlier, other anatomical evidence argues against Purkinje cell-dependent plasticity at pf*St/Bk synapses. Whereas Purkinje cells send collateral projections to basket cells (56), no such projections to stellate cells have been reported (31). This result might reflect a basic dichotomy between stellate and basket cells, or else Purkinje cells might exert their influence on stellate cells though intermediate cell types, such as Golgi cells (57). Such influence could in turn be transmitted to stellate cells via the ascending axons of granule cells, which previously have been postulated to exert a differential influence on their synaptic targets in the overlying layers (58). Another argument against the present hypothesis is that the number of pf*St/Bk synapses is less than 15% of the number of pf*Pkj synapses (59). Even so, this result still represents an enormous number of synaptic contacts for any given microzone. It is possible that such numerical disparities reflect the different postulated roles of the two sites of plasticity in motor learning. For instance, the task of initial motor adaptation, in which an improved motor strategy must be extrapolated from noisy data through a process of repeated association, might require more synapses than does simply storing such strategies once they have been fully acquired. Finally, it may be argued that it is possible to stabilize motor memories simply by making the changes in synaptic weight irreversible. Irreversible changes come at a cost, however, because they deplete the pool of synapses available for new learning. Evidence that several forms of cerebellar motor learning are reversible is also at variance with this idea (60–62).
The main experimental prediction of the present model is that in the cerebellar cortex, long-term motor memories are stored at pf*St/Bk synapses. At the behavioral level, this model predicts that even though asymptotic performance levels have been achieved, extensive further training may be necessary to transfer recently acquired motor memories to long-term storage. This result implies that the rate at which a motor memory is forgotten will depend not on the level of asymptotic performance attained but on the degree of overtraining after asymptotic performance levels have been reached. Finally, although the present analysis has been restricted to a single set of synapses in cerebellar cortex, it is not implausible that similar mechanisms could be involved in the transfer of memories to long-term storage at other sites in the central nervous system.
Acknowledgments
I gratefully acknowledge the useful comments and suggestions of Dianne Broussard, Keith Garcia, Michael Hausser, Paul Katz, David Marshak, John Moore, and Phillip Steele.
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
Abbreviations: pf*Pkj, parallel fiber onto Purkinje cell (i.e., pf*Pkj synapses); pf*St/Bk, parallel fiber onto stellate/basket cell; LTD, long-term depression; CS, conditioned stimulus; US, unconditioned stimulus.
References
- 1.Black J E, Isaacs K, Anderson B J, Alcantara A A, Greenough W T. Proc Natl Acad Sci USA. 1990;87:5568–5572. doi: 10.1073/pnas.87.14.5568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gilbert P, Thach W T. Brain Res. 1977;128:309–328. doi: 10.1016/0006-8993(77)90997-0. [DOI] [PubMed] [Google Scholar]
- 3.Ito M. Annu Rev Neurosci. 1982;12:85–102. doi: 10.1146/annurev.ne.12.030189.000505. [DOI] [PubMed] [Google Scholar]
- 4.McCormick D A, Thompson R F. J Neurosci. 1984;4:2811–2822. doi: 10.1523/JNEUROSCI.04-11-02811.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Optican L M, Robinson D A. J Neurophysiol. 1980;44:1058–1076. doi: 10.1152/jn.1980.44.6.1058. [DOI] [PubMed] [Google Scholar]
- 6.Robinson D A. J Neurophysiol. 1976;39:954–969. doi: 10.1152/jn.1976.39.5.954. [DOI] [PubMed] [Google Scholar]
- 7.Watanabe E. Brain Res. 1984;297:169–174. doi: 10.1016/0006-8993(84)90555-9. [DOI] [PubMed] [Google Scholar]
- 8.Martin T A, Keating J G, Goodkin H P, Bastian A J, Thach W T. Brain. 1996;119:1183–1198. doi: 10.1093/brain/119.4.1183. [DOI] [PubMed] [Google Scholar]
- 9.Sanes J N, Dimitrov B, Hallet M. Brain. 1990;113:103–120. doi: 10.1093/brain/113.1.103. [DOI] [PubMed] [Google Scholar]
- 10.Friston K J, Frith C D, Passingham R E, Liddle P F, Frackowiak R S J. Proc R Soc London. 1992;248:223–228. doi: 10.1098/rspb.1992.0065. [DOI] [PubMed] [Google Scholar]
- 11.Jenkins I H, Brooks D J, Nixon P D, Frackowiak R S J, Passingham R E. J Neurosci. 1994;14:3775–3790. doi: 10.1523/JNEUROSCI.14-06-03775.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marr D. J Physiol. 1969;202:437–470. doi: 10.1113/jphysiol.1969.sp008820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Albus J S. Math Biosci. 1971;10:25–61. [Google Scholar]
- 14.Bullock D, Fiala J, Grossberg S. Neural Networks. 1994;7:1101–1114. [Google Scholar]
- 15.Buonomano D V, Mauk M D. Neuro Comput. 1994;6:38–55. [Google Scholar]
- 16.Fujita M. Biol Cybern. 1982;45:195–206. doi: 10.1007/BF00336192. [DOI] [PubMed] [Google Scholar]
- 17.Gilbert P F C. Brain Res. 1974;70:1–18. doi: 10.1016/0006-8993(74)90208-x. [DOI] [PubMed] [Google Scholar]
- 18.Gluck M A, Reifsnider E S, Thompson R F. In: Neuroscience and Connectionist Theory. Gluck M A, Rumelhart D E, editors. Hillsdale, NJ: Erlbaum; 1990. pp. 131–186. [Google Scholar]
- 19.Kawato M, Gomi H. Biol Cybern. 1992;68:95–103. doi: 10.1007/BF00201431. [DOI] [PubMed] [Google Scholar]
- 20.Kenyon G T, Medina J F, Mauk M D. J Comput Neurosci. 1998;5:17–33. doi: 10.1023/a:1008874209991. [DOI] [PubMed] [Google Scholar]
- 21.Kenyon G T, Medina J F, Mauk M D. J Comput Neurosci. 1998;5:71–90. doi: 10.1023/a:1008830427738. [DOI] [PubMed] [Google Scholar]
- 22.Moore J W, Desmond J E, Berthier N E. Biol Cybern. 1989;62:17–28. doi: 10.1007/BF00217657. [DOI] [PubMed] [Google Scholar]
- 23.Sejnowski T J. J Math Biol. 1977;4:303–321. doi: 10.1007/BF00275079. [DOI] [PubMed] [Google Scholar]
- 24.Gibson A R, Robinson F R, Alam J, Houk J C. J Comp Neurol. 1987;260:362–377. doi: 10.1002/cne.902600304. [DOI] [PubMed] [Google Scholar]
- 25.Oscarsson O. In: The Inferior Olivary Nucleus: Anatomy and Physiology. Courville J, de Montigny C, Lamarre Y, editors. New York: Raven; 1980. pp. 279–289. [Google Scholar]
- 26.Ekerot C-F, Kano M. Brain Res. 1985;342:357–360. doi: 10.1016/0006-8993(85)91136-9. [DOI] [PubMed] [Google Scholar]
- 27.Ito M, Kano M. Neurosci Lett. 1982;33:253–258. doi: 10.1016/0304-3940(82)90380-9. [DOI] [PubMed] [Google Scholar]
- 28.Kano M, Kato M. Neurosci Res. 1988;1988:544–556. doi: 10.1016/0168-0102(88)90041-7. [DOI] [PubMed] [Google Scholar]
- 29.Berthier N E, Moore J W. Exp Brain Res. 1986;63:341–350. doi: 10.1007/BF00236851. [DOI] [PubMed] [Google Scholar]
- 30.Palkovits M, Magyar P, Szentagothai J. Brain Res. 1971;34:1–18. doi: 10.1016/0006-8993(71)90347-7. [DOI] [PubMed] [Google Scholar]
- 31.Palay S L, Chan-Palay V. Cerebellar Cortex. New York: Springer; 1974. [Google Scholar]
- 32.Kleim J A, Swain R A, Czerlanis C M, Kelly J L, Pipitone M A, Greenough W T. Neurobiol Learn Mem. 1997;67:29–33. doi: 10.1006/nlme.1996.3742. [DOI] [PubMed] [Google Scholar]
- 33.Ouardouz M, Lacaille J C. J Neurophysiol. 1995;73:810–819. doi: 10.1152/jn.1995.73.2.810. [DOI] [PubMed] [Google Scholar]
- 34.Ito M. The Cerebellum and Neural Control. New York: Raven; 1984. [Google Scholar]
- 35.Anderson G, Garwicz M, Hesslow G. Brain Res. 1988;472:450–456. doi: 10.1007/BF00250590. [DOI] [PubMed] [Google Scholar]
- 36.Angaut P, Sotelo C. Brain Res. 1989;479:361–365. doi: 10.1016/0006-8993(89)91641-7. [DOI] [PubMed] [Google Scholar]
- 37.Armstrong D M, Rawson J A. J Physiol. 1979;289:425–448. doi: 10.1113/jphysiol.1979.sp012745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sears L L, Steinmetz J E. Brain Res. 1991;545:114–122. doi: 10.1016/0006-8993(91)91276-7. [DOI] [PubMed] [Google Scholar]
- 39.Sakurai M. In: The Olivocerebellar System in Motor Control. Strata P, editor. Vol. 17. New York: Springer; 1989. pp. 221–230. [Google Scholar]
- 40.Salin P, Malenka R C, Nicoll R A. Neuron. 1995;16:797–803. doi: 10.1016/s0896-6273(00)80099-9. [DOI] [PubMed] [Google Scholar]
- 41.Shibuki K, Okada D. NeuroReport. 1992;3:231–234. doi: 10.1097/00001756-199203000-00003. [DOI] [PubMed] [Google Scholar]
- 42.Gormezano I, Schneiderman N, Deaux E, Fuentes I. Science. 1962;138:33–34. doi: 10.1126/science.138.3536.33. [DOI] [PubMed] [Google Scholar]
- 43.Rescorla R, Wagner A. In: Classical Conditioning II: Current Research and Theory. Black A, Prokasy W, editors. New York: Appleton-Century-Crofts; 1972. [Google Scholar]
- 44.Bloedel J R. Behav Brain Sci. 1992;15:666–678. [Google Scholar]
- 45.De Schutter E. Trends Neurosci. 1995;18:291–295. doi: 10.1016/0166-2236(95)93916-l. [DOI] [PubMed] [Google Scholar]
- 46.Llinas R, Welsh J P. Curr Opin Neurobiol. 1993;3:958–968. doi: 10.1016/0959-4388(93)90168-x. [DOI] [PubMed] [Google Scholar]
- 47.Welsh J P, Harvey J A. J Neurosci. 1989;9:299–311. doi: 10.1523/JNEUROSCI.09-01-00299.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gao J-H, Parsons L M, Bower J M, Xiong J, Li J, Fox P T. Science. 1996;272:545–547. doi: 10.1126/science.272.5261.545. [DOI] [PubMed] [Google Scholar]
- 49.Houk J C, Wise S P. Cerebral Cortex. 1995;2:95–110. doi: 10.1093/cercor/5.2.95. [DOI] [PubMed] [Google Scholar]
- 50.Moore J W, Desmond J E, Berthier N E, Blazis D E, Sutton R S. Behav Brain Res. 1986;21:143–154. doi: 10.1016/0166-4328(86)90092-6. [DOI] [PubMed] [Google Scholar]
- 51.Perrett S P, Ruiz B P, Mauk M D. J Neurosci. 1993;13:1708–1718. doi: 10.1523/JNEUROSCI.13-04-01708.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Raymond J L, Lisberger S G, Mauk M D. Science. 1996;272:1126–1131. doi: 10.1126/science.272.5265.1126. [DOI] [PubMed] [Google Scholar]
- 53.Thompson R F. Science. 1986;233:941–947. doi: 10.1126/science.3738519. [DOI] [PubMed] [Google Scholar]
- 54.Miles F A, Lisberger S G. Annu Rev Neurosci. 1981;4:273–299. doi: 10.1146/annurev.ne.04.030181.001421. [DOI] [PubMed] [Google Scholar]
- 55.Coenen O, Sejnowski T J, Lisberger S G. In: Advances in Neural Information Processing Systems 5. Giles C L, Hanson S J, Cowan J D, editors. San Mateo, CA: Morgan Kaufman; 1993. pp. 961–968. [Google Scholar]
- 56.O’Donoghue D L, King J S, Bishop G A. J Neurosci. 1989;9:2141–2150. doi: 10.1523/JNEUROSCI.09-06-02141.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schulman J A, Bloom F E. Brain Res. 1981;210:350–353. doi: 10.1016/0006-8993(81)90908-2. [DOI] [PubMed] [Google Scholar]
- 58.Bower J M, Kassel J. J Comp Neurol. 1990;302:768–778. doi: 10.1002/cne.903020409. [DOI] [PubMed] [Google Scholar]
- 59.Harvey R J, Napper R M A. Prog Neurobiol. 1991;36:437–463. doi: 10.1016/0301-0082(91)90012-p. [DOI] [PubMed] [Google Scholar]
- 60.Miles F A, Fuller J H. Brain Res. 1974;80:512–516. doi: 10.1016/0006-8993(74)91035-x. [DOI] [PubMed] [Google Scholar]
- 61.Napier R M, Macrae M, Kehoe E J. J Exp Psychol. 1992;18:182–192. doi: 10.1037//0097-7403.18.2.182. [DOI] [PubMed] [Google Scholar]
- 62.Smith M C, Gormezano I. Psychonom Sci. 1965;3:91–92. [Google Scholar]