

Abstract
Metacaspase (MCA) is an important enzyme in Trypanosoma brucei, absent from humans and differing significantly from the orthologous human caspases. Therefore MCA constitutes a new attractive drug target for antiparasitic chemotherapeutics, which needs further characterization to support the discovery of innovative drug candidates. A first series of inhibitors has been prepared on the basis of known substrate specificity and the predicted catalytic mechanism of the enzyme. In this Letter we present the first inhibitors of TbMCA2 with low micromolar enzymatic and antiparasitic activity in vitro combined with low cytotoxicity.
In 2000, metacaspases (E.C. 3.4.22) were identified as a new family of caspase-like proteins.1 Metacaspases, together with caspases and paracaspases, are endopeptidases that belong to family C14 in clan CD of the cysteine peptidases. They share the caspase fold2 and their active site comprises a conserved His-Cys catalytic dyad, the cysteine being the active site nucleophile. All metacaspases examined to date have a basic P1 specificity towards Arg/Lys, whereas caspases have an acidic P1 specificity towards Asp.2 This explains why metacaspases are not able to cleave known caspase substrates and are not inhibited by caspase inhibitors (zVAD, DEVD-CHO).3 Metacaspases are not present in mammals and have little overall sequence homology with human caspases, making them good potential drug targets. In Trypanosoma brucei, five metacaspases have been identified denoted TbMCA1-5. Interestingly, TbMCA1 and TbMCA4 genes encode a serine in place of a cysteine in the catalytic dyad, thus cannot function as cysteine peptidases, although they may still have activity through the use of the substituent serine as a nucleophile. Of the other three TbMCAs, TbMCA2 and 3 have demonstrated activity and TbMCA5 is expected to have similar activity.4 All three have been validated as drug targets by triple RNAi.5 Triple RNAi analysis (simultaneous down-regulation) showed TbMCA2, TbMCA3 and TbMCA5 to be essential in the bloodstream form (BSF), with parasites accumulating pre-cytokinesis, meaning cell division failed. However, this effect could not be confirmed with triple null mutants (Δmca2/3Δmca5). After an initial slow growth phase following sequential gene deletion, they grew well under both standard in vitro and in vivo conditions, suggesting compensatory activation of alternative pathways. However, mutant strains remained highly sensitive to changes to the standard conditions in vitro. Overall, it seems metacaspases fulfil an important role in BSF T. brucei, but the likelihood of overlapping functions means that therapeutic targeting would require inhibition of multiple TbMCAs and/or enzymes of the alternative pathways.
The functions of metacaspases in protozoa remain unclear. It has been proposed that in trypanosomatids metacaspases function as caspase-like enzymes and are involved in Programmed Cell Death (PCD).6,7,8 However, there are still doubts about the existence of PCD in trypanosomatids and other unicellular organisms9,10 and some authors oppose the PCD function of metacaspases in protozoa by pointing out their different substrate specificity, which makes a similar function to caspases unlikely.2 Helms et al.5 suggest that MCAs have a function associated with RAB11 vesicles that is independent of known recycling processes of RAB11-positive endosomes. Clearly, the precise role of MCAs remains to be elucidated. Nonetheless, the fact that T. brucei metacaspases seem to play a vital role in the parasite, and are absent in humans makes them attractive drug targets.11
In the absence of crystallographic data and full knowledge of the natural substrates, we describe the application of a rational design where the substrate specificity of TbMCA2 was used to design and synthesise the first specific MCA inhibitors. It is known that for peptides the S1 subsite selectivity of TbMCA2/3 is limited to Arg/Lys2 (Fig. 1A), leading to initial substrate-based inhibitor design with Arg/Lys in the P1 position. Previously, potent cysteine peptidase inhibitors have been obtained by replacing the scissile amide bond by an electrophile (P1′). Reversible competitive inhibitors were designed by using a nitrile12 or α-ketoheterocyclic warhead13. For the latter, the formation of a hydrogen bond between the sp2 nitrogen of the ketoheterocycle and protonated histidine from the His-Cys dyad was hypothesized. Irreversible inhibitors were developed with a vinylsulfone warhead. Irreversible peptidase inhibitors can be advantageous for the treatment of antiparasitic diseases, as these infections usually only require short courses of treatment; thus avoiding tissue accumulation and long term safety issues. In addition, irreversible inhibitors often show more efficient in vivo antiparasitic activities than reversible inhibitors.11
Figure 1.

A) Schematic representation of TbMCA inhibitors based on L-Arginine or L-Lysine. B) First set of TbMCA inhibitors derived from α-amino protected arginine with nitrile P1′ warhead and modifications in P1.
Our strategy was to develop a small diversity set of inhibitors in order to select a first hit from a screening assay on TbMCA2. Different parameters were tested: a set of warheads was investigated and peptidomimetic inhibitors were designed using a peptoid backbone. In addition, an attempt to influence basicity at P1 position was made. From this first screen a hit was selected which was then subjected to a range of optimization efforts. Several Arg mimetics were introduced in P1 and also P2 modification was addressed in order to obtain more potent inhibitors and gain insight in the scope of the active site of MCAs.
For a first set of inhibitors we started from the α-amino protected arginine with a nitrile warhead 1 (Fig. 1B). In the P2 position the protecting group benzyloxycarbonyl (Cbz) was used. Diversification was carried out at the level of the P1 position (Fig. 1B, 2 and 3). To circumvent possible membrane permeability issues which are known for highly basic residues such as amidines and guanidines, we selected functional groups in the P1 side chain with lower basicity. First, aromatic functions were introduced in the P1 tail (2 and 3). A second set of inhibitors was then designed with a peptoid backbone (Fig. 2, 4-10) and a variety of functional groups in the P1 position which were intended to have a similar basicity-lowering effect.
Figure 2.

TbMCA inhibitors with peptoid backbone, nitrile P1′ warhead and modifications in P1.
A third set of inhibitors (Fig. 3) was derived from α-amino protected arginine with a α-ketoheterocyclic warhead (P1′). Different inhibitors 11-14 were screened and resulted in our first hit 11 (IC50 TbMCA2 = 0.6 μM).
Figure 3.

TbMCA inhibitors derived from α-amino protected arginine with α-ketoheterocyclic P1′ warhead.
One inhibitor with a warhead known to have irreversible binding properties towards cysteine peptidases was included in our diversity set, derived from α-amino protected arginine with a vinylsulfone warhead 15 (Fig. 4).
Figure 4.
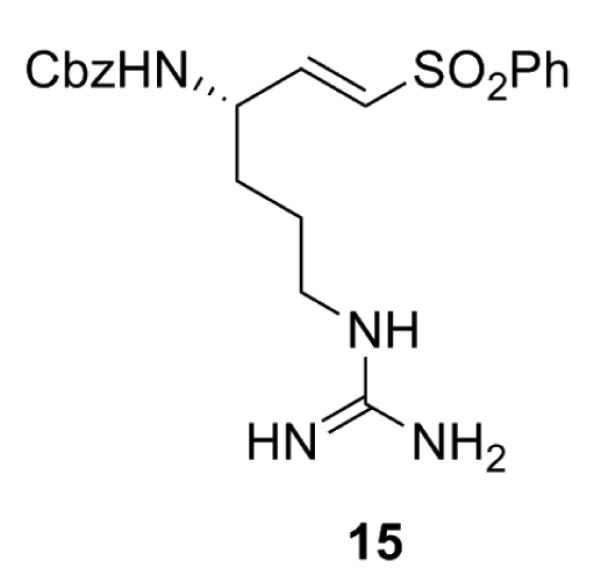
TbMCA inhibitor derived from α-amino protected arginine with α-vinylsulfone P1′ warhead.
The synthesis of inhibitors 1-15 is shown in schemes 1-6. For compound 1, commercially available Z-ArgPmc-OH (21) was converted to the corresponding amide (22) in the presence of N-hydroxysuccinimide, dicyclohexyl carbodiimide (DCC) and ammonia. The amide was then dehydrated with trifluoroacetic anhydride (TFAA) to the corresponding nitrile. TFA deprotection afforded target compound 1 (scheme 1).
Scheme 1.

Reagents: (a) 1. N-(OH)-Suc, DCC, THF, rt, 1 h; 2. NH3/MeOH 7N, rt, 1 h (68%); (b) TFAA, pyridine, THF, rt, 2 h; (c) TFA:DCM, rt, 1 h (19%).
Scheme 6.

Reagents: (a) N,O-dimethylhydroxylamine hydrochloride, TBTU, Et3N, DMF, 0°C, 3 h (95%); (b) TFA:DCM, rt, 1 h (99%); (c) tert-butyl (1H-pyrazol-1-yl)methylenedicarbamate, rt, 20 h (15%); (d) LiAlH4, THF, 0°C, 30 min (81%); (e) NaH, THF, 0°C, 1.5 h (14%); (f) TFA:DCM, rt, 1 h (70%).
4-Nitro-L-phenylalanine 23 was first protected with a Cbz group. Then the nitro group was reduced with SnCl2 and the obtained amine function was protected with a tert-butyloxycarbonyl (Boc) group (24). This intermediate was converted to the final compound 2 following the same synthetic steps as described for 1 (scheme 2).
Scheme 2.

Reagents: (a) benzylchloroformate, NaOH, water/dioxane, 0°C, 3 h (95%); (b) SnCl2, EtOH, reflux, 3 h (98%); (c) (Boc)2O, water, dioxane, Et3N, 0°C to rt, 12 h (17%); (d) 1. N-OH-Suc, DCC, THF, rt, 1 h; 2. NH3/MeOH 7 N, rt, 1 h (47%); (e) TFAA, pyridine, THF, rt, 3 h (8%); (f) TFA:DCM, rt, 1 h (65%).
For target compound 3, intermediate 25 was first Boc-deprotected in order to introduce a Boc-protected guanidine function, then a similar synthetic strategy was followed.
For target compound 4, synthesis was started from the aminoacetonitrile derivative 27 which was synthesised from tert-butyl 4-aminopiperidine-1-carboxylate 26 and bromoacetonitrile. After Cbz-protection of 27 followed by a final TFA deprotection, the desired compound was obtained (scheme 3).
Scheme 3.

Reagents: (a) bromoacetonitrile, K2CO3, 0°C to rt, 12 h (95%); (b) benzylchloroformate, NaOH, dioxane, 0°C, 3 h (43%); (c) TFA:DCM, rt, 1 h (26%).
Target compound 5 was synthesised from commercially available 4-aminobutanol (28) and bromoacetonitrile, followed by a Cbz-protection of the free amine (scheme 4). Compound 5 was then converted via a mesylate to intermediate 29 using a Finkelstein reaction.14 A nucleophilic substitution with different amino moieties afforded target compounds 6-10 (scheme 4).
Scheme 4.

Reagents: (a) bromoacetonitrile, K2CO3, 0°C to rt, 12 h (95%); (b) benzylchloroformate, DCM, Et3N, 0°C, 3 h (45%); (c) MsCl, Et3N, rt, 2 h (96%); (d) NaI, dry acetone, 60°C, 12 h (59%); (e) R = pyrrolidine (6), imidazole (7), piperidine (8), 1-(4-fluorophenyl)piperazine (9), 1-(bis(4-fluorophenyl)methyl)piperazine (10), DCM, rt, 1 h-12 h (6, 43%; 7, 99%; 8, 79%; 9, 17%; 10, 26%).
For compounds 11, 12 and 14 the synthesis described by Costanzo et al. was followed.13 For compound 13, the protocol reported by Lin et al. was used.15 The synthesis is shown for 11-14 starting from Z-Arg-Pmc-OH (21) (scheme 5).
Scheme 5.

Reagents: (a) N,O-dimethylhydroxylamine hydrochloride, TBTU, Et3N, DMF, 0°C, 12 h (96%); (b) 1. THF, n-BuLi, heterocycle, −78°C, 2 h; 2. TFA:DCM, rt, 1 h (11, 17%; 12, 29%; 13, 17%; 14, 26%).
To obtain compound 15, commercially available Z-Orn-Boc-OH (31) was converted to the corresponding Weinreb amide. The amine function was deprotected and converted to Boc-protected guanidine (32). Then the protected arginine Weinreb amide was reduced to the aldehyde (33) in which a Horner-Emmons reaction was executed in the presence of diethyl phenylsulfonylmethylphosphonate 34 to give the desired vinylogous arginine.16 A final deprotection in acidic medium provided target compound 15 (scheme 6).
All target compounds were biochemically evaluated by in vitro testing as competitive inhibitors of TbMCA2 (table 1). Six compounds showed significant inhibition (IC50 < 2.2 μM), compound 11 was the most potent inhibitor with an IC50 of 0.6 μM. In addition, compounds 1 and 11-14 were tested against TbMCA3 and the overall inhibition profile was similar to that for TbMCA2 (data not shown). TbMCA2 inhibitors would be expected to show inhibition of TbMCA3 as they share 89% sequence identity.17 As simultaneous inhibition of all catalytically active TbMCAs is probably necessary, inhibition of multiple TbMCAs by an inhibitor would be advantageous. The specificity of inhibitors 1 and 11-14 was assessed using human caspase-3, a member of the same peptidase family as the MCAs. Caspase-3 activity was unaffected by any of these inhibitors (data not shown), demonstrating their selectivity for TbMCAs.
Table 1.
Inhibition of TbMCA2 and T. b. brucei, T. cruzi, L. infantum and P. falciparum (IC50) for 1-15. Cytotoxicity was measured against human fibroblasts (MRC-5).
| Compound | IC50 Tb MCA2 (μM) |
IC50 T. b. brucei (μM) |
IC50 T. cruzi (μM) |
IC50 L. infantum (μM) |
IC50 P. falciparum (μM) |
IC50
MRC- 5 (μM) |
|---|---|---|---|---|---|---|
| 1 | 1.9 | > 64 | > 64 | > 64 | 50.2 | > 64 |
| 2 | > 100 | 30.3 | 28.6 | > 64 | 16.6 | > 64 |
| 3 | > 100 | > 64 | 27.2 | > 64 | 25.4 | > 64 |
| 4 | > 100 | 32.5 | 34.6 | > 64 | 28.0 | > 64 |
| 5 | > 100 | > 64 | > 64 | > 64 | > 64 | > 64 |
| 6 | > 100 | 52.1 | > 64 | > 64 | 20.5 | > 64 |
| 7 | > 100 | > 64 | 35.7 | > 64 | 17.2 | > 64 |
| 8 | > 100 | > 64 | > 64 | > 64 | 6.9 | > 64 |
| 9 | > 100 | 22.6 | 7.0 | > 64 | > 64 | > 64 |
| 10 | > 100 | 2.1 | 2.6 | 5.9 | 7.6 | 11.3 |
| 11 | 0.6 | 32.9 | 20.9 | > 64 | 8.0 | > 64 |
| 12 | 62 | > 64 | > 64 | > 64 | > 64 | > 64 |
| 13 | 2.2 | > 64 | 38.7 | > 64 | 24.7 | > 64 |
| 14 | 38.4 | 32.5 | 28.3 | > 64 | 7.1 | > 64 |
| 15 | 3.9 | 45.3 | 19.0 | > 64 | 3.4 | > 64 |
Next, a biological evaluation was performed in vitro against the following protozoa (table 1): T. b. brucei (BSF), T. cruzi and Leishmania infantum intracellular amastigotes, and Plasmodium falciparum (ring stage and schizont). Leishmania parasites have a single metacaspase gene18, whilst P. falciparum19 and T. cruzi20 both have 2 MCAs. Cytotoxicity was assessed by testing the compounds on human lung fibroblasts (MRC-5). Compound 1 has an IC50 value of 1.9 μM for TbMCA2 but showed no antiparasitic activity. Modifications of the P1 tail (2 and 3) in order to lower the basicity combined with the introduction of a peptoid backbone (4-10) resulted in inactive compounds. Introduction of α-ketoheterocyclic warheads with retention of the Arg P1 tail increased enzymatic inhibition, especially for benzothiazole (11, TbMCA2 IC50 = 0.6 μM) and thiazole (13, TbMCA2 IC50 = 2.2 μM). For 11 and 14, antiparasitic activity although very modest, was observed against P. falciparum. Finally, irreversible inhibitor 15 also was found to be a low micromolar inhibitor (TbMCA2 IC50 = 3.9 μM) with modest antiparasitic activity towards P. falciparum.
Compound 11 was considered as a valuable hit, the first to be developed towards TbMCA2, with low micromolar activity and without cytotoxicity. To obtain more potent compounds, optimisations were carried out at both P1 and P2 level (Fig. 5). At the P1 level, the arginine side chain was substituted for arginine mimetics. Many arginine mimetics are able to interact with carboxylic groups in the active site21 such as homoarginine 17 and less basic lysine 16 and ornithine. At the P2 level, the Cbz protecting group was substituted for a mimic of the P2 amino acid, which is known to be a hydrophobic uncharged aliphatic amino acid.22 This resulted in dipeptide analogues 18 and 19.
Figure 5.

TbMCA inhibitors derived from hit 11 with modifications at P1 and P2 level.
Compound 16 was synthesised as per the reported procedure by McGrath et al.23 Compound 17 was synthesised in the same way as 11-14 (scheme 5). For this compound, the amine function of N-ε-Boc-N-α-Z-L-Lysine was first deprotected and converted to a Boc-protected guanidine. The ornithine analogue could not be obtained due to formation of the inactive enamine after cyclisation. For compounds 18 and 19 the first synthetic steps are comparable to scheme 5: commercially available Boc-Arg-Pbf-OH was transformed to intermediate 35 (scheme 7). After a selective Boc deprotection and activation of Z-Leu/Z-Val in the presence of 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) and N-hydroxybenzotriazole (HOBt), a coupling reaction took place (36).24 A final TFA deprotection yielded compounds 18 and 19 (scheme 7).
Scheme 7.

(a) TFA:DCM (10%, 4 eq), rt, 3-5h (85%); (b) 1. Z-Leu/Z-Val, HOBt, EDC, DCM, rt, 2 h; 2. Et3N, rt, 12 h; (n = 1, 19%; n = 0, 13%) (c) TFA:DCM, rt, 1 h (18, 76%; 19, 63%).
Modification of P1 caused inhibition in the same range as the hit (16, TbMCA2 IC50 = 1.6 μM and 17, TbMCA2 IC50 = 4.0 μM) with modest antiparasitic activities. P2 modification (18 and 19) scored comparable enzymatic and antiparasitic activities (table 2).
Table 2.
Inhibition of TbMCA2 and T. b. brucei, T. cruzi, L. infantum and P. falciparum (IC50) for 16-19. Cytotoxicity was measured against human fibroblasts (MRC-5).
| Compound | IC50 Tb MCA2 (μM) |
IC50 T. b. brucei (μM) |
IC50 T. cruzi (μM) |
IC50 L. infantum (μM) |
IC50 P. falciparum (μM) |
IC50 MRC- 5 (μM) |
|---|---|---|---|---|---|---|
| 16 | 1.6 | 10.6 | 10.8 | 22.8 | 3.00 | 20.0 |
| 17 | 4.0 | 11.4 | 5.5 | 45.4 | 5.6 | 19.0 |
| 18 | 1.4 | 5.7 | 5.6 | 36.8 | 2.6 | 29.0 |
| 19 | 1.1 | 11.2 | 7.3 | > 64 | 3.3 | 32.6 |
We report the first MCA inhibitors with low micromolar activity. These compounds inhibit TbMCA2 and TbMCA3, possess modest antiparasitic activity, but have excellent selectivity when compared to mammalian caspases. Optimization efforts are ongoing.
Acknowledgments
This work was funded by a research project from the Research Foundation Flanders (FWO-Vlaanderen), the Medical Research Council (Grant numbers G9722968 and G0700127) and an SRDG grant (HR04013) from the Scottish Funding Council. M. Berg and M. Breugelmans have a Ph.D. grant from the Research Foundation Flanders (FWO-Vlaanderen). P. Van der Veken has a postdoctoral grant from the Research Foundation Flanders (FWO-Vlaanderen). The laboratory of Medicinal Chemistry and LMPH are partners of the Antwerp Drug Discovery Network (ADDN, www.addn.be).
References and Notes
- 1.Uren AG, O'Rourke K, Aravind L, Pisabarro MT, Seshagiri S, Koonin EV, Dixit VM. Mol. Cell. 2000;6:961. doi: 10.1016/s1097-2765(00)00094-0. [DOI] [PubMed] [Google Scholar]
- 2.Vercammen D, Declercq W, Vandenabeele P, Van Breusegem F. J. Cell Biol. 2007;179:375. doi: 10.1083/jcb.200705193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Figarella K, Rawer M, Uzcategui NL, Kubata BK, Lauber K, Madeo F, Wesselborg S, Duszenko M. Cell Death Differ. 2005;12:335. doi: 10.1038/sj.cdd.4401564. [DOI] [PubMed] [Google Scholar]
- 4.Moss CX, Westrop GD, Juliano L, Coombs GH, Mottram JC. FEBS Lett. 2007;581:5635. doi: 10.1016/j.febslet.2007.11.009. [DOI] [PubMed] [Google Scholar]
- 5.Helms MJ, Ambit A, Appleton P, Tetley L, Coombs GH, Mottram JC. J. Cell Sci. 2006;119:1105. doi: 10.1242/jcs.02809. [DOI] [PubMed] [Google Scholar]
- 6.Chowdhury I, Tharakan B, Bhat GK. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2008;151:10. doi: 10.1016/j.cbpb.2008.05.010. [DOI] [PubMed] [Google Scholar]
- 7.Debrabant A, Lee N, Bertholet S, Duncan R, Nakhasi HL. Int. J. Parasitol. 2003;33:257. doi: 10.1016/s0020-7519(03)00008-0. [DOI] [PubMed] [Google Scholar]
- 8.Nguewa PA, Fuertes MA, Valladares B, Alonso C, Pérez JM. Trends Parasitol. 2004;20:375. doi: 10.1016/j.pt.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 9.Ameisen JC. Cell Death Differ. 2002;9:367. doi: 10.1038/sj.cdd.4400950. [DOI] [PubMed] [Google Scholar]
- 10.Zangger H, Mottram JC, Fasel N. Cell Death Differ. 2002;9:1126. doi: 10.1038/sj.cdd.4401071. [DOI] [PubMed] [Google Scholar]
- 11.McKerrow JH, Rosenthal PJ, Swenerton R, Doyle P. Curr. Opin. Infect. Dis. 2008;21:668. doi: 10.1097/QCO.0b013e328315cca9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kaiser B, Richter M, Hauptmann J, Markwardt F. Pharmazie. 1991;46:128. [PubMed] [Google Scholar]
- 13.Costanzo MJ, Almond HR, Hecker LR, Schott MR, Yabut SC, Zhang HC, Andrade-Gordon P, Corcoran TW, Giardino EC, Kauffman JA, Lewis JM, de Garavilla L, Haertlein BJ, Maryanoff BE. J. Med. Chem. 2005;48:1984. doi: 10.1021/jm0303857. [DOI] [PubMed] [Google Scholar]
- 14.Enders D, Schusseler T. Tetrahedron Lett. 2002;43:3467. [Google Scholar]
- 15.Lin J, Deng H, Jin L, Pandey P, Quinn J, Cantin S, Rynkiewicz MJ, Gorga JC, Bibbins F, Celatka CA, Nagafuji P, Bannister TD, Meyers HV, Babine RE, Hayward NJ, Weaver D, Benjamin H, Stassen F, Abdel-Meguid SS, Strickler JE. J. Med. Chem. 2006;49:7781. doi: 10.1021/jm060978s. [DOI] [PubMed] [Google Scholar]
- 16.Konno H, Kubo K, Makabe H, Toshiro E, Hinoda N, Nosaka K, Akaji K. Tetrahedron. 2007;63:9502. [Google Scholar]
- 17.Mottram JC, Helms MJ, Coombs GH, Sajid M. Trends Parasitol. 2003;19:182. doi: 10.1016/s1471-4922(03)00038-2. [DOI] [PubMed] [Google Scholar]
- 18.Ambit A, Fasel N, Coombs GH, Mottram JC. Cell Death Differ. 2008;15:113. doi: 10.1038/sj.cdd.4402232. [DOI] [PubMed] [Google Scholar]
- 19.Wu Y, Wang X, Liu X, Wang Y. Genome Res. 2003;13:601. doi: 10.1101/gr.913403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kosec G, Alvarez VE, Agüero F, Sanchez D, Dolinar M, Turk B, Turk V, Cazzulo JJ. Mol. Biochem. Parasitol. 2006;145:18. doi: 10.1016/j.molbiopara.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 21.Peterlin-Masic L, Kikelj D. Tetrahedron. 2001;57:7073. [Google Scholar]
- 22.Results not published.
- 23.McGrath ME, Sprengeler PA, Hirschbein B, Somoza JR, Lehoux I, Janc JW, Gjerstad E, Graupe M, Estiarte A, Venkataramani C, Liu Y, Yee R, Ho JD, Green MJ, Lee CS, Liu L, Tai V, Spencer J, Sperandio D, Katz BA. Biochemistry. 2006;45:5964. doi: 10.1021/bi060173m. [DOI] [PubMed] [Google Scholar]
- 24.Costanzo MJ, Yabut SC, Almond HR, Jr., Andrade-Gordon P, Corcoran TW, De Garavilla L, Kauffman JA, Abraham WM, Recacha R, Chattopadhyay D, Maryanoff BE. J. Med. Chem. 2003;46:3865. doi: 10.1021/jm030050p. [DOI] [PubMed] [Google Scholar]
- 25.All newly synthesized compounds have full characterisation data (see Supplementary Data), for example, compound 13: MS (ESI) m/z 376.0 [M+H]+; LC-MS Rt 11.6 min, m/z 376.0 [M+H]+; 1H-NMR (400 MHz, MeOD) d δ 1.70-1.77 (m, 4H), 3.21-3.24 (m, 2H), 5.10 (s, 2H), 5.39-5.41 (m, 1H), 7.23-7.35 (m, 5H), 8.03 (d, J = 3.0 Hz, 1H), 8.09-8.11 (m, 1H); 13C-NMR (100 MHz, MeOD) d δ 26.4, 30.0, 41.8, 57.6, 67.8, 116.4 (q, CF3COOH), 128.8, 129.1, 129.5, 146.3, 158.7, 162.8 (q, CF3COOH), 166.1, 192.8