

Abstract
Interactions of the inhibitory receptor programmed death-1 (PD-1) with its ligands, programmed death ligand (PD-L)1 and PD-L2, regulate T-cell activation and tolerance. In this study, we investigated the role of PD-L1 and PD-L2 in regulating invariant natural killer T (iNKT)-cell-mediated airway hyperreactivity (AHR) in a murine model of asthma. We found that the severity of AHR and airway inflammation is significantly greater in PD-L2−/− mice compared with wild-type mice after either ovalbumin (OVA) sensitization and challenge or administration of α-galactosylceramide (α-GalCer). iNKT cells from PD-L2−/− mice produced significantly more interleukin (IL)-4 than iNKT cells from control mice. Moreover, blockade of PD-L2 interactions of wild-type iNKT cells in vitro with monoclonal antibodies (mAbs) resulted in significantly enhanced levels of IL-4 production. In contrast, PD-L1−/− mice showed significantly reduced AHR and enhanced production of interferon-γ (IFN-γ) by iNKT cells. iNKT-deficient Jα18−/− mice reconstituted with iNKT cells from PD-L2−/− mice developed high levels of AHR, whereas mice reconstituted with iNKT cells from PD-L1−/− mice developed lower levels of AHR compared with control. As PD-L2 is not expressed on iNKT cells but rather is expressed on lung dendritic cells (DCs), in which its expression is upregulated by allergen challenge or IL-4, these findings suggest an important role of PD-L2 on lung DCs in modulating asthma pathogenesis. These studies also indicate that PD-L1 and PD-L2 have important but opposing roles in the regulation of AHR and iNKT-cell-mediated activation.
Introduction
Bronchial asthma is an immunological disease resulting from Th2-driven inflammation in the airways. It is characterized by inflammation in the peribronchial space, with increased production of airway mucus, and by airway hyperreactivity (AHR), a cardinal feature of asthma. Although allergen-induced AHR is known to be dependent on CD4 + T cells and associated with increases in Th2 cytokines in the lung, the mechanisms by which asthma is controlled are not fully understood.T reg cells secreting interleukin (IL)-10 can inhibit airway inflammation and AHR, but other inhibitory pathways also exist. For example, the programmed death-1 (PD-1) pathway has been shown to modulate airway inflammation, as PD-1−/− mice have increased numbers of cells in bronchoal-veolar lavage (BAL) fluid after allergen challenge.1 Although PD-1 has been well characterized as a negative regulator of conventional CD4 + T cells, the relative roles of the PD-1 ligands, PD-L2 and PD-L1, in regulating activation and function of invariant natural killer T cells (iNKT) is not fully understood but may be critically important as we and others have shown iNKT to be required for the development of AHR. iNKT-cell-deficient mice failed to develop AHR and had substantially reduced eosinophilia after sensitization and challenge with allergen, although Th2 responses developed normally.2,3 The requirement for iNKT cells was specific, as the adoptive transfer of iNKT cells from wild-type (WT) mice reconstituted the development of AHR in iNKT-cell-deficient Jα18−/− mice. Other studies of iNKT cells in mice as well as studies in non-human primates2–9 strongly suggest that iNKT cells have an important role in asthma, although determining the precise role of iNKT cells in human asthma has been the focus of recent studies.10–13
NKT cells comprise a unique and relatively rare subset of lymphocytes that express markers of both αβTCR+ T cells and NK cells. Type I (or classical) NKT cells constitute a distinct subset of T cells expressing a highly restricted or conserved/invariant T-cell receptor (TCR) repertoire consisting of Vα14-Jα18 (in mice) or Vα24-Jα18 (in humans). These NKT cells are often referred to as iNKT cells.14 iNKT cells are CD4 + or CD4−/CD8− (double-negative, DN). Through their invariant TCRs, iNKT cells recognize exogenous and endogenous glycolipid antigens presented by the nonpolymorphic major histocompatibility complex class I-like protein, CD1d,15 which is widely expressed by many cell types, including intestinal and airway epithelial cells, T cells, hepatocytes, B cells, macrophages and dendritic cells (DCs). The activation of iNKT cells results in the rapid production of large quantities of cytokines, such as IL-4, IL-13, IL-10, and interferon-γ (IFN-γ).16,17 This capacity to produce cytokines rapidly is a manifestation of an innate-like immunity that endows the iNKT cells with the capacity to amplify adaptive immunity, and to regulate the development of polarized T cells.
As with conventional CD4 + T cells, optimal activation and function of iNKT cells is regulated by signals delivered through the TCR and co-stimulatory molecules. iNKT cell activation requires at least two distinct signals from antigen-presenting cells (APCs). The first signal, which confers specificity, is provided by the interaction of the TCR with CD1d complexes. A second co-stimulatory signal can be provided by APC ligands for molecules on T cells such as CD28 or inducible co-stimulator (ICOS), and engagement of co-inhibitory receptors, such as CTLA-4 and PD-1, modulates the response. TCR stimulation of iNKT cells in the absence of co-stimulation induces anergy or alters the cytokine profile of the immune response.18–20 The engagement of CD40 or CD28 has been shown to provide important co-stimulatory signals to iNKT cells and to modulate the production of IL-4 and IFN-γ.18,20,21 We have previously shown that ICOS co-stimulation is required for CD4 + iNKT cell function, homeostasis, and survival in the periphery, and has a major role in the induction of AHR by iNKT cells.22 Recently, the PD-1/PD-L1 pathway has been shown to enhance the α-galactosylceramide (α-GalCer)-mediated induction of NKT cell anergy 23,24
In this study, we examined the relative contribution of PD-L1 and PD-L2 to the development of allergic airway responses and polarization of iNKT cells in an in vivo murine model of asthma. Our results show that the severity of AHR is greatly enhanced in PD-L2−/− mice. Our data also suggest that the increased severity of AHR in the PD-L2−/− mice is due to higher production of IL-4 by iNKT cells from PD-L2−/− mice. Blockade of PD-L2 interactions of WT iNKT cells with monoclonal antibody (mAb) enhances IL-4 production and thus is consistent with the enhanced IL-4 production we observed in iNKT cells from PD-L2−/− mice. Moreover, PD-L2 expression on lung DCs was enhanced in the presence of IL-4. In contrast, the level of IFN-γ secreted by iNKT cells in PD-L1−/− mice is greatly enhanced compared with iNKT cells from WT mice. Together, these studies suggest that PD-L2 engagement by its receptor on iNKT cells selectively inhibits IL-4 production by iNKT cells and emphasize the critical role of PD-L2 in the lung environment.
Results
PD-L1 and PD-L2 modulate the severity of AHR
To determine the roles of PD-L1 and PD-L2 in the development of allergen-induced AHR, we examined the development of AHR in PD-L1−/− and PD-L2−/− mice.Figure 1a shows that when sensitized and challenged with antigen, BALB/c control mice developed severe AHR as expected, measured as airway resistance (RL) and dynamic compliance (Cdyn) in anesthetized, tracheostomized, and mechanically ventilated mice (Figure 1a), or as Penh in conscious mice (Figure 1b). PD-L2−/− mice developed more severe AHR than BALB/c mice, whereas PD-L1−/− mice developed very mild AHR when sensitized and challenged with antigen. The reduction in AHR in PD-L1−/− mice was associated with a significant reduction of eosinophils and macrophages in BAL fluid (Figure 1c) compared with WT BALB/c. We examined the lung histology of mice shown in Figure 1a by hematoxylin and eosin to determine cellular infiltration and periodic acid Schiff (PAS) staining to determine mucus production. Lung tissue of PD-L2−/− mice showed extensive cellular infiltration surrounding the airways and thickened membrane and mucus production. PD-L1−/− mice showed less infiltration of cells and did not show mucus production by periodic acid Schiff staining (Figure 1d). As deficiency of PD-L1 vs. PD-L2 had contrasting effects on the induction of AHR, we examined the development of AHR in mice that lacked both PD-L1 and PD-L2. We found that the development of AHR in PD-L1−/− /PD-L2−/− mice is comparable with that of WT mice (Supplementary Figure 1 online).
Figure 1.
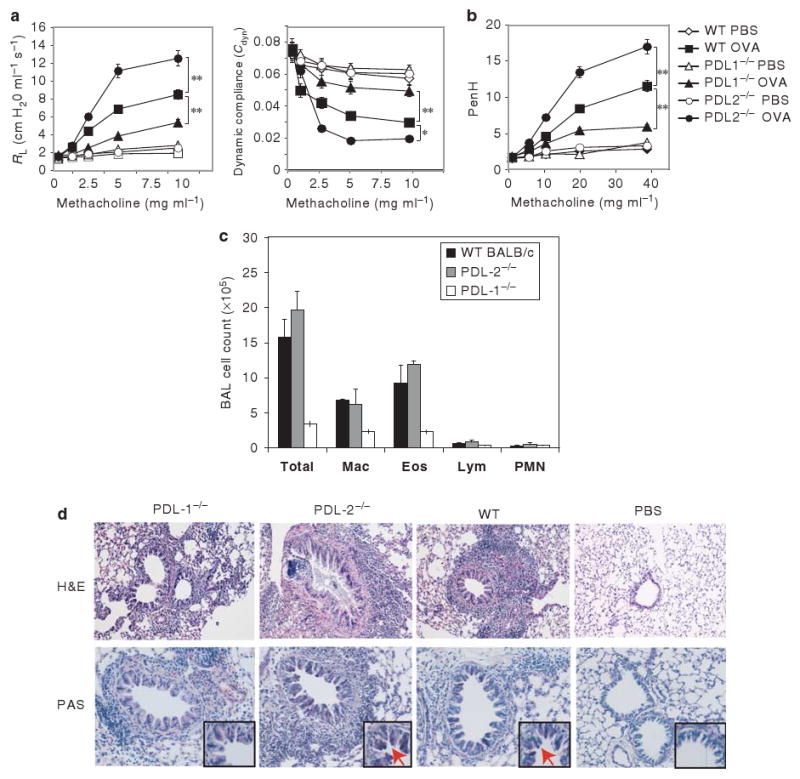
Increased airway inflammation and airway hyperreactivity (AHR) in PD-L2−/− but not PD-L1−/− mice. (a, b) BALB/c, PD-L1−/−, and PD-L2−/− mice were immunized with ovalbumin (OVA, 50μg) in alum IP. Mice were subsequently challenged with OVA IN on 3 consecutive days and assessed for AHR by measuring lung resistance (RL) and dynamic compliance (Cdyn) (a) or PenH (b). Data are the mean±s.e.m., representative of four experiments (n = 5) with * P<0.01 and ** P<0.001 (PD-L1−/− OVA vs. WT OVA and PD-L2−/− OVA vs. wild-type (WT) OVA). (c) Bronchoalveolar lavage (BAL) fluid from the mice in panel b was analyzed 24 h after AHR measurement. Results are shown as the total number of cells in BAL fluid. Total, total cell number; Mac, monocyte/macrophage; Eos, eosinophils; Lym, lymphocytes; PMN, neutrophils. (d) Lung histopathology of mice from panel b. Lung tissue from OVA challenged PD-L1−/−, PD-L2−/− or WT mice were stained with hematoxylin and eosin (H&E) (upper panel) and analyzed for cell infiltration. WT mice show numerous inflammatory cells surrounding the airways in the lumen. The degree of inflammation and cellular infiltration was much greater in PD-L2−/− mice and was less extensive in PD-L1−/− mice compared with OVA-sensitized WT mice. Phosphate-buffered saline (PBS) control mice show normal airway and the surrounding parenchyma. (lower panel) Lung tissue from the same mice were stained with periodic acid Schiff (PAS) and analyzed for the presence of mucus. The production of mucus was much greater in PD-L2−/− mice and minimal in PD-L1−/− mice compared with OVA-sensitized BALB/c. Arrows indicate the production of mucus in the lumen. Original magnifications, ×200; inset ×600.
Lung DCs express PD-L1 and PD-L2
As PD-L2 deficiency substantially increased the development of AHR, we investigated the expression of PD-L1 and PD-L2 on lung DCs and macrophages by flow cytometry after challenge of sensitized mice with ovalbumin (OVA) through the respiratory tract. PD-L1 and PD-L2 were expressed in moderate abundance on lung DCs in control mice that had received phosphate-buffered saline (PBS), and the levels of expression of both PD-L1 and PD-L2 on lung DCs were significantly increased upon challenge with OVA (Figure 2a). To further characterize the expression of PD-L2 in the lung, we examined by confocal microscopy the expression of PD-L2 on CD11c+ DCs from the lungs of mice sensitized and challenged with OVA (Figure 2b). PD-L2 (red) was expressed on the surface of all the lung DCs (CD11c+, green).
Figure 2.
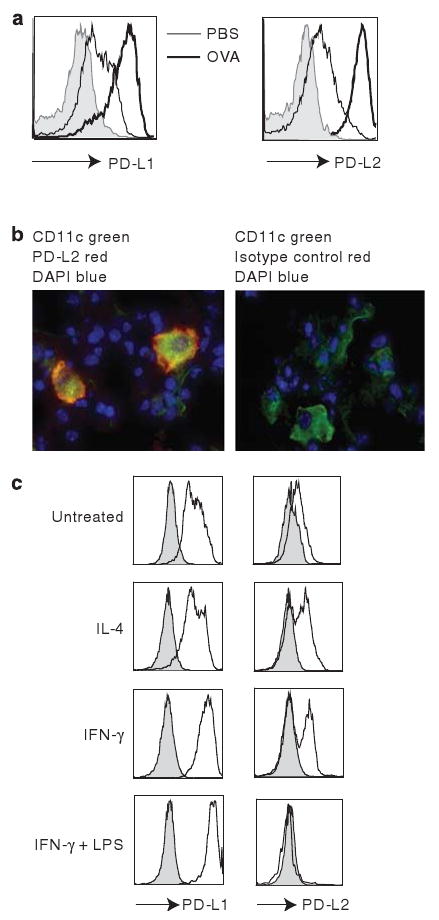
Differential cytokine regulation of PD-L1 and PD-L2 expression on lung dendritic cells (DCs). (a) DCs were positively selected from the lungs of wild type (WT) BALB/c mice that had received ovalbumin (OVA) in alum IP and been challenged with OVA (thick line) or PBS (thin line) IN on 3 consecutive days. The expression of PD-L1 and PD-L2 was analyzed on the CD11c+, I-Adhi population. (b) Laser confocal images of lung frozen sections from mice in panel a was stained with PD-L2 monoclonal antibody (mAb) and Cy3-conjugated anti-rat IgG (red), and FITC-CD11c mAb (green). The overlay results (orange) indicate that bronchial CD11c+ cells express PD-L2. (c) DCs purified from lungs of WT BALB/c mice were cultured in vitro with media, interleukin (IL)-4, interferon-γ (IFN-γ), or IFN-γ and lipopolysaccharide (LPS) for 24 h. The expression of PD-L1 and PD-L2 were analyzed on the CD11c+, I-Adhi population as indicated. The filled histogram represents the isotype control and is representative of three experiments.
Expression of PD-L1 and PD-L2 on peritoneal macrophages has been shown to be differentially regulated by exposure to Th1 or Th2 cytokine environments.25 As AHR is dependent on the presence of IL-4 and IL-13 in the lung microenvironment, we determined how the expression of PD-L1 and PD-L2 on lung DCs was modulated by the cytokine environment. DCs purified from lungs of naive mice were cultured with IL-4, IFN-γ, or IFN-γ plus lipopolysaccharide (LPS) in vitro. Freshly purified DCs from naive lung expressed PD-L1 in moderate abundance and low levels of PD-L2 (Figure 2c). Treatment with IL-4 upregulated the expression of PD-L2 but did not enhance PD-L1 expression. Treatment with IFN-γ strongly enhanced PD-L1 expression and weakly upregulated PD-L2 expression, whereas treatment with IFN-γ plus LPS further enhanced PD-L1 expression but downregulated PD-L2 expression (Figure 2c). These data suggest that the cytokine environment may have an important role in differentially regulating PD-L1 and PD-L2 expression and modulating inflammatory responses in the lung microenvironment.
PD-L1 and PD-L2 regulate iNKT-cell-dependent development of AHR
We have previously shown that iNKT cells are required for the development of AHR, as iNKT-cell-deficient Ja18−/− mice do not develop AHR, and adoptive transfer of iNKT cells from WT mice restores the development of AHR in Jα18−/− mice. To directly examine the roles of PD-L1 and PD-L2 in iNKT-cell-driven AHR, we used an α-GalCer model of AHR, which highlights and isolates the role of iNKT cells.5,22 WT mice challenged with intranasal (IN) α-GalCer, a glycolipid antigen that specifically activates CD1d-restricted iNKT cells, developed severe AHR in 24 h. After challenge with α-GalCer, PD-L2−/− mice developed more severe AHR, whereas PD-L1−/− mice showed less severe AHR compared with WT BALB/c as measured by PenH after challenge with increasing doses of methacholine (Figure 3a). To determine whether these differences in AHR of PD-L1−/− and/or PD-L2−/− mice were reflected in differences in the numbers of iNKT cells in these strains compared with WT mice, we compared the number of iNKT cells in the thymus, liver, and spleen, the principal organs where NKT cells are located in PD-L1−/−,PD-L2−/−,PD-L1−/−/PD-L2−/−, and WT BALB/c mice. We found no significant difference in the number of iNKT cells or in the proportions of DN and CD4 + iNKT cells in any organs examined in these strains (Figure 3b).
Figure 3.

PD-L1 and PD-L2 regulate invariant natural killer T (iNKT)-cell-dependent development of airway hyperreactivity (AHR). (a) BALB/c, PD-L1−/−, and PD-L2−/− mice received a single dose of α-galactosylceramide (α-GalCer) (1 μg). AHR was assessed 24 h after IN challenge with α-GalCer or vehicle control. Data are the mean±s.e.m. Penh, representative of three experiments (n = 6) with * P<0.01 (PD-L2−/− α-GalCer vs. WT α-GalCer) and ** P<0.001 (PD-L1−/− α-GalCer vs. WT α-GalCer). (b) Absolute number of iNKT cells in the liver, spleen, and thymus of BALB/c, PD-L1−/−, and PD-L2−/− mice. Liver, spleen and thymus cell suspensions prepared as described in Methods were stained with α-GalCer-loaded phycoerythrin (PE)-conjugated CD1d tetramers, anti-TCR Vβ-APC, and anti-CD4-Alexa 750. iNKT cell numbers were analyzed by gating on CD4+ and double-negative (DN) iNKT cells. Data are representative of three separate experiments.
Expression of PD-1, PD-L1, and PD-L2 on naive and activated iNKT
To ascertain whether differences in surface expression of PD-1 on iNKT cells in PD-L1−/− or PD-L2−/− mice compared with WT might reflect the differences in AHR in these strains, we compared the expression of PD-1 on resting or activated iNKT cells from PD-L1−/−, PD-L2−/−, and WT mice. Splenocytes from naive mice were activated for 24 or 48 h with α-GalCer, stained with α-GalCer-loaded CD1d tetramer, and analyzed for cell surface PD-1 expression. Surface expression of PD-1 was upregulated on activated iNKT cells and by 48 h PD-1 was expressed at comparably high levels on iNKT cells from WT, PD-L1−/−, and PD-L2−/− mice (Figure 4a).
Figure 4.
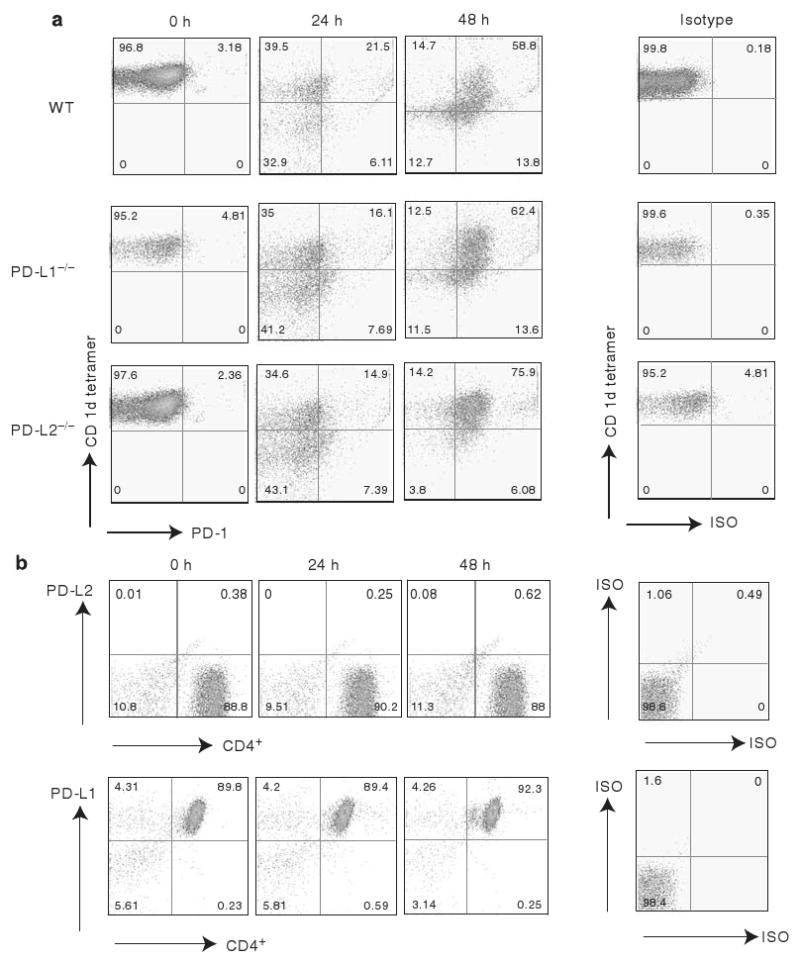
Surface expression of PD-1, PD-L1, and PD-L2 on naive and activated invariant natural killer T (iNKT) cells. (a) Positively selected iNKT cells from the spleen cells of naive BALB/c, PD-L1−/−, or PD-L2−/− mice were stained with α-galactosylceramide (α-GalCer)-loaded tetramer and Vβ-TCR monoclonal antibody (mAb) after 0, 24, and 48 h of activation in vitro and analyzed for the expression of PD-1. (b) iNKT cells from the spleen cells of naive BALB/c mice were stained with α-GalCer-loaded phycoerythrin (PE)-conjugated tetramer and mAbs against Vβ-TCR and CD4. Representative dot plots indicate the expression level of PD-L1 or PD-L2 on iNKT cell subsets.
We also examined iNKT cells from WT mice for surface expression of PD-L1 or PD-L2. Splenocytes from naive mice were stained with α-GalCer-loaded CD1d tetramer, and analyzed for cell surface expression of PD-L1 and PD-L2. We observed that the naive and activated iNKT cells do not express PD-L2, but PD-L1 is constitutively expressed at high levels on naive and activated iNKT cells (Figure 4b).
Cytokine profile of iNKT cells is regulated by PD-L1 and PD-L2 engagement
To determine the roles of PD-L1 and PD-L2 in regulating cytokine production by iNKT cells, we enriched iNKT cells from the spleens of BALB/c mice by negative selection and stimulated with α-GalCer in the presence of blocking mAbs. Culture of the iNKT cells with an anti-PD-L1 mAb, which blocks the interaction of PD-L1 with both PD-1 and B7-1 (9G2), resulted in greatly enhanced levels of IFN-γ production (Figure 5a), whereas culture in the presence of anti-PD-L1 mAb that blocks only the PD-L1-B7-1 interaction (2H11) had no effect on the production of IFN-γ or IL-4. Furthermore, culture of iNKT cells in the presence of PD-L2 blocking mAb significantly increased the level of IL-4 production but did not affect the production of IFN-γ. Blocking of PD-1 on iNKT cells with anti-PD-1 mAb 1A12 had no significant effect on cytokine production by iNKT cells.
Figure 5.

Cytokine production by invariant natural killer T (iNKT) cells is regulated by PD-L1 and PD-L2 co-inhibition. (a) Spleen cells from naive BALB/c mice were negatively enriched for iNKT cells and cultured with 103 α-galactosylceramide (α-GalCer)-loaded bone marrow-dendritic cells (BM-DCs) in the presence of blocking mAbs or control mAb. Supernatants were collected after 48 h and interleukin (IL)-4 and interferon-γ (IFN-γ) were measured by ELISA. Levels of cytokines in the presence of mAbs were compared with isotype controls with ** P<0.001. (b) Spleen cells from BALB/c, PD-L1−/−, and PD-L2−/− mice were stimulated with suboptimal amounts of α-GalCer (2.5 ng ml− 1). Supernatants were collected after 48 h and measured for cytokines by ELISA. Levels of cytokines produced by cells from PD-L1−/− and PD-L2−/− mice were compared with those of wild-type (WT) mice with ** P<0.001. (c) iNKT cells were negatively enriched from naive BALB/c splenocytes, and depleted of antigen-presenting cells as described in Methods and cultured with 103 α-GalCer-loaded BM-DCs from WT or PD-L2−/− mice. Supernatants were collected after 48 h and measured for IL-4 and IFN-γ cytokines by ELISA. Levels of cytokines from PD-L2−/− mice were compared with WT controls with ** P<0.001. ELISA data are representative of three separate experiments and are shown as mean±s.d. for triplicate samples.
To confirm the effects of PD-L1 and PD-L2 blocking antibodies on iNKT cells from WT mice, we examined cytokine production by iNKT cells from PD-L1−/− and PD-L2−/− mice. In the absence of PD-L1, iNKT cells produced higher levels of IFN-γ compared with iNKT cells from WT or PD-L2−/− mice. In contrast, iNKT cells from PD-L2−/− mice produced higher levels of IL-4 compared with WT or PD-L1−/− mice (Figure 5b). Thymic iNKT cells from PD-L2−/− mice also produced higher levels of IL-4 compared with WT mice (data not shown).
As PD-L2 is not expressed on iNKT cells (Figure 2) but rather is expressed on DCs and macrophages, we determined whether the profile of cytokine production by iNKT cells from PD-L2−/− mice would differ upon culture with DCs from WT or PD-L2−/− mice (Figure 5c). iNKT cells from either PD-L2−/− or WT mice produced significantly higher levels of IL-4 upon culture with DCs from PD-L2−/− mice than with DCs from WT mice. In contrast, higher levels of IFN-γ were produced when iNKT cells from either PD-L2−/− or WT mice were cultured with DCs from WT mice (Figure 5c). Together, these experiments confirm the importance of PD-L2 in inhibiting IL-4 production by iNKT cells.
Adoptive transfer of iNKT cells from PD-L1−/− or PD-L2−/− mice to Jα18−/− mice confers the donor AHR phenotype
We have previously shown that iNKT-cell-deficient Jα18−/− mice do not develop AHR and that adoptive transfer of iNKT cells from WT mice restores the development of AHR. In these studies, recipient iNKT-cell-deficient mice, which have conventional CD4+ T cells, are sensitized with OVA to prime Th2 responses before adoptive transfer of iNKT cells. To determine the roles of PD-L1 and PD-L2 in modulating iNKT cell responses during the induction of AHR, iNKT cells were positively selected from the spleen of WT, PD-L1−/−, or PD-L2−/− mice and adoptively transferred into OVA-sensitized Ja18−/− mice, which were then challenged with OVA to induce AHR. iNKT cells from WT BALB/c restored AHR, measured as RL and Cdyn in anesthetized, tracheostomized, and mechanically ventilated mice (Figure 6a), or as Penh (Figure 6b). In contrast, mice reconstituted with iNKT cells from PD-L1−/− mice failed to develop AHR or developed only mild AHR (Figure 6a and b). Mice reconstituted with iNKT cells from PD-L2−/− mice developed high levels of AHR (Figure 6a and b) similar to or greater than that observed with iNKT cells from WT BALB/c mice. Levels of AHR greater than those induced with WT iNKT cells are likely due to the partial activation of iNKT cells during the process of positive selection using α-GalCer-loaded CD1d tetramer. These findings are consistent with the patterns of cytokine production by iNKT cells from PD-L1−/− and PD-L2−/− mice and together suggest an important role for PD-L1 and PD-L2 in regulating the development of AHR.
Figure 6.

Reconstitution of Jα18−/− with invariant natural killer T (iNKT) cells from PD-L1−/− and PD-L2−/− mice. (a, b) iNKT cells were positively selected from the spleen of BALB/c, PD-L1−/−, and PD-L2−/− mice and adoptively transferred (3×106 cells) into ovalbumin (OVA)-sensitized Jα18−/− mice, which were then challenged with OVA IN on 3 consecutive days and assessed for AHR by measuring lung resistance (RL) and dynamic compliance (Cdyn) (a) or PenH (b). Non-recipient Jα18 and BALB/c mice were sensitized and challenged with OVA as control. Data are the mean±s.e.m., representative of three experiments (n= 5) with ** P<0.001 (recipients of PD-L1−/− vs. WT iNKT cells and recipients of PD-L2−/− vs. WT iNKT cells).
Discussion
Our findings suggest a novel mechanism by which PD-L2 distinctly regulates airway inflammation and AHR, in which the engagement of PD-L2 by its receptor on iNKT cells downregulates iNKT cell function and cytokine production. The analysis of PD-L2−/− mice shows that the loss of PD-L2 expression results in increased AHR and increased lung inflammation, and indicates that PD-L2 expression in the lung protects against the initiation and progression of iNKT cell-dependent airway inflammation. Our findings suggest that the severity of asthma is greatly enhanced in PD-L2−/− mice because of higher production of IL-4 by iNKT cells as PD-L2 engagement inhibited IL-4 production by iNKT cells. In contrast, PD-L1−/− mice showed reduced levels of AHR, minimal inflammation, and mucous secretion compared with WT mice, and enhanced production of IFN-γ by iNKT cells.
Several studies show the importance of iNKT cells in the induction of AHR in mice and humans,2,3,8,26 but the role of the co-inhibitory molecules PD-L1 and PD-L2 in the activation and modulation of iNKT cell-mediated AHR is still not understood. In this study we used ligand-deficient strains as an approach for understanding the functions of this immunoregulatory pathway. To focus on the role of iNKT cells, we used an α-GalCer model of AHR, as conventional CD4+ T cells do not respond to α-GalCer, but it is a potent activator of iNKT cells. We observed that upon challenge with α-GalCer, PD-L2−/− mice developed severe AHR compared with WT mice, whereas PD-L1−/− mice showed a reduced level of AHR and minimal airway inflammation. These disparate phenotypes were also observed after sensitization and challenge of mice with OVA in a conventional model of antigen-induced AHR. Deficiency of PD-L1 or PD-L2 expression did not affect the development of iNKT cells, as iNKT cell numbers in the thymus, liver, and spleen of PD-L1−/−, PD-L2−/−, and PDL1−/−/PDL2−/− were similar to those of WT mice. This is in contrast to the loss of ICOS expression, which we have shown affects NKT cell homeostasis.22 Moreover, surface expression of PD-1 on resting or activated splenic iNKT cells from PD-L1−/− and PD-L2−/− was similar to that of WT mice. These results suggest that PD-L2 preferentially modulates the effector phase of the asthmatic response, which includes the activation of iNKT cells in the lungs, through interaction with a receptor on iNKT cells.
To further examine the role of iNKT cells in mediating the phenotype of the PD-L1−/− and PD-L2−/− mice, we performed adoptive transfer studies. We and others have shown that the development of AHR in iNKT-cell-deficient Jα18−/− mice is restored by the transfer of iNKT cells from WT mice.2,27 OVA-sensitized Jα18−/− mice reconstituted with splenic iNKT cells from PD-L2−/− -deficient mice developed high levels of AHR. In contrast, Jα18−/− mice reconstituted with iNKT from PD-L1−/− -deficient mice developed only low levels of AHR. These results are consistent with the results observed in our in vitro experiments with iNKT cells purified from PD-L1−/− and PD-L2−/− mice.
Although iNKT cells have an important role in the development of AHR, the development of asthma, which is characterized by airway inflammation and mucus production, also requires the presence of allergen-specific CD4 + Th2 cells that respond to allergen. Th2 cells produce IL-4, IL-5, and IL-13 and interact with mast cells, basophils, and eosinophils to mediate airway inflammation.28 In this study, we showed that iNKT cell function and the development of AHR is regulated by PD-L1 and PD-L2. However, PD-L1 and/or PD-L2 engagement also regulates the responses of antigen-specific CD4 + T cells that are involved in airway inflammation. For example, during the OVA sensitization phase, immune deviation away from Th2 responses may reduce AHR and airway inflammation, as observed in the PD-L1−/− mouse, because CD4 + T cells from an OVA-sensitized PD-L1−/− mouse produce much higher levels of IFN-γ and less IL-4 than do CD4 + T cells from sensitized WT mouse upon in vitro re-stimulation with OVA (data not shown).
Although PD-L1 is expressed broadly on various hematopoietic and non-hematopoietic cell types, PD-L2 exhibits markedly restricted expression, being inducibly expressed only on DCs, macrophages, B1 B cells, and mast cells.29–31 PD-L2 expression was previously shown to be elevated on the lung and draining lymph node DCs after antigen challenge, 32 and upregulated on splenic DCs after in vivo activation of iNKT cells by the administration of α-GalCer IP.24 In this study, we observed that upon OVA challenge, all lung DCs showed increased expression of PD-L2. We also found that expression of PD-L2 was upregulated on lung DCs from naive mice upon culture with IL-4, whereas treatment with IFN-γ plus LPS inhibited PD-L2 expression, suggesting that the local cytokine microenvironment in the lungs defines the expression pattern of PD-L2. As PD-L2 inhibits IL-4 production and iNKT-cell-mediated AHR, these results suggest that PD-L2 in the lung during the course of a Th2 cytokine-driven lung inflammatory response constitutes a feedback loop that is upregulated by IL-4 and then acts to reduce continued IL-4 production, thereby modulating the severity of asthma. Loss of PD-L2 in this PD-L2/IL-4 circuit results in increased IL-4 and AHR. In contrast, the enhanced PD-L1 expression observed on lung DCs, macrophages, and B cells after OVA challenge 32 (and this study) inhibits IFN-γ production, which could mitigate AHR. Loss of PD-L1 in this PD-L1/IFN-γ circuit results in increased IFN-γ and reduced AHR. Previous work in cutaneous Leishmania infection has also shown differential effects of PD-L1 and PD-L2 on cytokine production and Th differentiation.33
Several other studies probed the role of PD-L2 in regulating airway inflammation using PD-L2 mAbs. Treatment with the blocking anti-PD-L2 mAb Ty25 during the challenge (effector), but not the sensitization phase of the response, augmented AHR and BAL eosinophilia, 32 which is consistent with our findings that mice lacking PD-L2 manifested increased AHR, and blocking iNKT cell function in vitro with PD-L2 mAb increased IL-4 production. Other studies found that administration of sHIgM1, a human IgM autoantibody that recognizes human and murine PD-L2, prevented lung inflammation and AHR by activating mouse DCs that induced deviation from Th2 to Th1 cytokine production.34,35 However, the in vivo effects of the sHIgM12 Ab may differ from other PD-L2 mAbs and the PD-L2/PD-1 interaction because this antibody regulates DC function by simultaneously recruiting and activating CD40 and TREM-2 on DCs,36 indicating that pathways other than PD-L2 are also involved.
PD-L1 and PD-L2 are ligands for PD-1; however, additional receptors for these B7 family members have been proposed 37–39 and a second ligand for PD-L1 has been molecularly identified.40 PD-L2 was reported to co-stimulate CD4 + T-cell proliferation and cytokine production and this co-stimulation was independent of PD-1.37,41 PD-L1 interacts not only with PD-1 but also with a second receptor, B7-1, on activated T cells, and this interaction negatively regulates T-cell expansion.40 We found that the addition of a mAb that blocks the PD-L1 interaction with both PD-1 and B7-1 (9G2) resulted in greatly enhanced levels of IFN-γ production by splenic iNKT cells stimulated with α-GalCer, but did not affect IL-4 production. However, culture in the presence of PD-L1 mAb that blocks only the PD-L1/B7-1 interaction (2H11) had no effect on the production of IFN-γ or IL-4, suggesting that the PD-1/PD-L1 interaction is primarily responsible for inhibiting iNKT cell production of IFN-γ. In contrast, iNKT cells activated in the presence of PD-L2 blocking mAb produced higher amounts of IL-4. Our observation that when activated in vitro with α-GalCer, iNKT cells from PD-L2−/− mice produced more IL-4, whereas those from PD-L1−/− mice produced more IFN-γ than WT controls is consistent with these mAb blocking studies. Together, these findings strongly suggest that PD-L2 specifically inhibits IL-4 production by iNKT cells, perhaps by binding to an additional receptor on iNKT cells other than PD-1. Alternatively, PD-L2 has a three-fold higher affinity for PD-1 than does PD-L140,42 and consequently may engage PD-1 in a different manner.
Studies of OVA-induced airway inflammation showed that PD-1- and BTLA-deficient mice displayed only a slight enhancement of acute airway inflammation, but showed a persistent inflammation, indicating an important role for these receptors in terminating the response.1 We found that the development of AHR in PDL1−/−/PDL2−/− mice measured at the acute phase of airway inflammation was similar to that of WT mice, which is consistent with the reported studies of the PD-1−/− mice. PD-1−/− mice are not directly comparable with PDL1−/−/PDL2−/− mice, as PD1−/− mice still have the interaction of PD-L1 with B7-1.
In summary, we show that PD-L1 and PD-L2 have important but distinct roles in modulating and polarizing iNKT cell function in AHR and airway inflammation. Understanding the complex mechanisms that regulate the development and effector function of Th2 and iNKT cells is crucial for developing new strategies for asthma therapies. Our finding gives impetus to the development of therapeutic approaches that exploit the PD ligands for controlling airway hyperresponsiveness and asthma.
Methods
Mice
Female BALB/cByJ mice (6–8 weeks old) were purchased from Jackson Laboratories (Bar Harbor, Maine). Ja18−/− mice (backcrossed to BALB/c) were a gift from M. Taniguchi/T. Nakayama (Chiba University, Chiba, Japan) and S. Balk (Brigham and Women's Hospital, Boston, MA). PD-L1−/−, PD-L2−/−, and PD-L1/PD-L2−/− mice backcrossed to BALB/cByJ mice as previously described.43 Mice were maintained and used according to institutional and the National Institutes of Health guidelines in a pathogen-free facility. The Animal Care and Use Committee, Children's Hospital Boston approved all animal protocols.
Generation of PD-L2 antibody
Antibodies to PD-L2 were generated by immunization of PD-L2-deficient mice 33 three times with murine PD-L2-human IgG1 fusion protein.31 After a final boost with a combination of human and mouse PD-L2-hIgG1, spleen cells were fused with X63-Ag8.653 myeloma cells, cloned, and hybridoma supernatants screened by ELISA, and cell surface staining of mouse and human PD-L2 transfected 300.19 and CHO cells 31 and for lack of reactivity with vector alone transfected cells. Clone 3.2 (mouse IgG1) was chosen for further analysis based on its capacity to recognize mouse and human PD-L2 equally well and block the interaction of PD-L2 with PD-1 (data not shown). Requests for mAb should be sent to gordon_freeman@dfci.harvard.edu.
Flow cytometry analysis
Cells were pre-incubated with anti-Fc receptor mAb 2.4G2 as well as with normal rat serum, and washed before staining. iNKT cells were identified using various antibody combinations that included phycoerythrin (PE)-conjugated CD1d:PBS-57 (α-GalCer analog)-loaded tetramer (NIH, the NIAID MHC Tetramer Core Facility, Atlanta, GA), TCRβ-PECy5 or allophycocyanin (clone H57-597, eBioscience, San Diego, CA), and CD4 allophycocyanin or allophycocyanin-Alexa750 (clone RM4-5) (eBioscience). mAbs against PD-L1 (conjugated to PE, clone M1H5), PD-L2 (conjugated to PE, clone Ty25), and (conjugated to PE-Cy7, clone RMP1-30), and CD11c (FITC) were all purchased from eBioscience. The cells were obtained on the FACS Canto 8 color flow cytometer (BD Biosciences, San Jose, CA) and 10,000 events within the iNKT cell gate were collected. The data were analyzed using FlowJo 6.2 software (Tree Star, Ashland, OR).
Confocal microscopy
From each lung sample, 4-μm frozen sections were cut, placed in acetone for 5 min, and dried at room temperature for 1 h. To reduce nonspecific binding of antibodies, the slides were placed in a solution of 10% goat serum, 10% bovine serum albumin in PBS for 45 min. After two rounds of wash with 1 × PBS, slides were stained for 1 h with 5 μg ml−1 of anti-PD-L2 mAb Ty25 (eBioscience), followed by 1:400 dilution of 2 mg ml−1 stock of Cy3-conjugated goat anti-rat Ig (Jackson ImmunoResearch, West Grove, PA). After two rounds of wash with 1 × PBS, directly conjugated mAb FITC-CD11c or FITC F4/80 (eBioscience) was used at a concentration of 10 μg ml−1 for 1 h. Nuclei were stained with 4′,6-diamidino-2-phenylindole, a fluorescent stain that binds strongly to DNA. Slides were analyzed for confocal microscopy using Nikon (Melville, NY) Eclipse TE2000.
Purification of iNKT cells
For isolation of splenic iNKT cells for transfer studies, cells were labeled with PE-conjugated CD1d tetramer followed by anti-PE microbeads and then sorted by Auto MACS according to the manufacturer's instruction. Purity of iNKT cells was >80%. For detection of iNKT cells in the liver, liver lymphocytes were isolated with 35% Percoll as described previously44 For in vitro studies, the iNKT cells were negatively selected from splenocytes using a cocktail of biotinylated mAbs against B220, CD19, CD62L, and CD11c, followed by incubation with anti-biotin microbeads. The samples were subsequently enriched using magnetic cell sorting. The enriched iNKT cell splenocytes were confirmed to contain an average of >10% pure iNKT cell population.
In vitro culture of NKT cells
NKT cells were magnetically enriched from the spleen of BALB/c, PD-L1−/−, or PD-L2−/− mice as described above. Total splenocytes or negatively enriched iNKT cells were cultured in round-bottom 96-well plates with 103 bone marrow-derived DCs from WT mice, prepared as previously described with some modifications.45,46 As indicated, in some wells, iNKT cells were treated with 20 μg ml−1 of anti-PD-L1 (10F.9G2), anti-PD-L1 (10F.2H11),40 anti-PD-L2 (mAb 3.2), or anti-PD-1 (mAb 29F.1A12)47 blocking mAbs. Where indicated, 2 × 105 negatively enriched iNKT cells were depleted of APCs and cultured with 103 α-GalCer-loaded BM-DCs from WT or PD-L2−/− mice. Supernatants were collected after 48 h and measured for cytokines by ELISA.
DC treatment in vitro
Naive BALB/c lungs were digested with collagenase and DNase I as described.5,22 CD11c+ DCs were positively selected from lungs and treated with IL-4 (20 ng ml−1) or LPS + IFN-γ (100 ng ml −1 each) for 24 h. Recombinant mouse IL-4 and IFN-γ were purchased from Peprotech (RockyHill, NJ). LPS was purchased from Sigma (St Louis, MO). Treated DCs were analyzed for the expression of PD-L1 and PD-L2 by flow cytometry.
Cytokine ELISAs
Cytokine secretion after in vitro challenge was determined by ELISA as previously described.2 The mAb pairs used were as follows (capture–detection): IFN-γ, R4-6A2–XMG1.2; IL-4, BVD4–BVD6.
Adoptive transfers
Purified iNKT cells (3 × 106) were adoptively transferred into Jα18−/− mice that were sensitized to OVA (50 μg OVA in alum IP, 1 week previously) and recipients were challenged with three consecutive doses of OVA IN (50 μg).
Induction of AHR and measurement of airway responsiveness
To induce AHR, mice were sensitized with 100 μg of LPS-free OVA antigen (Worthington, Lakewood, NJ) in alum administered IP. After 8 days, mice were exposed to OVA (50 μg, IN) or PBS for 3 consecutive days. AHR responses were assessed by methacholine-induced airflow obstruction in conscious mice placed in a whole-body plethysmograph (Buxco Electronics, Troy, NY) as described before.2,48 In some experiments, we assessed AHR by invasive measurement of airway resistance, in which anesthetized and tracheostomized mice were mechanically ventilated using a modified version of a described method.49 Aerosolized methacholine was administered in increasing concentrations of methacholine and we continuously computed RL and Cdyn by fitting flow, volume, and pressure to an equation of motion. For acute induction of AHR, α-GalCer (Axxora LLC, San Diego, CA) or vehicle control was administered IN (0.75 μg) to mice anesthetized with ketamine and xylazine or isofluorane. AHR was measured at 22–24 h after terminal IN challenge.
BAL fluid
After the measurement of AHR and killing, the lungs were lavaged twice with 1 ml of PBS plus 2% fetal calf serum and the fluid pooled as described previously.2 The relative number of different types of leukocytes was determined from slide preparations of BAL fluid stained with hematoxylin and eosin.
Supplementary Material
Acknowledgments
This study was supported by NIH Grants AI066020 (O.A.), AI56299 (G.J.F., A.H.S., D.T.U., R.D.K.), NIH AI56299, HHSN266200500030C, and the Foundation for the NIH through the Grand Challenges in Global Health initiative (G.J.F. and A.H.S.), NIH/NIAI/DAIT BAA-05-11 (G.J.F.) HL062348 (D.T.U.) and German Research Foundation (DFG) Grant ST0467/4-2 (P.S.).
Footnotes
Disclosure GJF has patents and receives patent royalties on the PD-1 pathway. Other authors have no conflict of interest.
Supplementary Material is linked to the online version of the paper at http://www.nature.com/mi
References
- 1.Deppong C, et al. Cutting edge: B and T lymphocyte attenuator and programmed death receptor-1 inhibitory receptors are required for termination of acute allergic airway infl ammation. J Immunol. 2006;176:3909–3913. doi: 10.4049/jimmunol.176.7.3909. [DOI] [PubMed] [Google Scholar]
- 2.Akbari O, et al. Essential role of NKT cells producing IL-4 and IL-13 in the development of allergen-induced airway hyperreactivity. Nat Med. 2003;9:582–588. doi: 10.1038/nm851. [DOI] [PubMed] [Google Scholar]
- 3.Lisbonne M, et al. Cutting edge: invariant V alpha 14 NKT cells are required for allergen-induced airway inflammation and hyperreactivity in an experimental asthma model. J Immunol. 2003;171:1637–1641. doi: 10.4049/jimmunol.171.4.1637. [DOI] [PubMed] [Google Scholar]
- 4.Jin N, et al. Airway hyperresponsiveness through synergy of {gamma}{delta} T cells and NKT cells. J Immunol. 2007;179:2961–2968. doi: 10.4049/jimmunol.179.5.2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Meyer EH, et al. Glycolipid activation of invariant T cell receptor + NK T cells is sufficient to induce airway hyperreactivity independent of conventional CD4+ T cells. Proc Natl Acad Sci USA. 2006;103:2782–2787. doi: 10.1073/pnas.0510282103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim JO, et al. Asthma is induced by intranasal coadministration of allergen and natural killer T-cell ligand in a mouse model. J Allergy Clin Immunol. 2004;114:1332–1338. doi: 10.1016/j.jaci.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 7.Kim DH, et al. 4-1BB engagement costimulates NKT Cell activation and exacerbates NKT cell ligand-induced airway hyperresponsiveness and inflammation. J Immunol. 2008;180:2062–2068. doi: 10.4049/jimmunol.180.4.2062. [DOI] [PubMed] [Google Scholar]
- 8.Pichavant M, et al. Ozone exposure in a mouse model induces airway hyperreactivity that requires the presence of natural killer T cells and IL-17. J Exp Med. 2008;205:385–393. doi: 10.1084/jem.20071507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matangkasombut P, et al. Direct activation of natural killer T cells induces airway hyperreactivity in nonhuman primates. J Allergy Clin Immunol. 2008;121:1287–1289. doi: 10.1016/j.jaci.2008.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Djukanovic R, Gadola SD. Virus infection, asthma, and chronic obstructive pulmonary disease. N Engl J Med. 2008;359:2062–2064. doi: 10.1056/NEJMcibr0806978. [DOI] [PubMed] [Google Scholar]
- 11.Matangkasombut P, et al. Natural killer T cells in the lungs of patients with asthma. J Allergy Clin Immunol. 2009;123:1181–1185. doi: 10.1016/j.jaci.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meyer EH, DeKruyff RH, Umetsu DT. T cells and NKT cells in the pathogenesis of asthma. Annu Rev Med. 2008;59:281–292. doi: 10.1146/annurev.med.59.061506.154139. [DOI] [PubMed] [Google Scholar]
- 13.Matangkasombut P, Pichavant M, Dekruyff RH, Umetsu DT. Natural killer T cells and the regulation of asthma. Mucosal Immunol. 2009;2:383–92. doi: 10.1038/mi.2009.96. [DOI] [PubMed] [Google Scholar]
- 14.Taniguchi M, Harada M, Kojo S, Nakayama T, Wakao H. The regulatory role of Valpha14 NKT cells in innate and acquired immune response. Annu Rev Immunol. 2003;21:483–513. doi: 10.1146/annurev.immunol.21.120601.141057. [DOI] [PubMed] [Google Scholar]
- 15.Bendelac A, Rivera MN, Park SH, Roark JH. Mouse CD1-specific NK1 T cells: development, specificity, and function. Annu Rev Immunol. 1997;15:535–562. doi: 10.1146/annurev.immunol.15.1.535. [DOI] [PubMed] [Google Scholar]
- 16.Park SH, Bendelac A. CD1-restricted T-cell responses and microbial infection. Nature. 2000;406:788–792. doi: 10.1038/35021233. [DOI] [PubMed] [Google Scholar]
- 17.Bendelac A, Savage PB, Teyton L. The biology of NKT cells. Annu Rev Immunol. 2007;25:297–336. doi: 10.1146/annurev.immunol.25.022106.141711. [DOI] [PubMed] [Google Scholar]
- 18.Fujii S, Liu K, Smith C, Bonito AJ, Steinman RM. The linkage of innate to adaptive immunity via maturing dendritic cells in vivo requires CD40 ligation in addition to antigen presentation and CD80/86 costimulation. J Exp Med. 2004;199:1607–1618. doi: 10.1084/jem.20040317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fujii S, Shimizu K, Kronenberg M, Steinman RM. Prolonged IFN-gamma-producing NKT response induced with alpha-galactosylceramide-loaded DCs. Nat Immunol. 2002;3:867–874. doi: 10.1038/ni827. [DOI] [PubMed] [Google Scholar]
- 20.Hayakawa Y, et al. Differential regulation of Th1 and Th2 functions of NKT cells by CD28 and CD40 costimulatory pathways. J Immunol. 2001;166:6012–6018. doi: 10.4049/jimmunol.166.10.6012. [DOI] [PubMed] [Google Scholar]
- 21.Uldrich AP, et al. NKT cell stimulation with glycolipid antigen in vivo : costimulation-dependent expansion, Bim-dependent contraction, and hyporesponsiveness to further antigenic challenge. J Immunol. 2005;175:3092–3101. doi: 10.4049/jimmunol.175.5.3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Akbari O, et al. ICOS/ICOSL interaction is required for CD4+ invariant NKT cell function and homeostatic survival. J Immunol. 2008;180:5448–5456. doi: 10.4049/jimmunol.180.8.5448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chang WS, et al. Cutting edge: programmed death-1/programmed death ligand 1 interaction regulates the induction and maintenance of invariant NKT cell anergy. J Immunol. 2008;181:6707–6710. doi: 10.4049/jimmunol.181.10.6707. [DOI] [PubMed] [Google Scholar]
- 24.Parekh VV, et al. PD-1/PD-L blockade prevents anergy induction and enhances the anti-tumor activities of glycolipid-activated invariant NKT cells. J Immunol. 2009;182:2816–2826. doi: 10.4049/jimmunol.0803648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Loke P, Allison J. PD-L1 and PD-L2 are differentially regulated by Th1 and Th2 cells. Proc Natl Acad Sci USA. 2003;100:5336–5341. doi: 10.1073/pnas.0931259100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim EY, et al. Persistent activation of an innate immune response translates respiratory viral infection into chronic lung disease. Nat Med. 2008;14:633–640. doi: 10.1038/nm1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Terashima A, et al. A novel subset of mouse NKT cells bearing the IL-17 receptor B responds to IL-25 and contributes to airway hyperreactivity. J Exp Med. 2008;205:2727–2733. doi: 10.1084/jem.20080698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wills-Karp M. Immunologic basis of antigen-induced airway hyperresponsiveness. Annu Rev Immunol. 1999;17:255–281. doi: 10.1146/annurev.immunol.17.1.255. [DOI] [PubMed] [Google Scholar]
- 29.Dong H, Zhu G, Tamada K, Chen L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med. 1999;5:1365–1369. doi: 10.1038/70932. [DOI] [PubMed] [Google Scholar]
- 30.Freeman GJ, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192:1027–1034. doi: 10.1084/jem.192.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Latchman Y, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol. 2001;2:261–268. doi: 10.1038/85330. [DOI] [PubMed] [Google Scholar]
- 32.Matsumoto K, et al. B7-DC regulates asthmatic response by an IFN-gamma-dependent mechanism. J Immunol. 2004;172:2530–2541. doi: 10.4049/jimmunol.172.4.2530. [DOI] [PubMed] [Google Scholar]
- 33.Liang SC, et al. PD-L1 and PD-L2 have distinct roles in regulating host immunity to cutaneous leishmaniasis. Eur J Immunol. 2006;36:58–64. doi: 10.1002/eji.200535458. [DOI] [PubMed] [Google Scholar]
- 34.Radhakrishnan S, Iijima K, Kobayashi T, Kita H, Pease LR. Dendritic cells activated by cross-linking B7-DC (PD-L2) block inflammatory airway disease. J Allergy Clin Immunol. 2005;116:668–674. doi: 10.1016/j.jaci.2005.04.038. [DOI] [PubMed] [Google Scholar]
- 35.Radhakrishnan S, et al. Blockade of allergic airway inflammation following systemic treatment with a B7-dendritic cell (PD-L2) cross-linking human antibody. J Immunol. 2004;173:1360–1365. doi: 10.4049/jimmunol.173.2.1360. [DOI] [PubMed] [Google Scholar]
- 36.Radhakrishnan S, et al. Indirect recruitment of a CD40 signaling pathway in dendritic cells by B7-DC cross-linking antibody modulates T cell functions. PLoS ONE. 2009;4:e5373. doi: 10.1371/journal.pone.0005373. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 37.Shin T, et al. Cooperative B7-1/2 (CD80/CD86) and B7-DC costimulation of CD4+ T cells independent of the PD-1 receptor. J Exp Med. 2003;198:31–38. doi: 10.1084/jem.20030242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu X, et al. B7DC/PDL2 promotes tumor immunity by a PD-1-independent mechanism. J Exp Med. 2003;197:1721–1730. doi: 10.1084/jem.20022089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dong H, et al. B7-H1 determines accumulation and deletion of intrahepatic CD8(+) T lymphocytes. Immunity. 2004;20:327–336. doi: 10.1016/s1074-7613(04)00050-0. [DOI] [PubMed] [Google Scholar]
- 40.Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity. 2007;27:111–122. doi: 10.1016/j.immuni.2007.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tseng SY, et al. B7-DC, a new dendritic cell molecule with potent costimulatory properties for T cells. J Exp Med. 2001;193:839–846. doi: 10.1084/jem.193.7.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Youngnak P, et al. Differential binding properties of B7-H1 and B7-DC to programmed death-1. Biochem Biophys Res Commun. 2003;307:672–677. doi: 10.1016/s0006-291x(03)01257-9. [DOI] [PubMed] [Google Scholar]
- 43.Keir ME, et al. Tissue expression of PD-L1 mediates peripheral T cell tolerance. J Exp Med. 2006;203:883–895. doi: 10.1084/jem.20051776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tupin E, Kronenberg M. Activation of natural killer T cells by glycolipids. Methods Enzymol. 2006;417:185–201. doi: 10.1016/S0076-6879(06)17014-7. [DOI] [PubMed] [Google Scholar]
- 45.Stockinger B, Hausmann B. Functional recognition of in vivo processed self antigen. Int Immunol. 1994;6:247–254. doi: 10.1093/intimm/6.2.247. [DOI] [PubMed] [Google Scholar]
- 46.Akbari O, DeKruyff RH, Umetsu DT. Pulmonary dendritic cells secreting IL-10 mediate T cell tolerance induced by respiratory exposure to antigen. Nat Immunol. 2001;2:725–731. doi: 10.1038/90667. [DOI] [PubMed] [Google Scholar]
- 47.Keir ME, Latchman YE, Freeman GJ, Sharpe AH. Programmed death-1 (PD-1):PD-ligand 1 interactions inhibit TCR-mediated positive selection of thymocytes. J Immunol. 2005;175:7372–7379. doi: 10.4049/jimmunol.175.11.7372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hansen G, Berry G, DeKruyff RH, Umetsu DT. Allergen-specific Th1 cells fail to counterbalance Th2 cell-induced airway hyperreactivity but cause severe airway inflammation. J Clin Invest. 1999;103:175–183. doi: 10.1172/JCI5155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Martin TR, Gerard NP, Galli SJ, Drazen JM. Pulmonary responses to bronchoconstrictor agonists in the mouse. J Appl Physiol. 1988;64:2318–2323. doi: 10.1152/jappl.1988.64.6.2318. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.