

Abstract
We previously synthesized a series of potent and selective A3 adenosine receptor (AR) agonists (North-methanocarba nucleoside 5′-uronamides) containing dialkyne groups on extended adenine C2 substituents. We coupled the distal alkyne of a 2-octadiynyl nucleoside by Cu(I)-catalyzed “click” chemistry to azide-derivatized G4 (fourth-generation) PAMAM dendrimers to form triazoles. A3AR activation was preserved in these multivalent conjugates, which bound with apparent Ki 0.1–0.3 nM. They were substituted with nucleoside moieties, solely or in combination with water-solubilizing carboxylic acid groups derived from hexynoic acid. A comparison with various amide-linked dendrimers showed that triazole-linked conjugates displayed selectivity and enhanced A3AR affinity. We prepared a PAMAM dendrimer containing equiproportioned peripheral azido and amino groups for conjugation of multiple ligands. A bifunctional conjugate activated both A3 and P2Y14 receptors (via amide-linked uridine-5′-diphosphoglucuronic acid), with selectivity in comparison to other ARs and P2Y receptors. This is the first example of targeting two different GPCRs with the same dendrimer conjugate, which is intended for activation of heteromeric GPCR aggregates. Synergistic effects of activating multiple GPCRs with a single dendrimer conjugate might be useful in disease treatment.
Keywords: G protein, coupled receptor, purines, alkyne, azide, radioligand binding, dendrimer
INTRODUCTION
Poly(amidoamine) (PAMAM) dendrimers are treelike polymers that have wide application in drug delivery in vivo (1, 2). We recently reported the first example of using a PAMAM dendrimeric scaffold for the polyvalent, nanoscale presentation of nucleosides and nucleotides to selectively activate adenosine receptors (ARs) and P2Y receptors to induce intracellular signal transduction (3–5). Both of these receptor families are cell-surface G protein–coupled receptors (GPCRs). The dendrimer carriers greatly multiply the size of the monomer agonist derivatives, and these multivalent conjugates are intended to bridge multiple binding sites present in GPCR dimers and other higher-order receptor aggregates. In general, we might expect the stability of the ligand in biological systems and its pharmacokinetic and pharmacodynamic properties to be enhanced in the conjugates in comparison to the monomeric ligands.
The A3AR is found at various levels of expression in neutrophils, eosinophils, cardiac myocytes, skeletal muscle, astrocytes, neurons, and other cell types (6) and has distinct effects on cell survival and response to stress. The A3AR agonist N6-(3-iodobenzyl)-5′-N-methylcarboxamidoadenosine (IB-MECA, CF101) 1 (Chart 1) and its 2-chloro derivative deregulate the NFκB pathway and are currently in clinical trials for the treatment of autoimmune inflammatory diseases and cancer, respectively (7–11). Anti-ischemic effects of A3AR agonists also suggest their use in protection of the heart, lung, nervous system, and skeletal muscle (6, 12, 13).
Chart 1.

Structures of adenosine A3AR agonist monomers (1–3) and the design of multivalent dendrimer conjugates in the(N) -methanocarba series, represented by the triazole derivative of general formula (4). Compound 3a was reported in ref. 9.
We previously synthesized a series of North (N)-methanocarba nucleoside 5′-uronamides as A3AR agonists of high affinity and selectivity (14). These selective agonists, including the anti-ischemic nucleoside MRS3558 2, contained a rigid bicyclo[3.1.0]hexane (methanocarba) ring system of the (N) conformation in place of the freely twisting ribose moiety (9, 11, 12). These agonists also contained an optimized N6-(3-chlorobenzyl) group, which enhanced A3AR selectivity in humans, mice, rats, and other species (14). Recently, we extended this series to include dialkyne groups on an extended adenine C2 substituent, e.g., 3a (21). We expected the proximal alkyne to promote receptor recognition and the distal alkyne to serve as a site for coupling to reporter groups while maintaining receptor recognition. Thus, we have selected a region on the nucleoside for attachment that is distal from the pharmacophore and is predicted by modeling to extend into the extracellular region of the A3AR (15).
Here, we coupled the distal alkyne of an A3AR agonist containing a 2-octadiyne group to azido-derivatized G4 (fourth-generation) PAMAM dendrimer carriers with Cu(I)-catalyzed alkyne-azide cycloaddition, a type of click chemistry (16–18), to form triazole derivatives, as in conjugate 3b, and determined the receptor-binding affinity and selectivity. This variation of click chemistry was used previously for coupling biologically active molecules to polymers (19). In our previous studies of dendrimer-bound ligands for GPCRs, the small molecular ligands were amide-linked to the carrier (3–5). For this study, we compared the amide linkages to PAMAM dendrimers with the use of azido groups for conjugation of alkyne derivatives.
We also included PAMAM dendrimers with mixed peripheral azido and amino groups in this study. The incorporation of azides for click chemistry along with the conventional terminal amino/carboxylic groups of the PAMAM dendrimers for amide coupling allowed the facile conjugation of ligands for two different GPCRs (Figure 1). We present the first example of coactivation of two GPCRs by a hybrid PAMAM dendrimer conjugate. The anti-inflammatory A3AR and the P2Y14 receptor, which is activated by endogenous uridine-5′-diphosphoglucose (UDPG) in the neuroimmune system (20), serve to illustrate this approach.
Figure 1.

Design of a bifunctional PAMAM dendrimer conjugate for the coactivation of two different GPCRs; shown here are agonists of the A3AR and the P2Y14 receptor. Both receptors are distributed in the immune system and may occur on the same cell (27). The stoichiometry of substitution of the dendrimer is not indicated.
EXPERIMENTAL PROCEDURES
Chemical Synthesis
Materials and Methods
All reactions were carried out under a nitrogen atmosphere. We purchased N-ethyl-N′-dimethylaminopropylcarbodiimide hydrochloride (EDC.HCl), UDPG acid trisodium salt (UDPGA), UDPG, G2.5 PAMAM (10 wt% solution in methanol), G3 PAMAM (20 wt% solution in methanol), PAMAM dendrimers (G4, G5.5, and G6, 5 wt% solution in methanol) with an ethylenediamine core from Sigma-Aldrich (St. Louis, MO). All other reagents and solvents, except those indicated, came from Sigma-Aldrich. We purchased dialysis membranes (Spectra/Pore Membrane, MWCO 3500, flat width 18 mm) from Spectrum Laboratories (Rancho Dominguez, CA). Synthesis of compound 3a is described elsewhere (21).
Nuclear magnetic resonance (NMR) spectra were recorded on a Bruker DRX-600 spectrometer with D2O as a solvent. The chemical shifts are expressed as relative ppm from HOD (4.80). We performed electrospray ionization mass spectrometry (MS) and matrix-assisted laser desorption ionization (MALDI) time-of-flight MS experiments on a Waters LCT Premier mass spectrometer at the Mass Spectrometry Facility, National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), NIH. We analyzed the azido dendrimer precursors on a Varian 500 MHz NMR and on a Bruker Vertex 70 FT-IR. Galbraith Laboratories (Knoxville, TN) performed the elemental analysis.
G4 PAMAM, Conjugated with 6-heptynoic acid (15)
We added freshly prepared aqueous sodium ascorbate (1 M, 42 μL, 42.1 μmol) to a mixture of G4 PAMAM dendrimer 13 (6.74 mg, 0.42 μmol) and 6-heptynoic acid (1.33 mg, 10.5 μmol) in a mixture of t-butanol (0.3 mL) and water (0.3 mL), then added 7.5% aqueous cupric sulfate (70 μL, 20.8 μmol). The reaction mixture was stirred at room temperature overnight, and the product was purified by dialysis in water. The mixture was lyophilized to yield compound 15 (5.45 mg, 68%) as a white foamy solid. MALDI-MS: calcd. 19,029; found 18,928.
G4 PAMAM, Conjugated with A3AR Agonist 3a and 6-heptynoic Acid (7)
We added freshly prepared aqueous sodium ascorbate (1 M, 31 μL, 31 μmol) to a mixture of compound 15 (5.45 mg, 0.28 μmol) and compound 3a (4.13 mg, 7.74 μmol) in a mixture of t-butanol (0.3 mL) and water (0.3 mL), then added 7.5% aqueous cupric sulfate (51 μL, 14.3 μmol). The reaction mixture was stirred at room temperature overnight, and the product was purified by dialysis in water. The mixture was lyophilized to yield compound 7 (5.8 mg, 60%) as a white foamy solid. 1H NMR (DMSO-d6, 600 MHz) δ 8.51 (s), 8.14 (s), 7.61 (s), 7.31–7.41 (m), 5.46 (s), 5.68–5.95 (m), 3.86 (s), 2.66 (s), 2.21 (s), 1.8 (s), 1.61 (bs), 1.11–1.29 (m). MALDI-MS: calcd. 33,852; found 34,088.
G4 PAMAM, Conjugated with A3AR Agonist 3a (6)
We followed the procedure used for compound 7 to synthesize compound 6 (69%) from 3a. 1H NMR (DMSO-d6, 400 MHz) δ 8.50 (bs), 8.15 (bs), 7.60 (s), 7.29–7.59 (m), 5.44 (s), 4.67–5.05 (m), 4.16 (s), 3.86 (s), 2.78 (s), 2.67 (s), 2.08–2.23 (m), 1.82 (s), 1.61 (s), 0.98–1.30 (m). MALDI-MS: calcd. 49,991; found 50,844.
G4 PAMAM, Conjugated with UDPGA (17)
We added EDC.HCl (12.5 mg, 65.2 μmol) to a solution of compound 5 (15.3 mg, 1.01 μmol) and UDPGA trisodium salt 16 (22.9 mg, 35.43 μmol) in dimethylsulfoxide (DMSO), then added 0.1 M 2-(N-morpholino)ethanesulfonic acid buffer (140 μL). We used 0.1 M HCl to adjust the pH of the reaction mixture to 4.5–5.0. The reaction mixture was stirred at room temperature overnight, and the product was purified by extensive dialysis followed by lyophilization to yield compound 17 (22 mg, 64%). 1H NMR (DMSO-d6, 400 MHz) δ 8.19 (s), 7.86–8.14 (m), 6.55 (m), 5.80 (s), 5.67 (s), 5.38–5.47 (m), 4.07 (s), 3.97 (s), 3.63 (br s), 3.25 (s), 3.12 (s), 2.91 (s), 2.69 (s), 2.24 (s). MALDI-MS: calcd. 33,060; found 33,036.
G4 PAMAM, Conjugated with UDPGA and A3AR Agonist 3a (8)
We followed the procedure used for compound 7 to synthesize compound 8 (66%) from compound 17. MALDI-MS: calcd. 50,092; found 50,610.
G2.5 PAMAM, Conjugated with A3AR Agonist 19 (9)
We followed the procedure used for compound 17 to synthesize compound 9 (54%) from dendrimer 18 and compound 19. 1H NMR (D2O, 600 MHz) δ 7.98 (br s), 6.88 (br s), 4.56 (br s), 3.85 (br s), 3.55 (br s), 3.45 (br s), 3.16–3.36 (m), 3.16 (br s), 3.08 (br s), 2.56–2.98 (m), 2.54 (s), 2.24–2.48 (m), 1.92 (br s), 1.68 (br s). MALDI-MS: calcd. 10,598; found 10,783.
G2.5 PAMAM, Conjugated with A3AR Agonist 20 (10)
We followed the procedure used for compound 17 to synthesize compound 10 (67%) from dendrimer 18 and compound 20. 1H NMR (DMSO-d6, 600 MHz) δ 8.11 (s), 7.85–8.01 (m), 7.58 (s), 7.36 (s), 7.18–7.32 (m), 5.06–5.21 (m), 4.88–4.93 (m), 4.65 (s), 4.59 (s), 3.86–3.91 (m), 3.07 (s), 2.65 (s), 2.39 (s), 2.21 (s), 1.85 (s), 1.74 (br s), 1.59 (br s), 1.21–1.33 (m), 1.03 (br s). MALDI-MS: calcd. 8370; found 8369.
G3 PAMAM, Conjugated with A3AR Agonist 22 (11)
We followed the procedure used for compound 17 to synthesize compound 11 (71%) from dendrimer 21 and compound 22. 1H NMR (DMSO-d6, 600 MHz) δ 8.88 (s), 8.11 (s), 7.73–8.05 (m), 7.58 (s), 7.32 (s), 7.13–7.28 (m), 5.43 (s), 5.03–5.21 (br s), 4.87–4.96 (s), 4.85 (br s), 4.67 (s), 4.61 (s), 3.89 (s), 3.06 (s), 2.82 (s), 2.65 (s), 2.34–2.46 (m), 2.19 (s), 1.83 (s), 1.69–1.78 (m), 1.61 (s), 1.29 (s). MALDI-MS: calcd. 13,694; found 13,304.
G4 PAMAM, Conjugated with A3AR Agonist 24 (12)
We added HATU (4.67 mg, 12.28 μmol) to a mixture of compound 24 (6.72 mg, 12.35 μmol) and dendrimer 23 (2.73 mg, 12.28 μmol) in dry DMF (0.5 mL), then added DIPEA (2.15 μL, 12.22 μmol). The reaction mixture was stirred at room temperature overnight, extensively dialyzed with water, then lyophilized to yield compound 12 (5.5 mg, 61%). 1H NMR (DMSO-d6, 400 MHz) δ 8.46 (br s), 8.10 (s), 7.79–7.93 (m), 7.60 (s), 7.27–7.36 (m), 5.45 (s), 4.67–4.93 (m), 3.87 (s), 3.08 (s), 2.86 (s), 2.66 (s), 2.41 (s), 2.19 (s), 1.78 (s), 1.61 (s), 1.23 (t, J = 24 Hz), 0.80–0.89 (m). MALDI-MS: calcd. 47,495; found 47,985.
Imidazole-1-sulfonyl azide hydrochloride (26)
A modification of a published procedure was used (22). A suspension of 6.53 g NaN3 (25, 100 mmol, Aldrich, >99.5%) and 100 mL acetonitrile (Aldrich, 99.8% anhydrous) in a 500-mL round-bottom flask was cooled in an ice water bath under a flow of nitrogen. We then added 8.15 mL sulfuryl chloride (100 mmol, TCI American, >98.0%) in 20 mL) anhydrous acetonitrile drop-wise to the ice-cooled suspension. The mixture was stirred overnight at room temperature, after which we cooled the white suspension to ~0°C with ice water and added 13.03 g imidazole (191 mmol, TCI America, >98.0%). We warmed the resulting white slurry to room temperature, then stirred it for 3 h. We then diluted it with ethyl acetate (200 mL) and separated and discarded the aqueous layer. We washed the organic layer twice with deionized water (2 × 100 mL), then twice with saturated aqueous NaHCO3 (2 × 100 mL). We then dried it over anhydrous MgSO4 overnight and filtered it. We added an ethanolic HCl solution (196 mmol) drop-wise to the filtrate while stirring at ice-water temperature. After we added ~3 mL, a large amount of white solid appeared. We kept the mixture in an ice bath for 15 min, then at room temperature for 30 min, after which we filtered it. We washed the filter cake with ethyl acetate (3 × 100 mL) to give 26 as a colorless solid (8.65 g, 49.9%), m.p. 99–101°C. IR (KBr) 2173, 1383 and 1160 cm−1. 1H NMR (500 MHz, D2O) d 7.61 (dd), 8.02 (dd), 9.39 (dd); 13C NMR (125. MHz, D2O) δ = 120.0, 123.5, 137.6. Element Analysis: Calculated C, 17.19; H, 1.92; N, 33.41. Found: C, 17.38; H, 1.98; N, 32.97.
Synthesis of Fully Azido-Derivatized PAMAM (G4) (13)
We added anhydrous methanol (10 mL, Aldrich, >99.8%) and 18.15 g G4 PAMAM dendrimer (23, Dendritech, Midland, MI, 14.93 w/w% in methanol, [NH2]=12.2 mmol) to a 250-mL round-bottom flask. We then added 3.12 g K2CO3 (Aldrich, >99.0%, 22.6 mmol) and 31.4 mg CuSO4.5H2O (Aldrich, 98.0%, 0.126 mmol), causing the solution to take on a pale bluish color. We next added 2.73 g imidazole-1-sulfonyl azide hydrochloride (26, 13.1 mmol) to the reaction mixture, and it was stirred at room temperature overnight. The mixture was then dialyzed against deionized water for 7 d, during which we changed the water every 2 h for the first 2 d and thereafter every 4 h. After lyophilization, we collected 2.51 g azide-derivatized G4 dendrimer. The proton NMR is shown in Figure 2. Element analysis: PAMAM-G4: calculated: C, 52.81; H, 8.48; N, 24.70, O, 14.01; fully azide-derivatized G4 PAMAM calculated: C, 47.03; H, 7.15; N, 33.31, O, 12.49; Found: C: 49.91; H: 7.55; N: 28.07, O: 13.12. The element analyses indicated that 98.0%of primary amines were replaced with azides.
Figure 2.

1H NMR spectra of G4-N3 (5, top), G4-NH2 from D2O (23, middle). Schematic indication of assignments of the methylene groups (bottom).
Synthesis of Partially Azido-Derivatized PAMAM (G4) (5)
We added anhydrous methanol (5 mL, Aldrich, >99.8%) and 5.13 g G4 PAMAM dendrimer (23, Dendritech, 14.93 w/w% in methanol, [NH2]=3.45 mmol) to a 100-mL round-bottom flask. We then added 0.803 g K2CO3 (Aldrich, >99.0%, 5.81 mmol) and 17.2 mg CuSO4.5H2O (Aldrich, 98.0%, 0.069 mmol); the solution took on a pale-bluish color. Next we added 0.43 g imidazole-1-sulfonyl azide hydrochloride (26, 2.05 mmol) to the reaction mixture, which was stirred at room temperature overnight. We then dialyzed the mixture against deionized water for 7 d, changing the water every 2 h during the first 2 d and every 4 h thereafter. After lyophilization, we collected 0.45 g partially azide-derivatized G4 dendrimer. Elemental analysis of the partially azide-derivatized G4 PAMAM found C: 49.91 H: 7.55; N: 28.07; O: 13.12. We compared results of the element analysis with the composition of the G4 PAMAM dendrimer and found that 47.2% of the primary amines were replaced with azides, corresponding to ~31 amines per dendrimer in 64 terminal positions.
Biological Methods: P2Y14 Receptor
Materials
We purchased 3-isobutyl-1-methyl-xanthine from Sigma-Aldrich and [3H]adenine from American Radiolabeled Chemicals (St. Louis, MO). We obtained all cell culture medium and serum from Gibco(Invitrogen, Carlsbad, CA).
Cell culture
We generated C6 glioma cells stably expressing the human P2Y 14-R (P2Y14-C6 cells) as previously described by Fricks et al. (23). We cultured P2Y14-C6 cells in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 5% fetal bovine serum, 1% Geneticin (Gibco), and 1% antibiotic-antimocotic (Gibco) at 37°C in a 5% CO2 environment.
Accumulation of adenosine 3′, 5′-cyclic phosphate (cAMP)
We grew P2Y14-C6 cells in 24-well plates and incubated them with 1 μCi [3H]adenine/well in serum-free DMEM for at least 2 h prior to assay. We initiated the assays by adding HEPES-buffered, serum-free DMEM containing 200 μM 3-isobutyl-1-methyl-xanthine and 30 μM forskolin, with or without drugs, and incubation continued for 15 min at 37°C. We terminated incubations by aspirating the medium and adding 450 μL ice-cold 5% trichloroacetic acid. We isolated [3H]cAMP by sequential Dowex and alumina chromatography and quantified it by liquid scintillation counting as previously described by Harden et al. (24).
Data analysis
We measured the concentrations of the dendrimer-ligand complexes by the concentration of the dendrimer, not the attached ligand. We determined EC50 and apparent Ki values with Prism software (GraphPad, San Diego, CA); they are presented as mean ± SE. The number of nucleoside moieties on a given dendrimer that are accessible to receptors is uncertain; the data are therefore shown as apparent Ki values. We repeated all experiments at least three times.
Biological Methods: ARs
Receptor-binding and functional assays
We obtained [3H]adenosine-5′-N-methyluronamide ([3H]NECA, 42.6 Ci/mmol) from PerkinElmer Life and Analytical Science (Boston, MA). We purchased [3H](2-[p-(2-carboxyethyl)phenyl-ethylamino]-5′-N-ethylcarboxamido-adenosine) ([3H]CGS21680, 40.5 Ci/mmol) and [125I]N6-(4-amino-3-iodobenzyl)adenosine-5′-N-methyluronamide ([125I]I-AB-MECA, 2200 Ci/mmol) from PerkinElmer. We prepared test compounds as 5-mM stock solutions in DMSO and stored them frozen at −20°C.
Cell culture and membrane preparation
We cultured Chinese hamster ovary (CHO) cells stably expressing the recombinant hA1 and hA3 and human embryonic kidney (HEK) 293 cells stably expressing the hA2AAR in DMEM and F12 (1:1) supplemented with 10% fetal bovine serum, 100 units/mL penicillin, 100 μg/mL streptomycin, and 2 μmol/mL glutamine. In addition, we added 800 μg/mL Geneticin to the A2A media and 500 μg/mL hygromycin to the A1 and A3 media. After harvesting the cells, we homogenized them with an electric homogenizer for 10 sec, pipetted them into 1-mL vials, and then stored them at −80°C until we conducted the binding experiments. We measured the protein concentration with a BCA Protein Assay Kit from Pierce Biotechnology (Rockford, IL) (25).
Binding assays
Each tube in the binding assay contained 50 μL of increasing concentrations of the test ligand in Tris-HCl buffer (50 mM, pH 7.5) containing 10 mM MgCl2, 50 μL of the appropriate agonist radioligand, and 100 μL membrane suspension, added sequentially. For the A1AR (20 μg protein/tube), we used the radioligand [3H]NECA (~3.5 nM, precise final concentration is calculated for each experiment). For the A2AAR (20 μg/tube), we used the radioligand[ 3H]CGS21680 (~10 nM). For the A3AR (21 μg/tube), we used the radioligand [125I]I-AB-MECA (~0.3 nM). We determined nonspecific binding with a final concentration of 10 μM unlabeled NECAdiluted with the buffer. We incubated the mixtures at 25°C for 60 min in a shaking water bath. We terminated binding reactions by filtration through Brandel GF/B filters under a reduced pressure with a M-24 cell harvester (Brandel, Gaithersburg, MD). We washed filters three times with 3 mL 50-mM ice-cold Tris-HCl buffer (pH 7.5). We then placed filters for A1 and A2AAR binding in scintillation vials containing 5 mL Hydrofluor scintillation buffer and counted with a PerkinElmer Liquid Scintillation Analyzer (Tri-Carb 2810TR). We counted filters for A3AR binding with a Packard Cobra II γ-counter (PerkinElmer). We determined the Ki values with GraphPad Prism for all assays.
Accumulation of cAMP
To determinecAMP production (26), we incubated protein kinase A with [3H]cAMP (2 nM) in K2HPO4/EDTA buffer (K2HPO4, 150 mM; EDTA, 10 mM), 20 μL cell lysate, and 30 μL 0.1 M HCl or 50 μL cAMP solution (0–16 pmol/200 μL for standard curve). We separated bound radioactivity by rapid filtration through Whatman GF/C filters and washed the filters once with cold buffer. We measured bound radioactivity with liquid scintillation spectrometry.
RESULTS
Chemical Synthesis
We prepared various PAMAM dendrimer derivatives of nucleosides and nucleotides (Table 1, 6–12) to act as multivalent ligands of GPCRs. The peripheral groups of the precursor dendrimers contained either all azido groups (Scheme 1), mixtures of azido and amino groups (Scheme 2), or the conventional all amino/carboxylate groups (Scheme 3). The synthetic methods used to partially or fully substitute terminal amino groups of a G4 PAMAM dendrimer with azides are shown in Scheme 4 (22).
Table 1.
Potency of a series of nucleoside-dendrimer conjugates at three subtypes of human ARs.
 | ||||
|---|---|---|---|---|
| Compd | Structure, R1, R2, or R3 | Affinity (Ki, nM) or % inhibitiona | ||
| A1 | A2 | A3 | ||
| Model compounds | ||||
|
1b IB-MECA |
51 | 2900 | 1.8 | |
| 2b,c | R1 = Cl | 260±60h | 2300±100 | 0.29±0.04 |
| 3ab,c | R1 = C≡C(CH2)4C≡CH | (31±2%) at 10 μM | 7040±143 0 |
29.4±9.8 |
| 4ab,c | R2 = (CH2)2NHCOCH3 | (12±4%) at 10 μM | 2440±320 | 22.3±1.6 |
| 4bb | R1 = C≡C(CH2)2CH3 | 1040±80 | (80%) at 10 μM | 0.82±0.20 |
| 4cb | R3 = C≡C(CH2)4CONH(CH2)2NHCOCH3 | 181±22 | (80%) at 10 μM | 2.88±0.54 |
| Dendrimer derivatives | ||||
| 5d |  |
NA | NA | NA |
| 6c |
 64 |
22.6±6.9 | 32.2±6.4 | 0.14±0.09 |
| 7c |
 24 28 |
35.1±7.5 | 34.4±1.8 | 0.32±0.03 |
| 8c |
 31 33 |
78±31 | 584±48 | 39.5±11.8 |
| 9 |
 8 |
ND | (27±7%) at 10 μM | 31.4±2.8 |
| 10 |
5 |
77 | (3±2%) at 1 μM | 41.7±6.4 |
| 11c |
 13 |
7.1 | 5750±1600 | 17.6±2.8 |
| 12 |
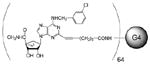 64 |
1.92±1.25 | 727±168 | 11.8±2.2 |
All experiments were done on CHO or HEK293 (A2A only) cells stably expressing one of four subtypes of human ARs. The binding affinity for A1, A2A, and A3ARs was expressed as Ki values (n = 3–5) and was determined by using agonist radioligands ([3H]NECA, [3H]CGS21680, or [125I]I-AB-MECA, respectively), unless noted. A percentage in parentheses refers to inhibition of radioligand binding at the indicated concentration. The concentrations of the dendrimer-ligand complexes were measured by the concentration of the dendrimer, not the ligand. Therefore, all binding Ki values of dendrimers are expressed as Kiapp values.
2, MRS3558; 3a, MRS5221; 4a, MRS5233; 6, MRS5246; 7, MRS5247; 8, MRS5259; 11, MRS5216.
Coumpound 5 is the dendrimer precursor of 8 and is similar to the dendrimer precursor of 6 and 7.
NA – not active in inhibition of radioligand binding (<10%) at 1 μM.
Scheme 1.

Use of click chemisry to synthesize PAMAM dendrimer derivatives 6 and 7, containing a functionalized 2-alkynyl A3AR agonist. An optional water-solubilizing group was added in 7, in the form of an aliphatic carboxylic acid.
Scheme 2.

Synthesis of two PAMAM dendrimer derivatives containing a functionalized agonist of the P2Y14 receptor derived by amide linkage of UDPGA 16. Conjugate 17 contains only the P2Y14 receptor agonist; conjugate 8 contains an A3AR agonist linked through a click reaction of its 2-alkynyl functionalized chain. The dendrimer precursor 5 contained a random distribution of peripheral amino and azido groups in a ratio of ~33:31.
Scheme 3.

Preparation of amide-linked nucleoside conjugates of PAMAM dendrimers.
Scheme 4.

Synthesis of azido-derivatized PAMAM dendrimers, which served as precursors for the GPCR ligand conjugates by reacting with imidazole-1-sulfonyl azide hydrochloride 26.
We recently introduced terminal alkynyl groups at the C2 position of (N)-methanocarba nucleoside derivatives to serve as sites on the A3AR agonists for cross-linking by click chemistry (21). Thus, a dialkyne was introduced at the C2 position in 2, and further reaction with azides generally occurred exclusively at the distal alkyne. Prior to coupling the nucleosides to dendrimer carriers, we explored in detail the structure-activity relationship at the distal region of an alkyne-functionalized chain at the 2 position and the resultant click products (21).
The synthetic route to the multivalent dendrimer conjugates of nucleosides for activation of the A3AR that contain 5′-N-methyluronamide (N)-methanocarba 2-alkynyl triazoles 6 and 7 is shown in Scheme 1. Click chemistry efficiently coupled the A3AR agonist 3a to a PAMAM dendrimer fully substituted with peripheral azido groups. We used two versions of this azido-functionalized dendrimeric scaffold: 15, containing extra appended carboxylic acid groups for increasing water solubility, and 13, lacking these groups. We added the carboxylic acid moieties to the azido dendrimer 13 by partial reaction of the peripheral azido groups with hexynoic acid 14. Thus, we prepared two nucleoside-containing dendrimer derivatives 6 and 7. Derivative 6 was fully substituted with the adenosine analogue, with 64 nucleoside moieties per dendrimer molecule. Derivative 7 was partially (28 out of 64) substituted with the adenosine analogue and contained 24 carboxylic acid moieties. 12 azido groups per molecule of 7 remained unreacted.
In the next phase of our study, we combined different GPCR ligands, i.e., the A3AR and another receptor, on the same dendrimer carrier. We selected the P2Y14 receptor, which is activated by naturally occuring uridine-5′-diphosphoglucose (UDPG), for this purpose because, like the A3AR, it occurs on immune cells and because we recently introduced high-potency dendrimer conjugates for this subtype (5, 27). Each ligand would provide selectivity for a particular GPCR subtype, and the overall selectivity of the conjugate would be a function of the union of the sets of selectivity of the attached nucleosides. We sequentially coupled alkyne and carboxylic acid derivatized ligands for the two receptors to a novel multifunctional PAMAM dendrimer. Thus, alkynes reacted exclusively with azido terminal groups and carboxylic acids with terminal amino groups on the same dendrimer to randomly (but in a defined ratio) produce a multivalent, binary conjugate.
We proceeded to synthesize a multivalent, binary conjugate of a mixed azido/amino dendrimer 5, as shown in Scheme 2. In the dendrimer precursor 5, we determined, through NMR (28) (Figure 2) and elemental analysis, that the ratio of amines to azides was ~31:33. Therefore, of the 64 terminal groups on each G4 dendrimer, ~33 would be available for cycloaddition, and the remaining terminal groups would be available for amide formation. We first coupled the amino groups of 5 to the functionalized, P2Y14-selective nucleotide UDPGA 16, which we previously showed to be a suitable functionalized congener for chain attachment without loss of receptor activation (5). We then coupled the azido groups of 17 to the functionalized nucleoside 3a, which is a suitable functionalized congener for chain attachment without loss of binding affinity at the A3AR (21). The product of this second coupling reaction was the mixed dendrimer 8, in which the average ratio of content of UDGPA and nucleoside moieties was ~31:33 per G4 dendrimer. Thus, conjugate 8 would be expected to potently activate both A3 and P2Y14 receptors and perhaps maintain selectivity over other ARs and P2Y receptors.
To compare alkyne-azide click chemistry with the more standard amide formation as a means of linking A3AR-selective agonists to PAMAM dendrimers, we prepared various dendrimer derivatives starting with our previously reported amino and carboxylic-functionalized congeners 19, 20, 22, and 24 (14, 15). The reaction sequences are shown in Scheme 3. We included a variety of dendrimer sizes and sites of conjugation on the nucleoside.
The synthesis of the PAMAM dendrimer intermediate 5 containing equiproportioned peripheral azido and amino groups for conjugation of multiple ligands is shown in Scheme 4. We used the reagent imidazole-1-sulfonyl azide hydrochloride 26 for the conversion of peripheral amino groups to azido groups (22). We prepared a fully azido-derivatized G4 dendrimer 13 similarly and used it to synthesize conjugates 6 and 7.
Quantification of Pharmacological Activity
We tested the dendrimer nucleoside conjugates similarly in binding assays at three hAR subtypes; we used standard radioligands (29–31) and membrane preparations from CHO cells (A1 and A3) or HEK293 cells (A2A) stably expressing a hAR subtype (Table 1) (32, 33). We used the previously reported agonist molecular probes of the A3AR (1, 2, 3a, and 4) for comparison in the biological assays (15, 21).
We included the parent dendrimer 5, which contained both amino and azido groups, as a control; it was inactive in AR binding at 1 μM. We considered compound 4a to be a model compound for the attachment of the 2-octadiyne derivative 3a to azido-bearing PAMAM dendrimers by click chemistry. As reported previously, the triazole-containing monomer 4a maintained both high potency and selectivity (at least 100-fold) for the A3AR (21).
The multivalent dendrimer derivatives 6 and 7 obtained from 3a through click conjugation were considerably more potent than 4a in binding assays at the A1, A2A, and A3 ARs. The affinity of 6 and 7 in binding to the A3AR was particularly high, with Ki values of 0.1–0.3 nM (Figure 3). The fully substituted dendrimer 6 was only 2.3-fold more potent at the A3AR than the less heavily loaded 7, which was substituted with nucleosides at 44% of the theoretically available terminal positions. The selectivity of these dendrimer conjugates for the A3AR in comparison to the A1AR was still present (161- and 110-fold for 6 and 7, respectively) but diminished in comparison to the monomer 4a (>500-fold). However, the A3AR selectivity of 6 (230-fold) and 7 (180-fold) in comparison to the A2AAR was similar to that of 4a. Thus, the presence of the carboxylic acid chain in 7 had only minor effects on the observed AR affinity and selectivity, but it increased the observed aqueous solubility. Both 6 and 7 were readily soluble in DMSO at a concentration of 1 mM, but when we diluted the DMSO stock solutions with phosphate-buffered saline for a final calculated concentration of 30 μM, we observed some cloudiness in 6 but not in 7.
Figure 3.


Inhibition of radioligand binding by dendrimer nucleoside conjugates in membranes of CHO cells expressing the human A1 (A) and A3 (C) ARs and HEK-293 cells expressing the human A2AAR (B). Inhibition curves at three ARs are shown for the dendrimer-nucleoside conjugates 6 and 7, in which the linkage to an azide-derivatized dendrimer was formed by click cycloaddition, for the mixed nucleoside/nucleotide conjugate 8 and for the amide-linked nucleoside conjugate 12. Both 6 and 7 were highly selective for the hA3AR in comparison to the hA1 and hA2AARs.
Dendrimer derivative 8 contained an additional covalently bound GPCR ligand, a nucleotide designed to activate the P2Y14 receptor. We found that combining strategically functionalized nucleosides and nucleotideson the same dendrimer carrier —for example, to activate both adenosine and P2Y receptors—provided high-affinity recognition at both GPCRs. However, the Ki value of 8 in binding to the A3AR (39.5 nM) indicated that it was considerably less potent than the conjugates 6 and 7, which contained only the A3AR agonist ligand. The A3AR selectivity of 8 was only 2-fold in comparison to the A1AR and 15-fold in comparison to the A2AAR. Thus it was much less A3AR selective than were 6 and 7.
A functional assay of adenylate cyclase inhibition carried out in C6 cells expressing the human P2Y14 receptor demonstrated that potency at this receptor was enhanced in the dendrimer conjugate 8 over that of the monomer, with an EC50 value of 2.24 ± 0.80 nM (Figure 4). Both UDPG and UDPGA displayed potencies in the same assay of 200–300 nM (5). This degree of potency enhancement at the P2Y14 receptor was consistent with previous results of conjugation of UDPGA derivatives to PAMAM carriers (5). Dendrimer 17, which contained only the ligand of the P2Y14 receptor (i.e., 31 nucleotides bound per G4 dendrimer) and not the A3AR-activating moiety, displayed an EC50 value at the P2Y14 receptor of 2.41 ± 0.77 nM. Curiously, nucleotide-containing dendrimers 8 and 17 also interacted with at least one other subtype of P2Y receptor, but nevertheless with selectivity for the P2Y14 receptor. Dendrimers 8 and 17 displayed EC50 values in activation of phospholipase C (PLC) by the human P2Y2 receptor of 1.39 ± 0.54 μM and 733 ± 223 nM (n = 3), respectively, assayed as described in stably transfected 1321N1 astrocytoma cells (34). The native agonist of the P2Y14 receptor, UDPG, also activated the human P2Y2 receptor, with an EC50 value of 10 μM (34).
Figure 4.

Activation of the Gi-coupled P2Y14 receptor stably expressed in C6 cells by the mixed nucleoside/nucleotide G4 dendrimer conjugate 8 (MRS5259) and the nucleotide G4 dendrimer conjugate 17 (MRS2949). Inhibition of forskolin (FSK)-stimulated cAMP is shown in comparison to the effects of UDPG (n = 3).
PAMAM dendrimer conjugates (9–12) of carboxylic- and amino-functionalized congeners of A3AR-selective agonists consistently displayed lower affinity in A3AR binding than did the click-linked dendrimers 6 and 7. The amide-linked conjugates included derivatives of both terminal carboxylic acid–functionalized dendrimers 9 and 10 and terminal amine-functionalized dendrimers 11 and 12, ranging from generation 2.5 to 4. The affinities of these dendrimers at the hA3AR were in the range of 12–42 nM, and the nature of the residual terminal groups of the dendrimer carrier had little effect. We did not observe the subnanomolar affinity seen in click-linked nucleoside-dendrimer conjugates. Thus, the high affinity is partially the result of the linkers and the coupling chemistry used, which in 6 and 7 produce a triazole rather than an amide linkage.
The selectivity of the amide-linked dendrimers in comparison to the A2AAR was high (×-fold, for compound): >300, 9; ≫24, 10; 327, 11; 62, 12. However, there was a substantial gain in affinity at the A1AR over the corresponding monomeric nucleosides. Thus, 10 and 11 lost selectivity for the A3AR in comparison to the A1AR. Compound 12 was reversed in its selectivity (6-fold more potent at the A1AR than at the A3AR). Conversely, we considered compound 4b to be a model nucleoside derivative for the amide-linked dendrimer derivatives 9 and 12, and it was clearly A3AR selective. Compound 4c (63-fold selective for the A3AR versus the A1AR) was a model for N6-functionalized dendrimer conjugates 10 and 11, although it was a higher homologue with 4 methylenes. Thus, both model compounds 4b and 4c were more potent and selective in binding to the A3AR than were the corresponding amide-linked dendrimers.
We determined binding at the rat A3AR expressed in CHO cells for selected compounds. The Ki values of the N6-chain derivatized, amide-linked dendrimers 10 and 11 were 62.0 ± 36.5 and 4.94 ± 1.96 nM (n = 3), respectively. The triazole-linked adenosine conjugate 7 with water-solubilizing groups displayed a Ki value of 0.08 ± 0.02 nM in binding to the rat A3AR. Thus, there was a relatively low degree of species-dependent variation in the observed A3AR affinity (4-fold greater in rat than human).
We tested the nucleoside conjugates in a functional assay at the A3AR (inhibition of forskolin-stimulated cAMP production in A3AR-expressing CHO cells) as shown in Table 2. These multivalent derivatives displayed A3AR agonist properties with varying degrees of maximal inhibition of forskolin-stimulated cAMP production at 0.1–10 μM (concentration selected to be in >100-fold excess of the Ki value for each). In general, the relative efficacy was similar to that of NECA (1 μM), taken as a reference standard. The dendrimer control was inactive. The EC50 value of the amide-linked adenosine conjugate 11 was 2.93 ± 0.37 nM, determined with a full curve in CHO cells stably expressing the human A3AR (Figure 5). Conjugate 11 was a full agonist in the A3AR-mediated inhibition of cAMP production. We also determined EC50 values for cAMP inhibition with full curves for the triazole-linked adenosine conjugate with water-solubilizing groups 7 (0.12 nM) and 8 (1.36 nM).
Table 2.
Maximal efficacy of (N)-methanocarba adenosine derivatives in a functional assay at the A3AR.
| Compound | Concentration (μM) | % Inhibition of cAMP accumulationa at the hA3AR |
|---|---|---|
| IB-MECA | 1.0 | 99±6 |
| Cl-IB-MECA | 1.0 | 97 |
| NECA | 1.0 | 100 |
| 3a | 10 | 60.2±17.0 |
| 4ab | 10 | 93.6±17.7 |
| 5 | 1.0 | 1.7±1.4 |
| 6 | 0.1 | 98.7±14.3 |
| 7 | 0.1 | 91.4±20.2 |
| 8 | 0.1 | 111±20 |
| 9c | 1.0 | 88.2±15.1 |
| 10 | 1.0 | 91.3±12.3 |
| 11 | 1.0 | 101±10 |
| 12 | 0.1 | 92.1±21.2 |
The efficacy at the human A3AR was determined by inhibition of forskolin-stimulated cyclic AMP production in AR-transfected CHO cells, as described in the text. Percent inhibition is shown at the indicated concentration, in comparison to the maximal effect of a full agonist NECA at 1 μM. Data are expressed as mean ± standard error (n = 3–5). ND, not determined.
Values from Tosh et al. (21).
EC50 values were determined with full curves for compounds 7 (0.12 ± 0.03 nM), 8 (1.36 nM), and 11 (2.93 ± 0.37 nM).
Figure 5.

Activation of the Gi-coupled A3AR stably expressed in CHO cells by 11. The agonist effect measured is inhibition of forskolin-stimulated cAMP production, in comparison to the maximal effect of a full agonist NECA at 1 μM. Curve is representative of three determinations.
DISCUSSION
We used the Cu(I)-catalyzed alkyne-azide click reaction for linking functionalized congeners of AR ligands (21,35) to other moieties, specifically PAMAM dendrimer derivatives. In this study, we compared different chemical strategies for tethering GPCR ligands to macromolecular carriers. Dendrimer derivatives prepared by click chemistry were particularly potent and selective in binding to the A3AR. Multivalent nucleoside-dendrimer conjugates 6 and 7 showed much greater A3AR affinity than the corresponding monomer, the model nucleoside 4a. Furthermore, amide-linked nucleoside conjugates were considerably weaker than click-linked conjugates in binding to the A3AR. This illustrates the dramatic effect that both the multivalency and the intermediate structures contained within the functionalized chain can have on interaction with the target receptor (35). A comparison with various amide-linked dendrimers showed that the triazole linkage had a general A3AR affinity–enhancing effect. Click chemistry was previously used to link small molecules to dendrimeric structures, but ours was the first application for studying PAMAM dendrimer derivatives of small-molecular GPCR ligands. Recently, peptide ligands were conjugated to dendrimers by click chemistry for activation of a GPCR (18).
Other studies have focused on PAMAM dendrimers as reversible drug carriers, for example, to deliver anticancer drugs that dissociated at the site of action (36). Our approach derivatizes a small molecule but allows it to remain active while covalently attached to the carrier (35). The binding sites of the receptors are accessible from the extracellular medium. Thus, activation of the desired receptor would not benefit from either drug release or cellular internalization of the dendrimer. The flexible dendrimer conjugate is available to spread over the cell surface and to adopt a conformation that can bridge multiple binding sites (37).
The efficiency of the click reaction readily allowed complete substitution in the case of one of the dendrimer derivatives, i.e., 6. In previous experiences with amide coupling of functionalized nucleosides, we encountered difficulty in the complete substitution of amine-terminated dendrimers (3, 4). Click chemistry proved to be a more efficient coupling technique and allowed orthogonal reaction control over the coupling chemistry when two distinct receptor ligands were bound to the same dendrimer carrier (as in 8).
This study presents the first example of targeting two different GPCRs with the same dendrimer conjugate, intended for activation of heteromeric aggregates of GPCRs. Each ligand provided selectivity for a particular GPCR subtype, and the overall selectivity of the conjugate was a function of the union of the sets of selectivity of the attached nucleosides. Specifically, the dendrimer conjugate 8 contained ligands for both the A3AR and the P2Y14 receptor. The principle of attachment of UDPG derivatives to PAMAM dendrimers to enhance potency was introduced previously (5). This bifunctional dendrimer potently activated both A3 and P2Y14 receptors, although not to the exclusion of other ARs.
The bifunctional dendrimer 8 and its nucleotide-dendrimer precursor 17 both activated one other P2Y receptor examined, the hP2Y2 receptor, with intermediate potency. UDPG itself activates the hP2Y2 receptor with an EC50 of 10 μM for stimulation of PLC in stably transfected 1321N1 cells (34). Therefore, the potency at both subtypes of P2Y receptors was much greater in the dendrimer conjugates than in the monomer, but the desired selectivity for the P2Y14 receptor was maintained. Thus, both covalently tethered GPCR ligands, which were previously shown to be suitable functionalized congeners for coupling to carriers, retained their activity as the mixed dendrimer conjugate 8. This conjugate had reduced A3AR affinity in the presence of tethered nucleotide moieties, but not reduced P2Y14 potency due to the tethered nucleoside moieties. This is the first example of targeting two different GPCRs with the same dendrimer conjugate, which is intended for activation of heteromeric aggregates of GPCRs.
Many examples exist of two small molecules linked with a spacer group to make binary conjugates for activation of two different GPCRs (35, 38, 39). Some researchers have systematically varied the length of the spacer to achieve the correct fit to the two adjacent binding sites. This multivalent approach was taken in part because of uncertainty about the geometric orientation of protomer binding sites in a GPCR dimer. Flexible dendritic carriers facilitate empirical testing of a variety of spatial orientations of the same conjugate. This obviates the need to probe the optimal geometry a priori of a single spacer chain to achieve simultaneous binding to two sites.
Both A3 and P2Y14 receptors are distributed in the immune system and appear to occur on the same cell, as was shown for RBL-2H3 mast cells (27). Further experiments will be needed to determine whether heteromers composed of A3 and P2Y14 receptors exist and whether this conjugate is capable of bridging the binding sites on both protomers. This approach of mixing receptor ligands on the same dendrimer carrier can be extended to many other combinations of GPCRs.
We know that GPCRs tend not to be evenly or randomly distributed over the surface of a given cell; we demonstrated this for the A3AR expressed heterologously in CHO cells (4). In general, GPCRs may exist as complexes with other receptors and associated proteins. The receptors to which a given GPCR pairs or forms a higher-order aggregate can dramatically affect its pharmacology, and the binding of agonist to both protomers does not have equivalent pharmacological effects (40). We lack effective ligand tools for separating the effects of one receptor dimeric combination from another. This approach of mixing ligands for two different receptors (35, 38, 39, 41) promises to be a means of achieving selectivity for receptors in a given tissue, based on association or aggregation of GPCRs, in comparison to another tissue in which the same receptor may be alternatively paired. At this early stage, we do not know the topological requirements for selectively targeting these combinations of receptors, because we are just beginning to detect their existence. The multivalent binding to other types of cell surface receptors has been explored through structural modification of the carrier polymer (42).
Multivalent conjugates of GPCR ligands provide quantitatively and qualitatively different pharmacological properties than do monomers. Prosthetic groups for targeting or therapeutic purposes or reporter groups for diagnostic purposes might be introduced. Specific combinations of receptor ligands on the same dendrimer carrier might achieve synergistic effects and prove useful for disease treatment, such as inflammatory conditions, to which the current example of adenosine and P2Y receptor ligands apply. Various combinations may be proposed for targeted therapeutic applications, but the linking strategy to achieve the desired potency and selectivity still requires development. We are working on prototypical examples that could be applied to other combinations of dendrimer-bound GPCR ligands.
Supplementary Material
Acknowledgments
We thank Dr. John Lloyd and Dr. Noel Whittaker (NIDDK) for the mass spectral determinations. We thank Dr. Bruce Liang (Univ. of Connecticut) for helpful discussion. This research was supported by the Intramural Research Program of the NIH, NIDDK. Research conducted at the Center for Nanophase Materials Sciences at Oak Ridge National Laboratory is sponsored by the Scientific User Facilities Division, Office of Basic Energy Sciences, U.S. Department of Energy.
Abbreviations
- AR
adenosine receptor
- DMSO
dimethylsulfoxide
- EDC
N-ethyl-N′-dimethylaminopropylcarbodiimide
- GPCR
G protein–coupled receptor
- IBMX
3-isobutyl-1-methylxanthine
- AR
adenosine receptor
- cAMP
adenosine 3′,5′-cyclic phosphate
- CHO
Chinese hamster ovary
- IB-MECA
N6-(3-iodobenzyl)-5′-N-methylcarboxamidoadenosine
- DMEM
Dulbecco’s modified Eagle’s medium
- EDTA
ethylenediaminetetraacetic acid
- GPCR
G protein–coupled receptor
- HATU
2-(1H-7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyl uronium hexafluorophosphate methanaminium
- HEK
human embryonic kidney
- I-AB-MECA
2-[p-(2-carboxyethyl)phenyl-ethylamino]-5′-N-ethylcarboxamidoadenosine
- MS
mass spectrometry
- NECA
5′-N-ethylcarboxamidoadenosine
- NMR
nuclear magnetic resonance
- DIPEA
diisopropylethylamine
- DMF
N,N-dimethylformamide
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- PAMAM
polyamidoamine
- PLC
phospholipase C
- UDPG
uridine-5′-diphosphoglucose
- UDPGA
uridine-5′-diphosphoglucuronic acid.
Footnotes
Supporting Information Available: 1H NMR and mass spectra of representative compounds 6 and 11. This material is available free of charge via the Internet at http://pubs.acs.org/BC.
References
- 1.Nwe K, Brechbiel MW. Growing applications of “click chemistry” for bioconjugation in contemporary biomedical research. Cancer Biother Radiopharm. 2009;24:289–302. doi: 10.1089/cbr.2008.0626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tomalia DA, Reyna LA, Svenson S. Dendrimers as multi-purpose nanodevices for oncology drug delivery and diagnostic imaging. Biochem Soc Transact. 2007;35:61–67. doi: 10.1042/BST0350061. [DOI] [PubMed] [Google Scholar]
- 3.Kim Y, Hechler B, Klutz A, Gachet C, Jacobson KA. Toward multivalent signaling across G protein-coupled receptors from poly(amidoamine) dendrimers. Bioconjugate Chem. 2008;19:406–411. doi: 10.1021/bc700327u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Klutz AM, Gao ZG, Lloyd J, Shainberg A, Jacobson KA. Enhanced A3 adenosine receptor selectivity of multivalent nucleoside-dendrimer conjugates. J Nanobiotechnol. 2008;6:12. doi: 10.1186/1477-3155-6-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Das A, Zhou Y, Ivanov AA, Carter RL, Harden TK, Jacobson KA. Enhanced potency of nucleotide-dendrimer conjugates as agonists of the P2Y14 receptor: Multivalent effect in G protein-coupled receptor recognition. Bioconjugate Chem. 2009;20:1650–1659. doi: 10.1021/bc900206g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jacobson KA, Gao ZG. Adenosine receptors as therapeutic targets. Nature Rev Drug Disc. 2006;5:247–264. doi: 10.1038/nrd1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gessi S, Merighi S, Varani K, Cattabriga E, Benini A, Mirandola P, Leung E, Mac Lennan S, Feo C, Baraldi S, Borea PA. Adenosine receptors in colon carcinoma tissues and colon tumoral cell lines: focus on the A3 adenosine subtype. J Cell Physiol. 2007;211:826–836. doi: 10.1002/jcp.20994. [DOI] [PubMed] [Google Scholar]
- 8.Yang H, Avila MY, Peterson-Yantorno K, Coca-Prados M, Stone RA, Jacobson KA, Civan MM. The cross-species A3 adenosine-receptor antagonist MRS 1292 inhibits adenosine-triggered human nonpigmented ciliary epithelial cell fluid release and reduces mouse intraocular pressure. Current Eye Res. 2005;30:747–754. doi: 10.1080/02713680590953147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hua X, Chason KD, Fredholm BB, Deshpande DA, Penn RB, Tilley SL. Adenosine induces airway hyperresponsiveness through activation of A3 receptors on mast cells. J Allergy Clin Immunol. 2008;122:107–113. doi: 10.1016/j.jaci.2008.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fishman P, Jacobson KA, Ochaion A, Cohen S, Bar-Yehuda S. The anti-cancer effect of A3 adenosine receptor agonists: A novel, targeted therapy. Immun Endoc Metab Agents in Med Chem. 2007;7:298–303. doi: 10.2174/187152207781369878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Silverman MH, Strand V, Markovits D, Nahir M, Reitblat T, sMolad Y, Rosner I, Rozenbaum M, Mader R, Adawi M, Caspi D, Tishler M, Langevitz P, Rubinow A, Friedman J, Green L, Tanay A, Ochaion A, Cohen S, Kerns WD, Cohn I, Fishman-Furman S, Farbstein M, Yehuda SB, Fishman P. Clinical evidence for utilization of the A3 adenosine receptor as a target to treat rheumatoid arthritis: Data from a phase II clinical trial. J Rheumatol. 2008;35:41–48. [PubMed] [Google Scholar]
- 12.Zheng J, Wang R, Zambraski E, Wu D, Jacobson KA, Liang BT. A novel protective action of adenosine A3 receptors: Attenuation of skeletal muscle ischemia and reperfusion injury. Am J Physiol; Heart and Circ Physiol. 2007;293:3685–3691. doi: 10.1152/ajpheart.00819.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang M, Hu H, Zhang X, Lu W, Lim J, Eysteinsson T, Jacobson KA, Laties AM, Mitchell CH. The A3 adenosine receptor attenuates the calcium rise triggered by NMDA receptors in retinal ganglion cells. Neurochem Int. 2010;56:35–41. doi: 10.1016/j.neuint.2009.08.011. in press. doi:10/1016/j.neuint.2009.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Melman A, Gao ZG, Kumar D, Wan TC, Gizewski E, Auchampach JA, Jacobson KA. Design of (N)-methanocarba adenosine 5′-uronamides as species-independent A3 receptor-selective agonists. Bioorg Med Chem Lett. 2008;18:2813–2819. doi: 10.1016/j.bmcl.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tosh DK, Chinn M, Ivanov AA, Klutz AM, Gao ZG, Jacobson KA. Functionalized congeners of A3 adenosine receptor-selective nucleosides containing a bicyclo[3.1.0]hexane ring system. J Med Chem. 2009;52:7580–7592. doi: 10.1021/jm900426g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Agard NJ, Baskin JM, Prescher JA, Lo A, Bertozzi CR. A comparative study of bioorthogonal reactions with azides. ACS Chem Biol. 2006;1:644–648. doi: 10.1021/cb6003228. [DOI] [PubMed] [Google Scholar]
- 17.Camponovo J, Hadad C, Ruiz J, Cloutet E, Gatard S, Muzart J, Bouquillon S, Astruc D. “Click” glycodendrimers containing 27, 81, and 243 modified xylopyranoside termini. J Org Chem. 74:5071–5074. doi: 10.1021/jo900554b. [DOI] [PubMed] [Google Scholar]
- 18.Yim C-B, Boerman OC, de Visser M, de Jong M, Dechesne AC, Rijkers DTS, Liskamp RMJ. Versatile conjugation of octreotide to dendrimers by cycloaddition (“Click”) chemistry to yield high-affinity multivalent cyclic peptide dendrimers. Bioconjugate Chem. 2009;20:1323–1331. doi: 10.1021/bc900052n. [DOI] [PubMed] [Google Scholar]
- 19.van Dijk M, Rijkers DTS, Liskamp RMJ, van Nostrum CF, Hennink WE. Synthesis and applications of biomedical and pharmaceutical polymers via click chemistry methodologies. Bioconjugate Chem. 2009;20:2001–2016. doi: 10.1021/bc900087a. [DOI] [PubMed] [Google Scholar]
- 20.Abbracchio MP, Burnstock G, Boeynaems JM, Barnard EA, Boyer JL, Kennedy C, Fumagalli M, King BF, Gachet C, Jacobson KA, Weisman GA. International Union of Pharmacology. Update of the P2Y G protein-coupled nucleotide receptors: from molecular mechanisms and pathophysiology to therapy. Pharmacol Rev. 2006;58:281–341. doi: 10.1124/pr.58.3.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tosh DK, Chinn M, Yoo L, Kang DW, Luecke H, Gao ZG, Jacobson KA. 2-Dialkynyl derivatives of (N)-methanocarba nucleosides: “Clickable” A3 adenosine receptor-selective agonists. Bioorg Med Chem. 2010 doi: 10.1016/j.bmc.2009.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goddard-Borger ED, Stick RV. An efficient, inexpensive, and shelf-stable diazotransfer reagent: Imidazole-1-sulfonyl azide hydrochloride. Org Lett. 2007;9:3797–3800. doi: 10.1021/ol701581g. [DOI] [PubMed] [Google Scholar]
- 23.Fricks I, Maddiletti S, Carter R, Lazarowski ER, Nicholas RA, Jacobson KA, Harden TK. UDP is a competitive antagonist at the human P2Y14 receptor and a full agonist at the rat P2Y14 receptor. J Pharm Exp Therap. 2008;325:588–594. doi: 10.1124/jpet.108.136309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harden TK, Scheer AG, Smith MM. Differential modification of the interaction of cardiac muscarinic cholinergic and beta-adrenergic receptors with a guanine nucleotide binding component(s) Mol Pharmacol. 1982;21:570–580. [PubMed] [Google Scholar]
- 25.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 26.Nordstedt C, Fredholm BB. A modification of a proteinb inding method for rapid quantification of cAMP in cell-culturesupernatants and body fluid. Anal Biochem. 1990;189:231–234. doi: 10.1016/0003-2697(90)90113-n. [DOI] [PubMed] [Google Scholar]
- 27.Gao ZG, Ding Y, Jacobson KA. UDP-glucose acting at P2Y14 receptors is a mediator of mast cell degranulation. Biochem Pharmacol. 2010;79:873–879. doi: 10.1016/j.bcp.2009.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gomez MV, Guerra J, Velders AH, Crooks RM. NMR Characterization of fourth-generation PAMAM dendrimers in the presence and absence of palladium dendrimer-encapsulated nanoparticles. J Am Chem Soc. 2009;131:341–350. doi: 10.1021/ja807488d. [DOI] [PubMed] [Google Scholar]
- 29.Klotz KN, Lohse MJ, Schwabe U, Cristalli G, Vittori S, Grifantini M. 2-Chloro-N6-[3H]cyclopentyladenosine ([3H]CCPA)-a high affinity agonist radioligand for A1 adenosine receptors. Naunyn Schmiedebergs Arch Pharmacol. 1989;340:679–683. doi: 10.1007/BF00717744. [DOI] [PubMed] [Google Scholar]
- 30.Jarvis MF, Schutz R, Hutchison AJ, Do E, Sills MA, Williams M. [3H]CGS 21680, an A2 selective adenosine receptor agonist directly labels A2 receptors in rat brain tissue. J Pharmacol Exp Ther. 1989;251:888–893. [PubMed] [Google Scholar]
- 31.Olah ME, Gallo-Rodriguez C, Jacobson KA, Stiles GL. 125I-4-Aminobenzyl-5′-N-methylcarboxamidoadenosine, a high affinity radioligand for the rat A3 adenosine receptor. Mol Pharmacol. 1994;45:978–982. [PMC free article] [PubMed] [Google Scholar]
- 32.Englert M, Quitterer U, Klotz KN. Effector coupling of stably transfected human A3 adenosine receptors in CHO cells. Biochem Pharmacol. 2002;64:61–65. doi: 10.1016/s0006-2952(02)01071-7. [DOI] [PubMed] [Google Scholar]
- 33.Jacobson KA, Park KS, Jiang J-l, Kim YC, Olah ME, Stiles GL, Ji Xd. Pharmacological characterization of novel A3 adenosine receptor-selective antagonists. Neuropharmacology. 1997;36:1157–1165. doi: 10.1016/s0028-3908(97)00104-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ko H, Das A, Carter RL, Fricks IP, Zhou Y, Ivanov AA, Melman A, Joshi BV, Kovác P, Hajduch J, Kirk KL, Harden TK, Jacobson KA. Molecular recognition in the P2Y14 receptor: Probing the structurally permissive terminal sugar moiety of uridine-5′-diphosphoglucose. Bioorg Med Chem. 2009;17:5298–5311. doi: 10.1016/j.bmc.2009.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jacobson KA. Functionalized congener approach to the design of ligands for G protein–coupled receptors (GPCRs) Bioconjugate Chem. 2009;20:1816–1835. doi: 10.1021/bc9000596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kojima C, Kono K, Maruyama K, Takagishi T. Synthesis of polyamidoamine dendrimers having poly(ethyleneglycol) grafts and their ability to encapsulate anticancer drugs. Bioconjugate Chem. 2000;11:910–917. doi: 10.1021/bc0000583. [DOI] [PubMed] [Google Scholar]
- 37.Ivanov AA, Jacobson KA. Molecular modeling of a PAMAM- CGS21680 dendrimer bound to an A2A adenosine receptor homodimer. Bioorg Med Chem Lett. 2008;18:4312–4315. doi: 10.1016/j.bmcl.2008.06.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Portoghese PS, Ronsisvalle G, Larson DL, Yim CB, Sayre LM, Takemori AE. Opioid agonist and antagonist bivalent ligands as receptor probes. Life Sci. 1982;31:1283–1286. doi: 10.1016/0024-3205(82)90362-9. [DOI] [PubMed] [Google Scholar]
- 39.Morphy R, Rankovic Z. Designed multiple ligands. An emerging drug discovery paradigm. J Med Chem. 2005;48:6523–6565. doi: 10.1021/jm058225d. [DOI] [PubMed] [Google Scholar]
- 40.Han Y, Moreira IS, Urizar E, Weinstein H, Javitch JA. Allosteric communication between protomers of dopamine class A GPCR dimers modulates activation. Nature Chem Biol. 2009;5:688–695. doi: 10.1038/nchembio.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Soriano A, Ventura R, Molero A, Hoen R, Casadó V, Cortés A, Fanelli F, Albericio F, Lluís C, Franco R, Royo M. Adenosine A2A receptor-antagonist/dopamine D2 receptor-agonist bivalent ligands as pharmacological tools to detect A2A-D2 receptor heteromers. J Med Chem. 2009;52:5590–5602. doi: 10.1021/jm900298c. [DOI] [PubMed] [Google Scholar]
- 42.Cairo CW, Jason E, Gestwicki JE, Kanai M, Kiessling LL. Control of multivalent interactions by binding epitope density. J Am Chem Soc. 2002;124:1615–1619. doi: 10.1021/ja016727k. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.