

Abstract
We undertook a two-stage genome-wide association study of Alzheimer's disease involving over 16,000 individuals. In stage 1 (3,941 cases and 7,848 controls), we replicated the established association with the APOE locus (most significant SNP: rs2075650, p= 1.8×10−157) and observed genome-wide significant association with SNPs at two novel loci: rs11136000 in the CLU or APOJ gene (p= 1.4×10−9) and rs3851179, a SNP 5′ to the PICALM gene (p= 1.9×10−8). Both novel associations were supported in stage 2 (2,023 cases and 2,340 controls), producing compelling evidence for association with AD in the combined dataset (rs11136000: p= 8.5×10−10, odds ratio= 0.86; rs3851179: p= 1.3×10−9, odds ratio= 0.86). We also observed more variants associated at p< 1×10−5 than expected by chance (p=7.5×10−6), including polymorphisms at the BIN1, DAB1 and CR1 loci.
Alzheimer's disease (AD) is the most common form of dementia, is highly heritable (heritability of up to 76%) but genetically complex1. Neuropathologically, the disease is characterized by extracellular senile plaques containing β-amyloid (Aβ) and intracellular neurofibrillary tangles containing hyperphosphorylated τ protein1. Four genes have been definitively implicated in its etiology. Mutations of the amyloid precursor protein (APP) gene and the presenilin 1 and 2 genes (PSEN1, PSEN2) cause rare, Mendelian forms of the disease usually with an early-onset. However, in the more common form of AD, only apolipoprotein E (APOE) has been established unequivocally as a susceptibility gene1. Aiming to identify novel AD loci, several genome-wide association studies (GWAS) have been conducted prior to the present study. All have identified strong evidence for association to APOE, but less convincing evidence implicating other genes2-9. This outcome is consistent with the majority of findings from GWAS of other common phenotypes, where susceptibility alleles typically have effect sizes with odds ratios (OR) of 1.5 or less, rather than of the magnitude for APOE (OR~3). Detecting such modest effects requires much larger samples than those that have been applied in the GWAS of AD to date10, which have all included fewer than 1,100 cases. Based upon the hypothesis that risk alleles for AD are likely to confer ORs in the range seen in other common diseases, we undertook a more powerful GWAS than has been undertaken to date.
We established a collaborative consortium from Europe and the USA from which we were able to draw upon a combined sample of up to 19,000 subjects (before quality control) and conducted a two-stage study. In Stage 1, 14,639 subjects were genotyped on Illumina platforms. 5,715 samples were genotyped for the present study using the Illumina 610-quadchip; genotypes for the remaining subjects were either made available to us from population control datasets or through collaboration and were genotyped on the Illumina HumanHap550 or the HumanHap300 BeadChips. Prior to association analysis, all samples and genotypes underwent stringent quality control, which resulted in the elimination of 53,383 autosomal SNPs and 2,850 subjects. Thus, in Stage 1, we tested 529,205 autosomal SNPs for association in up to 11,789 subjects (3,941 AD cases, 7,848 controls of which 2,078 were elderly screened controls, see Supplementary Table 1). The genomic control inflation factor (λ)11 was 1.037, suggesting little evidence for residual stratification.
In addition to the known association with the APOE locus, GWA analysis identified two novel loci at a genome-wide level of significance (see Fig. 1). Table 1 shows SNPs which were genome-wide significant (GWS) in stage 1; 13 GWS SNPS map within or close to the APOE locus on chromosome 19 (p< 3×10−8 − 2×10−157) and the top 5 are shown in Table 1 (see Supplementary Table 2 for the complete list). The other two SNPs represent novel associations. One of the novel SNPs (rs11136000) is located within an intron of clusterin (CLU, also known as APOJ) on chromosome 8 (p= 1.4×10−9, OR=0.840); the other SNP (rs3851179) is 88.5 kb 5′ to PICALM on chromosome 11 (p = 1.9×10−8, OR=0.849). Note that there was no significant difference in allele frequencies between elderly, screened controls and population controls for these two SNPs. In stage 2, the two novel GWS SNPs were genotyped in an independent sample comprising 2,023 AD cases and 2,340 age-matched, cognitively screened controls (Supplementary Table 3). Both were independently associated in this sample (rs11136000: one-tailed p= 0.017, OR= 0.905; rs3851179: one-tailed p= 0.014, OR= 0.897). Meta-analysis of the stage 1 and 2 datasets also produced highly significant evidence of association (rs11136000: p= 8.5×10−10, OR = 0.861 and rs3851179: p= 1.3×10−9, OR= 0.859, two-tailed, Table 1) for CLU and PICALM loci respectively. We sought further evidence from the Translational Genomics Research Institute (TGEN) study9 and the Li et al. study8, two publicly available AD GWAS datasets, but neither of the novel GWS SNPs had been genotyped or could be imputed. As secondary analyses, we tested each novel finding for interaction with APOE status and for association with age at onset. No significant interactions of the novel SNPs with APOE status were observed influencing AD risk (rs11136000xAPOE-ε4 interaction p= 0.674; rs3851179xAPOE-ε4 interaction p=0.735). Although we observed significant effects of the GWS SNPs on age at onset, these were limited to SNPs at the APOE locus (data not shown).
Figure 1.
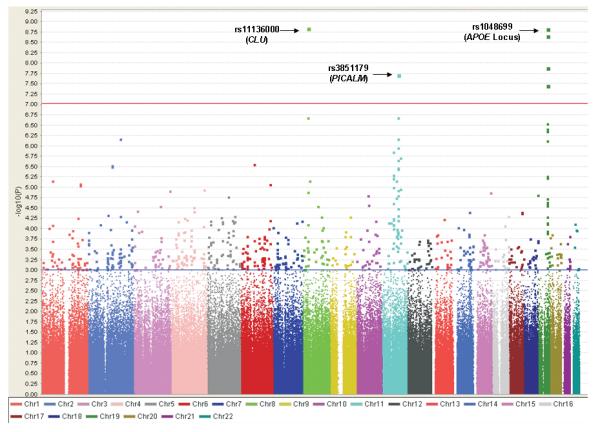
Scatterplot of chromosomal position (x-axis) against −log 10 GWAS P-value (y-axis). The y-axis scale has been limited to 9.25 (p = 5.6×10−10), although highly significant association was observed with SNPs in the vicinity of the APOE locus (e.g. rs2075650 with p = 1.8×10−157). The threshold for genome-wide significance (p ≤ 9.4×10−8) is indicated by the red horizontal line. 761 SNPs with p ≤ 1×10−3 lie above the blue horizontal line and are listed in Supplementary Table 2. The plot was produced using Haploview v4.048.
Table 1.
SNPs showing genome-wide significant association with AD in stage 1 of the GWAS. 5 of the13 genome-wide significant SNPs at the APOE locus are shown (see Supplementary Table 2 for the complete list). P-values in the extension sample and the combined sample are also shown for the two SNPs unlinked to the APOE locus (highlighted in bold).
| SNP | Chr | Closest RefSeq Gene |
Location Relative to Gene |
MAF | GWAS: 3941 cases 7848 controls |
GWAS OR (95% CI) |
Extension: 2023 cases 2340 controls |
Extension OR (95% CI) |
Combined: 5964 cases 10188 controls |
Combined OR (95% CI) |
Population Attributable Risk (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
|
|||||||||
| P-value (two-tailed) |
P-value (one-tailed) |
P-value (two-tailed) |
|||||||||
| rs2075650 * | 19 | TOMM40 | Intron | 0.15 | 1.8×10−157 | 2.53 (2.37-2.71) | 18.7% | ||||
| rs157580 | 19 | TOMM40 | Intron | 0.39 | 9.6×10−54 | 0.63 (0.59-0.66) | |||||
| rs6859 | 19 | PVRL2 | 3′ UTR | 0.43 | 6.9×10−41 | 1.46 (1.38-1.54) | |||||
| rs8106922 | 19 | TOMM40 | Intron | 0.40 | 5.4×10−39 | 0.68 (0.64-0.72) | |||||
| rs405509 | 19 | APOE | 5′ | 0.52 | 4.9×10−37 | 0.70 (0.66-0.74) | |||||
| rs11136000 | 8 | CLU | Intron | 0.40 | 1.4×10−9 | 0.84 (0.79-0.89) | 0.017 |
0.91 (0.83-0.99) |
8.5×10−10 |
0.86 (0.82-0.90) |
8.9% |
| rs3851179 | 11 | PICALM | 5′ | 0.37 | 1.9×10−8 | 0.85 (0.80-0.90) | 0.014 |
0.90 (0.82-0.99) |
1.3−10−9 |
0.86 (0.82-0.90) |
9.3% |
Chr = Chromosome; MB = position in megabases; MAF = minor allele frequency in controls; OR = odds ratio for the minor allele; 95% CI = 95% confidence interval; UTR= untranslated region.
rs2075650 is in linkage disequilibrium with rs429358, the APOE ε4 SNP (r2 = 0.48).
In a preliminary attempt to attribute the source of the association to a functional variant, we used publicly available data to identify additional SNPs at each locus that were correlated through linkage disequilibrium (LD) with either novel GWS SNP or that might plausibly have functional effects (see Supplementary Table 4). A synonymous SNP (rs7982) in the CLU gene was in strong LD (r2=0.95 in our extension sample) with the GWS SNP and showed a similar level of evidence for association with AD in the whole sample (meta-p =8×10−10; stage 1 genotypes were imputed). This SNP is in exon 5 of the CLU gene, which codes for part of the beta chain of the protein and may influence a predicted exon splicing enhancer. We note that Tycko and colleagues12 previously published a negative association study on the CLU gene, analyzing 4 SNPs in an AD case-control sample of African-American, Hispanic and Caucasian/non-Hispanic individuals. Although they identified rs7982 through mutation screening (referred to as VB in their study), the SNP was not tested for association in their sample. The 4 SNPs that were analyzed were rare in Caucasians (minor allele frequency <2%) and there was very limited power to detect association in their Caucasian sub-sample of 53 AD cases and 43 controls. As these 4 SNPs were not genotyped in the present GWAS, there is no overlap between the two studies.
Several potentially functional SNPs were identified at the PICALM locus. Of these, two showed good evidence for association; rs561655, which is within a putative transcription factor binding site and rs592297, which is a synonymous SNP in exon 5 of the gene that may influence a predicted exon splicing enhancer. However, neither of these SNPs showed the strength of evidence for association observed for rs3851179, the GWS SNP at the PICALM locus (i.e. rs561655: meta-p= 1×10−7 and rs592297: meta-p= 2×10−7). A number of SNPs in LD with rs3851179 and showing moderate evidence of association in the GWAS (p<1×10−4) were also followed up, most notably rs541458. This SNP is 8kb 5′ to the PICALM gene and was directly genotyped in the extension sample, the TGEN study and the Li et al. study, with p<0.05 in each. Following meta-analysis, this is one of our most significant SNPs (meta-p= 8×10−10) and is also supported by the study of Amouyel et al. published in this issue (p=3×10−3). Further genetic analyses will be required to characterize the true nature of the associations observed at these loci, which is beyond the scope of this paper.
We also tested whether the observed number of significant associations observed in the GWAS exceeded what would be expected by chance. Having removed SNPs within the APOE, CLU and PICALM loci (see Methods) we focused on those which showed most evidence for association (p< 1×10−5). Approximately 13 independent signals were observed; less than 4 would be expected by chance (p= 7.5×10−6). Table 2 shows the loci implicated and provides strong evidence for association with the complement receptor 1 (CR1) gene, particularly in the context of the results of the Amouyel et al. study published in this issue. Also noteworthy are the bridging integrator 1 (BIN1) gene, which produces a protein involved in synaptic vesicle endoctyosis13, and the disabled homolog 1 (DAB1) gene, whose product is involved with tyrosine phosphorylation and microtubule function in neurons14. These data thus provide strong evidence that there are several genes associated with AD which remain to be identified. We have also tested over 100 variants highlighted by previous AD GWA studies for association in our sample (see Supplementary note for full discussion). These are summarized in Supplementary Table 5.
Table 2.
SNPs showing association with AD at P≤ 1×10−5 (excluding SNPs at the APOE, CLU and PICALM loci).
| SNP | Chr | MB | Closest RefSeq Gene |
Location Relative to Gene |
GWAS P-value |
OR | 95% CI |
|---|---|---|---|---|---|---|---|
| rs11894266 | 2 | 170.3 | SSB | 5′ | 6.9×10−7 | 0.86 | 0.81-0.91 |
| rs610932 | 11 | 59.7 | MS4A6A | 3′UTR | 1.4×10−6 | 0.87 | 0.82-0.92 |
| rs10501927 | 11 | 99.3 | CNTN5 | Intronic | 2.0×10−6 | 1.18 | 1.10-1.26 |
| rs9446432 | 6 | 72.4 | Intergenic | 2.8×10−6 | 1.28 | 1.15-1.41 | |
| rs7561528 | 2 | 127.6 | BIN1 | 5′ | 3.0×10−6 | 1.16 | 1.09-1.24 |
| rs744373 | 2 | 127.6 | BIN1 | 5′ | 3.2×10−6 | 1.17 | 1.09-1.25 |
| rs662196 | 11 | 59.7 | MS4A6A | Intronic | 5.2×10−6 | 0.88 | 0.83-0.93 |
| rs583791 | 11 | 59.7 | MS4A6A | Intronic | 5.3×10−6 | 0.88 | 0.83-0.93 |
| rs676309 | 11 | 59.8 | MS4A4E | 5′ | 6.3×10−6 | 1.14 | 1.08-1.20 |
| rs1157242 | 8 | 37.2 | Intergenic | 7.0×10−6 | 1.17 | 1.10-1.26 | |
| rs1539053 | 1 | 57.9 | DAB1 | Intronic | 7.1×10−6 | 0.88 | 0.83-0.93 |
| rs11827375 | 11 | 76.0 | C11orf30 | 3′ | 7.2×10−6 | 1.23 | 1.12-1.35 |
| rs1408077 | 1 | 205.9 | CR1 | Intronic | 8.3×10−6 | 1.17 | 1.09-1.25 |
| rs9384428 | 6 | 156.5 | Intergenic | 8.5×10−6 | 1.14 | 1.08-1.21 | |
| rs6701713 | 1 | 205.9 | CR1 | Intronic | 8.7×10−6 | 1.17 | 1.09-1.25 |
| rs3818361 | 1 | 205.9 | CR1 | Intronic | 9.2×10−6 | 1.17 | 1.09-1.25 |
Chr = Chromosome; MB = position in megabases; OR = odds ratio for the minor allele; 95% CI = 95% confidence interval; UTR= untranslated region.
Until now, the APOE-ε4 allele was the only consistently replicated genetic risk factor for AD. It is therefore intriguing that we find compelling evidence for association with CLU, a gene that encodes another major brain apolipoprotein15, suggesting that susceptibility genes are not randomly distributed through functional pathways. The predominant form of clusterin is a secreted heterodimeric glycoprotein of 75-80kDa. The single copy gene spans about 16kb on chromosome 8p21-p12 and encodes an mRNA of approximately 2kb that translates into a 449 amino acid primary polypeptide chain. Clusterin is expressed in all mammalian tissues16 and there is strong evidence that CLU levels are elevated in a number of pathological conditions involving injury or chronic inflammation of the brain17. In Alzheimer's disease brain, CLU expression is reported to be increased in affected cortical areas and is present in amyloid plaques and in the cerebrospinal fluid of AD cases17-20.
Clusterin is a multi-functional molecule. It interacts with the soluble form of Aβ in animal models of disease and binds soluble Aβ in a specific and reversible manner, forming complexes that have been shown to cross the blood-brain barrier21-24. Interestingly, APOE also appears to act as a molecular chaperone for Aβ and influences when it aggregates and deposits25 as well as influencing Aβ conformation and toxicity26,27. In a similar way, clusterin appears to regulate both the toxicity and conversion of Aβ into insoluble forms28-32. Furthermore, APOE and CLU have been shown to cooperate in suppressing Aβ deposition33 and APOE and CLU may critically modify Aβ clearance at the blood brain barrier, suggesting a role for clusterin in the amyloidogenic pathway34. Levels of APOE protein appear to be inversely proportional to APOE-ε4 allele dose levels, i.e. expression levels are reduced in ε4 homozygotes compared with heterozygotes. Conversely, CLU levels are increased in proportion to APOE-ε4 allele dose suggesting an induction of clusterin in individuals with low APOE levels35. Thus, the strong statistical evidence for the involvement of this gene in AD has additional support in terms of biological plausibility.
The second gene locus to show compelling evidence for association with AD is PICALM (phosphatidylinositol-binding clathrin assembly protein; also known as CALM: clathrin assembly lymphoid-myeloid leukemia gene). PICALM is ubiquitously expressed in all tissue types with prominent expression in neurons, where it is nonselectively distributed at the pre- and post synaptic structures. It has been shown that like BIN1, PICALM is involved in clathrin-mediated endocytosis (CME), an essential step in the intracellular trafficking of proteins and lipids such as nutrients, growth factors and neurotransmitters36-38. Of relevance to AD, PICALM appears to be involved in directing the trafficking of VAMP2. VAMP2 is a SNARE protein that plays a prominent role in the fusion of synaptic vesicles to the presynaptic membrane in neurotransmitter release, a process that is crucial to neuronal function39. AD brains show a reduced number of synapses and stereological and biochemical analysis has shown that this reduction in synaptic density correlates with cognitive defects better than the accumulation of plaques and tangles40. More recent analysis indicates synapses within AD brains may be dysfunctional even before they visibly degenerate41. Therefore, we can hypothesize that genetically directed changes in PICALM function result in perturbations at the synapse, possibly through synaptic vesicle cycling, thereby increasing risk for AD. Alternatively, PICALM could influence risk of AD through APP processing via endocytic pathways resulting in changes in Aβ levels. Cell culture experiments have shown that full length APP is retrieved from the cell surface by CME42 and inhibition of endocytosis reduces APP internalization and reduces Aβ production and release43,44. Increased synaptic activity is known to lead to the elevated endocytosis of synaptic vesicle proteins and Cirrito et al. have since provided evidence in vivo that the increased CME, triggered by increased synaptic activity, drives more APP into endocytotic compartments resulting in an increase of Aβ production and release45. Thus, as for clusterin, the strong statistical evidence for the involvement of PICALM in AD has support in terms of biological plausibility.
The power of the present study to detect loci of the effect sizes observed in the GWAS for rs11136000 and rs3851179 at a genome-wide significance level is 0.74 and 0.57, respectively. However, it is widely acknowledged that effect sizes of significant loci obtained from genome-wide studies are over-estimates of the true effects46. In the extension study, rs11136000 had an OR of 0.905, while rs3851179 had an OR of 0.897. Assuming that these better reflect the true effect sizes associated with these loci, the GWAS has power to detect genome-wide significant association of 0.026 and 0.041 respectively. Thus it is likely that there are other genes of similar effect sizes that did not reach genome-wide significance. Indeed, we observed many SNPs which, while failing to reach this stringent level of statistical significance, might still reflect true associations with disease, e.g. variants at the CR1 locus, also highlighted by the results of Amouyel and colleagues (this issue). As with other GWAS research into complex disorders, such as Type 2 diabetes where 18 susceptibility loci have now been confirmed through GWAS meta-analysis of 54,000 subjects47, further larger GWAS are required to identify new susceptibility variants for AD.
Methods
Sample ascertainment and diagnostic criteria
The study comprised a Stage 1 discovery sample of 4,957 AD cases and 9,682 controls and a Stage 2 follow-up sample of 2,023 AD cases and 2,340 controls. See the Supplementary note for a complete description of the study subjects.
Stage 1 Genotyping
Genotyping was performed at the Sanger Institute, UK. 200ng of input DNA per sample were used and prepared for genotyping using the Illumina Infinium™ system (Illumina® Inc., San Diego, CA, USA). Manufacturer's protocols were followed throughout. The Illuminus algorithm for cluster analysis was used for genotype calling49.
Stage 1: Individual Quality Control
4,113 AD cases and 1,602 controls were genotyped on the Illumina 610-quad chip as part of this study (the 610 group). In addition, 844 AD cases and 8,080 controls previously genotyped using either the Illumina HumanHap550 or Illumina HumanHap300 were included in the analysis. These genotypes were generated as part of 7 different studies, making 8 separate groups in total: 1) 610; 2) Mayo; 3) 1958 birth cohort (Sanger); 4) 1958 birth cohort (T1DGC); 5) ALS control; 6) Coriell control; 7) Heinz Nixdorf Recall (HNR) study; 8) KORA F4. As we used genotype data from multiple sources, it was important to apply stringent QC filters, as differential genotyping error rates between groups could result in spurious associations when the data are combined50,51. These filters were applied separately to each of these 8 groups to remove poorly performing samples using tools implemented in PLINK v1.0552.
We removed 1,469 individuals with missing genotype rates > 0.01. We also applied a filter based on mean autosomal heterozygosity, excluding 578 individuals with values above or below empirically determined thresholds. 71 individuals with inconsistencies between reported gender and genotype-determined gender and 22 individuals with ambiguous genotype-determined gender were removed. All individuals passing these QC filters were examined for potential genetic relatedness by calculating identity by descent (IBD) estimates for all possible pairs of individuals in PLINK, and removing one of each pair with an IBD estimate ≥ 0.125 (the level expected for first cousins). IBD estimates were calculated using SNPs that were common to the Illumina 610, 550 and 300 chips with a genotype missing data rate ≤ 0.01, Hardy-Weinberg P ≥ 1×10−5 and a minor allele frequency ≥ 0.01. As a result, 506 individuals were excluded (note that this includes 311 individuals that were included in both the Coriell and ALS control group).
We also sought to detect non-European ancestry. To this end, genotype data from SNPs typed in all cohorts was merged with genotypes at the same SNPs from 210 unrelated European (CEU), Asian (CHB and JPT) and Yoruban (YRI) samples from the HapMap project. Subsequent to removing SNPs in extensive regions of linkage disequilibrium (chr5:44-51.5 Mb; chr6: 25-33.5 Mb; chr8: 8-12 Mb; chr11: 45-57 Mb)53, we further pruned SNPs if any pair within a 50-SNP window had r2 > 0.2. Genome-wide average identity by state (IBS) distance was calculated in PLINK between each pair of individuals in the resulting dataset, based on 57,966 SNPs (all with a genotype missing data rate ≤ 0.01, Hardy-Weinberg P ≥ 1×10−5 and a minor allele frequency ≥ 0.01). The resulting matrix of IBS distances was used as input for classical multi-dimensional scaling (MDS) in R v2.7.1. When the first two dimensions were extracted and plotted against each other, three clusters were observed corresponding to the European, Asian and Yoruban samples. Sixteen samples appeared to be ethnic outliers from the European cluster and were excluded from further analysis.
We assessed population structure within the data using principal components analysis (PCA) as implemented in EIGENSTRAT54 to infer continuous axes of genetic variation. Eigenvectors were calculated based on the previously described LD-pruned subset of 57,966 SNPs common to all arrays. The EIGENSTRAT program also identifies genetic outliers, which are defined as individuals whose ancestry is at least 6 standard deviations from the mean on one of the top ten axes of variation. As a result, 188 outliers were identified and excluded. Following sample QC 3,941 AD cases and 7,848 controls were included in the analysis.
Stage 1: SNP Quality Control
Due to unresolved genotype-calling issues with a proportion of SNPs on the sex chromosomes, only autosomal SNPs were included in this analysis (with the exception of rs5984894, an X chromosome SNP that has previously been associated with AD by Carrasquillo et al.5; see Supplementary Table 5). Individuals were genotyped on either the Illumina 610-quad as part of this project, or were previously genotyped on the Illumina HumanHap550 or the Illumina HumanHap300 array, and the genotypes made available to us. Note that SNPs had already been filtered out of some groups prior to inclusion in this study. Moreover, where different versions of the same array were used (e.g. HumanHap550v1 used to genotype the 1958 birth cohort (Sanger) cohort compared with the HumanHap550v3 array used to genotype the 1958 birth cohort (T1DGC)), only SNPs common to both versions were considered as present on that array. As such, SNPs included in our analysis fell into 4 different categories; 1) 266,714 SNPs common to all 3 arrays and genotyped in all individuals; 2) 202,516 SNPs common to the 610 and 550 arrays, but not present or without genotypes in individuals typed on the 300 array; 3) 7,744 SNPs common to the 610 and 300 arrays, but not present or without genotypes in individuals typed on the 550 array; 4) 105,614 SNPs with genotypes only in the 610 data.
We assessed the effects of different missing data rate and Hardy-Weinberg filters, aiming to remove poorly performing SNPs without excluding markers that may show genuine association with AD. For each of the 4 SNP categories, markers were excluded if they had a minor allele frequency (MAF) < 0.01 or a Hardy-Weinberg P ≤ 1×10−5, in either cases or controls. SNPs with a MAF ≥ 0.05 were excluded if they had a genotype missing rate of > 0.03 in either cases or controls; for SNPs with a MAF between 0.01 and 0.05, a more stringent genotype missing rate threshold of 0.01 was employed. As a result of this basic SNP QC 43,542 SNPs were excluded.
Ten principal components (PCs) were extracted using EIGENSTRAT, as previously described. To determine if the PCs could assuage any population structure within our sample, we performed logistic regression tests of association with AD, sequentially including between 0 and 10 of the top PCs as covariates. The impact of including the PCs was evaluated by calculating the genomic control inflation factor, λ11. We found that including the first 4 PCs as covariates had the maximum impact on λ.
To minimize inter-chip and inter-cohort differences that could result in an inflation of type I error rate, minor allele frequencies were compared between controls in the different groups using logistic regression analysis, incorporating the top 4 PCs as covariates as previously described. Comparisons were only performed between individuals from the same geographical region (i.e. British Isles, Germany or USA). For each of the 4 categories of SNPs, a quantile-quantile (Q-Q) plot was produced for each cohort control comparison, and the significance threshold employed to exclude SNPs was based on where the observed χ2 statistics departed from the null expectation. A further 9,828 SNPs were excluded as a result of these comparisons. Thus, a total of 529,218 autosomal SNPs were analyzed for association with AD in this study.
Stage 1 Statistical Analysis
SNPs were tested for association with AD using logistic regression, assuming an additive model. Covariates were included in the logistic regression analysis to allow for geographical region and chip, i.e. to distinguish between 1) individuals from the British Isles, 2) individuals from Germany, 3) individuals from the US typed on the 610 or 550 chip, 4) individuals from the US typed on the 300 chip. It was not possible to include a covariate for each chip as only controls were genotyped on the 550 chip. Similarly, it was not possible to include a covariate for each of the 8 groups, as only two included both cases and controls (610 and Mayo groups). The first 4 PCs extracted from EIGENSTRAT were also included as covariates, as previously described. Following analysis, 130 cluster plots were visually inspected for SNPs with a p-value ≤ 1×10−4. Thirteen SNPs showing poorly formed clusters were excluded. Thus our analysis was based on 529,205 SNPs, and a conservative genome-wide significance threshold of 0.05/529205 = 9.4×10−8 was employed. The overall genomic control inflation factor, λ, was calculated to be 1.037. Population attributable risk (the expected reduction in disease load following removal of a risk factor) was calculated for GWS SNPs according to the formula: PAR = Fcon(OR-1)/[Fcon(OR-1)+1], where Fcon is the frequency of the risk allele in controls and OR is the odds ratio associated with the risk allele55.
Expected number of significant SNPs
We assessed our results to determine if we observed more significant SNPs than would be expected by chance. We first removed SNPs within 500 kb either side of risk SNPs, i.e. rs429358 (the APOE ε4 SNP), rs11136000 (CLU) and rs3851179 (PICALM). We thus excluded 170 “APOE” SNPs, 290 “CLU” SNPs and 257 “PICALM” SNPs. Of the 528,448 remaining SNPs we estimated 397,224.7 “independent” tests using the algorithm we described in 56. Of 16 SNPs significant at a significance level α=10−5 (excluding APOE, CLU and PICALM SNPs, see Table 2) we estimated 12.6 “independent” tests. We calculated the mean (N*α =397224.7*10−5 ≈ 4.0) and variance (N*α*(1-α) = 3.97) of the expected number of significant tests at α=10−5 level using the binomial distribution. Thus the probability of observing 12.6 significant tests is P = 7.5×10−6.
Stage 2 Genotyping and Statistical Analysis
We genotyped SNPs in cases and controls from 5 European cohorts (described in Supplementary Table 3). Putative functional SNPs were identified using PupaSuite57. Genotyping was performed at Cardiff using the MassARRAY and iPlexGOLD systems (Sequenom, San Diego, CA) according to manufacturer's recommendations. All genotyped SNPs had genotype call frequency rates >90% in the follow-up sample, and no SNPs had HWE P-value ≤ 0.05 in cases or controls. SNPs were tested for association with AD using logistic regression, assuming an additive model. Covariates were included in the logistic regression analysis to allow for each cohort, i.e. 1) Belgium, 2) MRC, 3) ART, 4) Bonn, 5) Greek.
Meta-analysis
We included genotype data from stages 1 and 2 in a meta-analysis for SNPs at the CLU and PICALM loci. In addition, we employed genotype data from the TGEN study and the Li et al. study, two publicly available AD GWAS datasets. The TGEN sample is comprised of 861 AD cases and 550 controls genotyped on the Affymetrix 500K chip. The Li et al. discovery sample is comprised of 753 AD cases and 736 controls also genotyped on the Affymetrix 500K chip. If a SNP of interest was not genotyped in our GWAS or the TGEN dataset, an attempt was made to impute genotypes in PLINK, using the 60 HapMap CEU founders as a reference panel. Only imputed SNPs with an information content metric value greater than 0.8 were included in analysis (see PLINK website). Individual level data were not available for the Li study and so SNPs could not be imputed. Where individual level data was available, SNPs were tested for association with AD using logistic regression, assuming an additive model. Covariates were included in the logistic regression analysis to allow for geographical region and chip as in Stage 1 and for cohort as in Stage 2. Covariates included for the TGEN sample distinguished between samples from the Netherlands Brain Bank and samples from the USA. Meta-analyses incorporating data from the Li et al. study are based on Mantel-Haenzsel χ2 tests. Results of the meta-analysis are shown in Table 1 and Supplementary Table 4.
Secondary Analyses
We also tested the GWS SNPs for relationships with age at onset (AAO). To this end, age at onset (in years) was employed as the dependent variable in a linear regression analysis and an additive model was assumed. AAO data was available for 2,856 AD cases. Covariates were included in the logistic regression analysis to allow for geographical region and chip, i.e. to distinguish between 1) cases from the British Isles, 2) cases from Germany, 3) cases from the US typed on the 610 chip, 4) cases from the US typed on the 300 chip.
In addition, we stratified our sample based on presence/absence of at least 1 APOE ε4 allele. We had APOE genotype data for 6045 individuals; our ε4-positive sample consisted of 2,203 AD cases and 632 controls; our ε4-negative sample consisted of 1,446 cases and 1,764 controls. We performed genome-wide tests for association with AD in each sub-sample, but no SNP achieved genome-wide significance (see Supplementary Tables 6 and 7).
URLs
VIB Genetic Service Facility, http://www.vibgeneticservicefacility.be
TGEN, http://www.tgen.org
Haploview, http://www.broad.mit.edu/mpg/haploview
PLINK, http://pngu.mgh.harvard.edu/~purcell/plink
The GWAS data will be made available to bona fide researchers within 6 months (contact the corresponding authors for access).
Supplementary Material
Acknowledgements
We thank the patients and families who took part in this research. Cardiff University was supported by the Wellcome Trust, Medical Research Council (MRC), Alzheimer's Research Trust (ART) and the Welsh Assembly Government. ART supported sample collections at the Institute of Psychiatry, the South West Dementia Bank, Universities of Cambridge, Nottingham, Manchester and Belfast. The Belfast group acknowledges support from the Alzheimer's Society, Ulster Garden Villages, N.Ireland R&D Office and the Royal College of Physicians/Dunhill Medical Trust. The MRC and Mercer's Institute for Research on Ageing supported the Trinity College group. The South West Dementia Brain Bank acknowledges support from Bristol Research into Alzheimer's and Care of the Elderly. The Charles Wolfson Charitable Trust supported the OPTIMA group. Ammar Al-Chalabi and Christopher Shaw thank the MNDA and MRC for support. DCR is a Wellcome Trust Senior Clinical Research Fellow. Washington University was funded by NIH grants, Barnes Jewish Foundation and the Charles and Joanne Knight Alzheimer's Research Initiative. The Mayo GWAS was supported by NIH grants, the Robert and Clarice Smith and Abigail Van Buren AD Research Program and the Palumbo Professorship in AD Research. Patient recruitment for the MRC Prion Unit/UCL Department of Neurodegenerative Disease collection was supported by the UCLH/UCL Biomedical Centre. LASER-AD was funded by Lundbeck SA. The Bonn group was supported by the German Federal Ministry of Education and Research (BMBF), Competence Network Dementia and Competence Network Degenerative Dementia, and by the Alfried Krupp von Bohlen und Halbach-Stiftung. The KORA F4 studies were financed by Helmholtz Zentrum München; German Research Center for Environmental Health; BMBF; German National Genome Research Network and the Munich Center of Health Sciences. The Heinz Nixdorf Recall cohort was funded by the Heinz Nixdorf Foundation (Dr. jur. G.Schmidt, Chairman) and BMBF. Coriell Cell Repositories is supported by NINDS and the Intramural Research Program of the National Institute on Aging. We acknowledge use of DNA from the 1958 Birth Cohort collection, funded by the MRC and the Wellcome Trust which was genotyped by the Wellcome Trust Case Control Consortium and the Type-1 Diabetes Genetics Consortium, sponsored by the National Institute of Diabetes and Digestive and Kidney Diseases, National Institute of Allergy and Infectious Diseases, National Human Genome Research Institute, National Institute of Child Health and Human Development and Juvenile Diabetes Research Foundation International. The Antwerp site was supported by the VIB Genetic Service Facility, the Biobank of the Institute Born-Bunge, the Special Research Fund of the University of Antwerp; the Fund for Scientific Research-Flanders, the Foundation for Alzheimer Research, the Interuniversity Attraction Poles program P6/43 of the Belgian Federal Science Policy Office. K.S. is a postdoctoral fellow and K.B. a PhD fellow (Fund for Scientific Research-Flanders). We thank Robert Brown, John Landers, Donald Warden, Donald Lehmann, Nigel Leigh, James Uphill, Jon Beck, Tracy Campbell, Steffi Klier, Gary Adamson, Julia Wyatt, Marc Lucia Perez, T. Meitinger, P. Lichtner, G. Eckstein, Neill Graff-Radford, Ronald Petersen, Dennis Dickson, G. Fischer, Horst Bickel, Michael Hüll, Holger Jahn, Hanna Kaduszkiewicz, Christian Luckhaus, Steffi Riedel-Heller, Stefanie Wolf, Siegfried Weyerer, the Helmholtz Zentrum München genotyping staff, Eric Reiman, TGEN and the NIMH AD Genetics Initiative. We thank Advanced Research Computing @Cardiff (ARCCA) who facilitated data analysis.
Footnotes
Accession numbers
GenBank: CLU isoform 1 mRNA, NM_001831.2; CLU isoform 2 mRNA, NM_203339.1; PICALM isoform 1 mRNA, NM_007166.2; PICALM isoform 2 mRNA, NM_001008660.1.
References
- 1.Avramopoulos D. Genetics of Alzheimer's disease: recent advances. Genome Med. 2009;1:34. doi: 10.1186/gm34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abraham R, et al. A genome-wide association study for late-onset Alzheimer's disease using DNA pooling. BMC Med Genomics. 2008;1:44. doi: 10.1186/1755-8794-1-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beecham GW, et al. Genome-wide association study implicates a chromosome 12 risk locus for late-onset Alzheimer disease. Am J Hum Genet. 2009;84:35–43. doi: 10.1016/j.ajhg.2008.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bertram L, et al. Genome-wide association analysis reveals putative Alzheimer's disease susceptibility loci in addition to APOE. Am J Hum Genet. 2008;83:623–32. doi: 10.1016/j.ajhg.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carrasquillo MM, et al. Genetic variation in PCDH11X is associated with susceptibility to late-onset Alzheimer's disease. Nat Genet. 2009;41:192–8. doi: 10.1038/ng.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Coon KD, et al. A high-density whole-genome association study reveals that APOE is the major susceptibility gene for sporadic late-onset Alzheimer's disease. J Clin Psychiatry. 2007;68:613–8. doi: 10.4088/jcp.v68n0419. [DOI] [PubMed] [Google Scholar]
- 7.Grupe A, et al. Evidence for novel susceptibility genes for late-onset Alzheimer's disease from a genome-wide association study of putative functional variants. Hum Mol Genet. 2007;16:865–73. doi: 10.1093/hmg/ddm031. [DOI] [PubMed] [Google Scholar]
- 8.Li H, et al. Candidate single-nucleotide polymorphisms from a genomewide association study of Alzheimer disease. Arch Neurol. 2008;65:45–53. doi: 10.1001/archneurol.2007.3. [DOI] [PubMed] [Google Scholar]
- 9.Reiman EM, et al. GAB2 alleles modify Alzheimer's risk in APOE epsilon4 carriers. Neuron. 2007;54:713–20. doi: 10.1016/j.neuron.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.WTCCC Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–78. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Devlin B, Roeder K. Genomic control for association studies. Biometrics. 1999;55:997–1004. doi: 10.1111/j.0006-341x.1999.00997.x. [DOI] [PubMed] [Google Scholar]
- 12.Tycko B, et al. Polymorphisms in the human apolipoprotein-J/clusterin gene: ethnic variation and distribution in Alzheimer's disease. Hum Genet. 1996;98:430–6. doi: 10.1007/s004390050234. [DOI] [PubMed] [Google Scholar]
- 13.Cousin MA, Robinson PJ. The dephosphins: dephosphorylation by calcineurin triggers synaptic vesicle endocytosis. Trends Neurosci. 2001;24:659–65. doi: 10.1016/s0166-2236(00)01930-5. [DOI] [PubMed] [Google Scholar]
- 14.Hiesberger T, et al. Direct binding of Reelin to VLDL receptor and ApoE receptor 2 induces tyrosine phosphorylation of disabled-1 and modulates tau phosphorylation. Neuron. 1999;24:481–9. doi: 10.1016/s0896-6273(00)80861-2. [DOI] [PubMed] [Google Scholar]
- 15.Jenne DE, Tschopp J. Clusterin: the intriguing guises of a widely expressed glycoprotein. Trends Biochem Sci. 1992;17:154–9. doi: 10.1016/0968-0004(92)90325-4. [DOI] [PubMed] [Google Scholar]
- 16.Jones SE, Jomary C. Clusterin. Int J Biochem Cell Biol. 2002;34:427–31. doi: 10.1016/s1357-2725(01)00155-8. [DOI] [PubMed] [Google Scholar]
- 17.Calero M, et al. Apolipoprotein J (clusterin) and Alzheimer's disease. Microsc Res Tech. 2000;50:305–15. doi: 10.1002/1097-0029(20000815)50:4<305::AID-JEMT10>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 18.Giannakopoulos P, et al. Possible neuroprotective role of clusterin in Alzheimer's disease: a quantitative immunocytochemical study. Acta Neuropathol. 1998;95:387–94. doi: 10.1007/s004010050815. [DOI] [PubMed] [Google Scholar]
- 19.Liang WS, et al. Altered neuronal gene expression in brain regions differentially affected by Alzheimer's disease: a reference data set. Physiol Genomics. 2008;33:240–56. doi: 10.1152/physiolgenomics.00242.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McGeer PL, Kawamata T, Walker DG. Distribution of clusterin in Alzheimer brain tissue. Brain Res. 1992;579:337–41. doi: 10.1016/0006-8993(92)90071-g. [DOI] [PubMed] [Google Scholar]
- 21.Ghiso J, et al. The cerebrospinal-fluid soluble form of Alzheimer's amyloid beta is complexed to SP-40,40 (apolipoprotein J), an inhibitor of the complement membrane-attack complex. Biochem J. 1993;293(Pt 1):27–30. doi: 10.1042/bj2930027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Golabek A, Marques MA, Lalowski M, Wisniewski T. Amyloid beta binding proteins in vitro and in normal human cerebrospinal fluid. Neurosci Lett. 1995;191:79–82. doi: 10.1016/0304-3940(95)11565-7. [DOI] [PubMed] [Google Scholar]
- 23.Zlokovic BV, et al. Brain uptake of circulating apolipoproteins J and E complexed to Alzheimer's amyloid beta. Biochem Biophys Res Commun. 1994;205:1431–7. doi: 10.1006/bbrc.1994.2825. [DOI] [PubMed] [Google Scholar]
- 24.Zlokovic BV, et al. Glycoprotein 330/megalin: probable role in receptor-mediated transport of apolipoprotein J alone and in a complex with Alzheimer disease amyloid beta at the blood-brain and blood-cerebrospinal fluid barriers. Proc Natl Acad Sci U S A. 1996;93:4229–34. doi: 10.1073/pnas.93.9.4229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bales KR, et al. Apolipoprotein E is essential for amyloid deposition in the APP(V717F) transgenic mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 1999;96:15233–8. doi: 10.1073/pnas.96.26.15233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Castano EM, et al. Fibrillogenesis in Alzheimer's disease of amyloid beta peptides and apolipoprotein E. Biochem J. 1995;306(Pt 2):599–604. doi: 10.1042/bj3060599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ma J, Yee A, Brewer HB, Jr., Das S, Potter H. Amyloid-associated proteins alpha 1-antichymotrypsin and apolipoprotein E promote assembly of Alzheimer beta-protein into filaments. Nature. 1994;372:92–4. doi: 10.1038/372092a0. [DOI] [PubMed] [Google Scholar]
- 28.Boggs LN, et al. Clusterin (Apo J) protects against in vitro amyloid-beta (1-40) neurotoxicity. J Neurochem. 1996;67:1324–7. doi: 10.1046/j.1471-4159.1996.67031324.x. [DOI] [PubMed] [Google Scholar]
- 29.DeMattos RB, et al. Clusterin promotes amyloid plaque formation and is critical for neuritic toxicity in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2002;99:10843–8. doi: 10.1073/pnas.162228299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lambert MP, et al. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A. 1998;95:6448–53. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matsubara E, Soto C, Governale S, Frangione B, Ghiso J. Apolipoprotein J and Alzheimer's amyloid beta solubility. Biochem J. 1996;316(Pt 2):671–9. doi: 10.1042/bj3160671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oda T, et al. Clusterin (apoJ) alters the aggregation of amyloid beta-peptide (A beta 1-42) and forms slowly sedimenting A beta complexes that cause oxidative stress. Exp Neurol. 1995;136:22–31. doi: 10.1006/exnr.1995.1080. [DOI] [PubMed] [Google Scholar]
- 33.DeMattos RB, et al. ApoE and clusterin cooperatively suppress Abeta levels and deposition: evidence that ApoE regulates extracellular Abeta metabolism in vivo. Neuron. 2004;41:193–202. doi: 10.1016/s0896-6273(03)00850-x. [DOI] [PubMed] [Google Scholar]
- 34.Bell RD, et al. Transport pathways for clearance of human Alzheimer's amyloid beta-peptide and apolipoproteins E and J in the mouse central nervous system. J Cereb Blood Flow Metab. 2007;27:909–18. doi: 10.1038/sj.jcbfm.9600419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bertrand P, Poirier J, Oda T, Finch CE, Pasinetti GM. Association of apolipoprotein E genotype with brain levels of apolipoprotein E and apolipoprotein J (clusterin) in Alzheimer disease. Brain Res Mol Brain Res. 1995;33:174–8. doi: 10.1016/0169-328x(95)00097-c. [DOI] [PubMed] [Google Scholar]
- 36.Dreyling MH, et al. The t(10;11)(p13;q14) in the U937 cell line results in the fusion of the AF10 gene and CALM, encoding a new member of the AP-3 clathrin assembly protein family. Proc Natl Acad Sci U S A. 1996;93:4804–9. doi: 10.1073/pnas.93.10.4804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tebar F, Bohlander SK, Sorkin A. Clathrin assembly lymphoid myeloid leukemia (CALM) protein: localization in endocytic-coated pits, interactions with clathrin, and the impact of overexpression on clathrin-mediated traffic. Mol Biol Cell. 1999;10:2687–702. doi: 10.1091/mbc.10.8.2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yao PJ, Petralia RS, Bushlin I, Wang Y, Furukawa K. Synaptic distribution of the endocytic accessory proteins AP180 and CALM. J Comp Neurol. 2005;481:58–69. doi: 10.1002/cne.20362. [DOI] [PubMed] [Google Scholar]
- 39.Harel A, Wu F, Mattson MP, Morris CM, Yao PJ. Evidence for CALM in directing VAMP2 trafficking. Traffic. 2008;9:417–29. doi: 10.1111/j.1600-0854.2007.00694.x. [DOI] [PubMed] [Google Scholar]
- 40.Masliah E, et al. Altered expression of synaptic proteins occurs early during progression of Alzheimer's disease. Neurology. 2001;56:127–9. doi: 10.1212/wnl.56.1.127. [DOI] [PubMed] [Google Scholar]
- 41.Fitzjohn SM, et al. Age-related impairment of synaptic transmission but normal long-term potentiation in transgenic mice that overexpress the human APP695SWE mutant form of amyloid precursor protein. J Neurosci. 2001;21:4691–8. doi: 10.1523/JNEUROSCI.21-13-04691.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nordstedt C, Caporaso GL, Thyberg J, Gandy SE, Greengard P. Identification of the Alzheimer beta/A4 amyloid precursor protein in clathrin-coated vesicles purified from PC12 cells. J Biol Chem. 1993;268:608–12. [PubMed] [Google Scholar]
- 43.Carey RM, Balcz BA, Lopez-Coviella I, Slack BE. Inhibition of dynamin-dependent endocytosis increases shedding of the amyloid precursor protein ectodomain and reduces generation of amyloid beta protein. BMC Cell Biol. 2005;6:30. doi: 10.1186/1471-2121-6-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Koo EH, Squazzo SL. Evidence that production and release of amyloid beta-protein involves the endocytic pathway. J Biol Chem. 1994;269:17386–9. [PubMed] [Google Scholar]
- 45.Cirrito JR, et al. Endocytosis is required for synaptic activity-dependent release of amyloid-beta in vivo. Neuron. 2008;58:42–51. doi: 10.1016/j.neuron.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Goring HH, Terwilliger JD, Blangero J. Large upward bias in estimation of locus-specific effects from genomewide scans. Am J Hum Genet. 2001;69:1357–69. doi: 10.1086/324471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zeggini E, et al. Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nat Genet. 2008;40:638–45. doi: 10.1038/ng.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–5. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 49.Teo YY, et al. A genotype calling algorithm for the Illumina BeadArray platform. Bioinformatics. 2007;23:2741–6. doi: 10.1093/bioinformatics/btm443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Clayton DG, et al. Population structure, differential bias and genomic control in a large-scale, case-control association study. Nat Genet. 2005;37:1243–6. doi: 10.1038/ng1653. [DOI] [PubMed] [Google Scholar]
- 51.Moskvina V, Craddock N, Holmans P, Owen MJ, O'Donovan MC. Effects of differential genotyping error rate on the type I error probability of case-control studies. Hum Hered. 2006;61:55–64. doi: 10.1159/000092553. [DOI] [PubMed] [Google Scholar]
- 52.Purcell S, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Price AL, et al. Long-range LD can confound genome scans in admixed populations. Am J Hum Genet. 2008;83:132–5. doi: 10.1016/j.ajhg.2008.06.005. author reply 135-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Price AL, et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–9. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 55.Schlesselman JJ. Case-Control Studies: Design, Conduct, Analysis. University Press; Oxford: 1982. [Google Scholar]
- 56.Moskvina V, Schmidt KM. On multiple-testing correction in genome-wide association studies. Genet Epidemiol. 2008;32:567–73. doi: 10.1002/gepi.20331. [DOI] [PubMed] [Google Scholar]
- 57.Conde L, et al. PupaSuite: finding functional single nucleotide polymorphisms for large-scale genotyping purposes. Nucleic Acids Res. 2006;34:W621–5. doi: 10.1093/nar/gkl071. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.