

Abstract
The methylated DNA immunoprecipitation microarray (MeDIP-chip) is a genome-wide, high-resolution approach to detect DNA methylation in whole genome or CpG (cytosine base followed by a guanine base) islands. The method utilizes anti-methylcytosine antibody to immunoprecipitate DNA that contains highly methylated CpG sites. Enriched methylated DNA can be interrogated using DNA microarrays or by massive parallel sequencing techniques. This combined approach allows researchers to rapidly identify methylated regions in a genome-wide manner, and compare DNA methylation patterns between two samples with diversely different DNA methylation status. MeDIP-chip has been applied successfully for analyses of methylated DNA in the different targets including animal and plant tissues (1, 2). Here we present a MeDIP-chip protocol that is routinely used in our laboratory, illustrated with specific examples from MeDIP-chip analysis of breast cancer cell lines. Potential technical pitfalls and solutions are also provided to serve as workflow guidelines.
Keywords: DNA methylation, epigenetics, MeDIP-chip, microarray, cancer
1. Introduction
Epigenetic modification involves DNA methylation, covalent modification of histones and small inhibitory RNA molecules known as microRNAs (miRNAs) (3). DNA methylation is a heritable, enzyme-induced modification without changing the nucleotide sequence of the DNA base pairs. DNA methylation involves transfer of a methyl-group to the 5-carbon on the cytosine in a CpG dinucleotide via DNA methyltransferases (DNMT1, DNMT3A, and DNMT3B) (4). Most of the CpG dinucleotides are unevenly distributed within the genome and regions high in these dinucleotides termed CpG islands. The haploid human genome consists of 29,848,753 CpGs and about 6% of all CpGs are located in the CpG islands (5). These islands are frequently located in the 5′-untranslated region and the first exon of approximately 60% of all genes. Methylation of CpG islands affects the transcriptional activation of genes. It is generally accepted that a high level of promoter CpG island methylation results in gene silencing (6). In the normal genome, DNA methylation is essential for proper development, chromosomal integrity, maintenance of gene expression states, and X chromosome inactivation (7, 8). In primary human tumors, methylation patterns are severely altered. This includes hypermethylation of CpG islands and genome-wide hypomethylation (9, 10). Because DNA methylation has significant effects on gene function and expression, detection of DNA methylation becomes an active area of research for the understanding of normal biological processes and tumorigenesis.
There are several methods available for the determination of methylation patterns and the quantitative assessment of methylation levels in sample tissues (11). These include methods for interrogating the combined methylation status of several CpG sites in a single gene (methylation-specific polymerase chain reaction or MSP and MethyLight; 12, 13), methods for interrogating methylation status of individual CpG sites present in a gene (combined bisulfite restriction analysis or COBRA and methylation-specific single-nucleotide primer extension or MS-SNuPE; 14, 15), methods for interrogating multiple CpG loci in many genes (methylation-specific oligonucleotide microarray or MSO microarray; 16), and methods for high-throughput, genome-wide epigenetic analysis (differential methylation hybridization or DMH; 17).
A large number of currently available techniques used for studying global methylation differences involve the use of methylation sensitive restriction enzyme(s) thereby limiting these approaches to profile genomic regions containing these restriction site motifs. In this regard, the methylated DNA immunoprecipitation (MeDIP)-chip approach is less biased and therefore more appealing (2, 18). The general strategy for the MeDIP-chip procedure is outlined in Fig. 10.1. Genomic DNA is sheared to low molecular weight fragments (average 400 bp) (see Note 1). Methylated DNAs are immunoprecipitated with the anti-methyl-cytosine antibody, and may be PCR-amplified if source material is limited (see Note 2). Input and methylated DNA can be subsequently labeled with fluorescent dyes Cy3 (green) and Cy5 (red), pooled, denatured, and hybridized to a microarray slide containing all the annotated human CpG islands (n = 27,800) or other whole genome or promoter microarray designs (see Note 3). The slide is scanned using a GenePix 4000B scanner and each image is analyzed with the GenePix Pro 6.0 image analysis software. MeDIP-chip has proven to be an efficient and robust method for analyzing DNA methylation at a genome-wide scale. Recently, several different companies have provided array designs to fit customer needs including whole-genome survey sets and promoter sets so that you can obtain comprehensive data from your methylation samples.
Fig. 10.1.

Overview of the MeDIP protocol. The genomic DNA is sonicated into small fragments and then immunoprecipitated with an antibody directed against 5-methylcy-tidine. Input DNA and methylated DNA (black circle) can be differentially labeled with Cy5 (red) and Cy3 (green) and co-hybridized as a two-color experiment on microarrays, or used for data validation.
2. Materials
2.1. Preparation of Genomic DNA
Qiagen QIAamp DNA Mini kit (Valencia, CA).
2.2. Sonication of Genomic DNA
Bioruptor model 200 (Diagenode, Sparta, NJ).
Microcentrifuge (Thermo Scientific, Waltham, MA).
Water bath at 37°C, 50°C and 95°C (Thermo Scientific).
2.3. Immunoprecipitation of methylated DNA (MeDIP)
Mouse monoclonal 5-methylcytidine antibody, MAb-5MECYT-500 (Diagenode, Sparta, NJ).
Dynabeads Protein G (Invitrogen, Carlsbad, CA).
Phase lock, heavy tubes (Eppendorf, Westbury, NY).
Magnetic rack for 1.5-ml tubes (Invitrogen).
Rotating/rocking platform (Fisher Scientific, Pittsburgh, PA).
Proteinase K solution (Invitrogen).
10x IP buffer: 100 mM Na-Phosphate, pH 7.0, 1.4 M NaCl, 0.5% Triton X-100.
TE buffer: 10 mM Tris-HCl, pH 7.5, 1 mM EDTA.
Digestion buffer: 50 mM Tris, pH 8.0, 10 mM EDTA, 0.5% SDS.
2.4. Purification of Methylated DNA
Glycogen (Roche, Indianapolis, IN).
Phenol:chloroform:isoamyl alcohol (Fluka, St. Louis, MO).
5 M NaCl.
TE buffer: 10 mM Tris-HCl, pH 7.5, 1 mM EDTA.
NanoDrop ND-3300 Fluorospectrometer (Thermo Scientific).
Thermocycler (Applied Biosystems, Foster City, CA).
2.5. Analysis by CpG Island Microarrays
Cy3/Cy5 labeling kit (GE Healthcare, Piscataway, NJ).
Agilent Technologies Human CpG island microarray G4492A (Santa Clara, CA).
2.6. Data Validation (Components for a Combined Bisulfite Restriction Analysis or COBRA)
Universal Methylated DNA Standard (Zymo Research, Orange, CA) or prepare methylated positive control using SssI methylase (New England Biolabs, Ipswich, MA).
EZ DNA Methylation-Gold Kit (Zymo Research).
PCR primers reflecting the bisulfite-converted genome sequence of region of interest (Illumina, San Diego, CA).
AmpliTaq Gold DNA Polymerase (Applied Biosystems, Foster City, CA).
Methylation sensitive restriction enzyme such as BstUI (New England Biolabs).
Agarose (Fisher Scientific).
3. Methods
3.1. Preparation of Genomic DNA
3.2. Sonication of Genomic DNA
Sonicate purified genomic DNA using Diagenode Bioruptor 200, as follows: Dilute 20 μg genomic DNA in 300–450 μl IP buffer (use 10x IP buffer to adjust the buffer concentration to 1x strength) in a 1.5 ml eppendorf tube. As we have observed inconsistencies in DNA fragmentation patterns between runs, we resort to careful control of the sonication conditions. If that is not your experience, you can skip the following section.
Prior to the beginning of the sonication process, remove all the ice particles using a strainer and add a predetermined amount of fresh ice. Bring the water level to a preset mark.
Turn sonicator on 30 s then off 30 s for 20 times. We replenish ice after the first two cycles and replace both fresh ice and ice-cold water after 4th cycle.
Load 4 μl on a 2% agarose gel to verify fragment size of DNA (mean size should be 200–800 bp; average 400 bp) (see Note 6 and Fig. 10.2).
Aliquot 100–150 μl sonicated DNA to three eppendorf tubes; each tube contains 6–7 μg DNA.
Fig. 10.2.
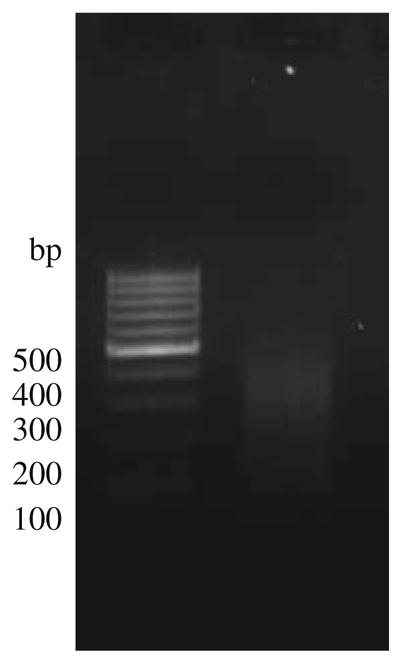
The gel represents sonicated genomic DNA isolated from MCF-7 run on a 2% agarose gel. Most of the sheared fragments have a size between 200 and 800 bp.
3.3. Immunoprecipitation of Methylated DNA (MeDIP)
Heat-denature the samples (DNA) for 10 min in boiling water, and immediately cool on ice for 10 min.
Save one tube of the heat-denatured DNA, and store at −20°C for use as an input control.
Add 10 μg of antibody (monoclonal mouse anti-5-methyl cytidine) (see Notes 7–8).
Incubate the mixture overnight on a rotating platform at 4°C.
Add 50 μl of Dynabeads Protein G to the DNA-antibody mixture.
Incubate 2 h on a rotating platform at 4°C.
The washing steps are simplified by the use of an Invitrogen magnetic rack. Once the magnetized Dynabeads are tightly held by the magnet in the rack, remove unbound DNA-antibody mixture. Remove tubes from the rack. Add 1 ml of 1x IP buffer and flick gently to wash. Replace the tubes to the rack and repeat this washing step three times.
Resuspend the beads in 250 μl digestion buffer.
Add 5 μl Proteinase K (20 mg/ml stock).
Incubate overnight on a rotating platform at 50 °C.
3.4. Purification of Methylated DNA
For 200 μl volume, add 400 μl phenol:chloroform:isoamyl alcohol, vortex for 30 s. For a clean separation of the two phases, employ a 2 ml heavy Eppendorf phaselock tube whereby the organic phase is located above the gel and can be cleanly removed (follow instructions provided by Eppendorf).
Transfer the aqueous supernatant to a new tube.
Add 1.5 μl glycogen (20 mg/ml stock) and mix well.
Add 16 μl 5 M NaCl and then 800 μl 100% ethanol.
Precipitate in −80°C freezer for 30 min or overnight.
Centrifuge at 20,000g for 10 min at 4°C. Carefully remove the supernatant. Wash pellets by adding 500 μl of 80% EtOH. Vortexing to resuspend pellet and spin again at 20,000g for 5 min at 4°C.
Resuspend the DNA pellet in 70 μl TE buffer.
Measure DNA concentration (see Note 9).
Save 5 μl of immunoprecipitated DNA to check for enrichment in the immunoprecipitation samples using gene-specific qPCR (see Note 10).
3.5. Analysis by CpG Island Microarrays
Cy3/Cy5 labeling of MeDIP-enriched and input DNA, array hybridization, and array washing were performed using a previously published protocol (19).
An example of a commercially available microarray suitable for MeDIP-chip experiment is described here. Agilent Technologies Human CpG island microarray (G4492A) contains 45–60 mers oligonucleotide probes tiling all the CpG islands as defined by the University of California-Santa Cruz Genome Browser. This slide format has 195,000 probes covering a total of 27,800 human CpG islands. The average spacing between probes is 116 bp. Probes are selected for uniqueness within the genome (repeat sequence masked) and predicted hybridization properties according to standard Agilent probe design criteria was used in the design with the exception of theoretical Tm window restriction. This restriction cannot be used due to high GC content in CpG islands. As such, the standard Tm restriction is lifted to achieve the appropriate spacing.
Other appropriate microarrays for MeDIP analysis include NimbleGen HD2 Whole Genome Tiling Array Sets and the University Health Network HCG12K Human CpG Island Microarray (for additional designs, see Note 3).
As the Agilent Cp Gisland microarray is our plat form of choice, we use the Agilent DNA Analytics program (version 4.0.76) to identify regions that are enriched by the 5-methylcytidine antibody. For researchers who do not have quick access to biostatisticians to assist in data analysis, this is the most reasonable approach.
3.6. Data Validation
The combined bisulfite restriction analysis (COBRA) is a preferred way to validate MeDIP-chip data. This protocol is as much a gold standard in DNA methylation analysis as bisulfite sequencing (14). The steps that follow represent useful insight for smooth adaptation of this protocol.
Positive methylation control can be purchased but can also be prepared by following the protocol that comes with SssI methylase. Because the methyl-donor SAM is quite labile, we spike the reaction mixture with another dose of SAM half way through the recommended reaction time.
Blood DNA and sperm DNA are good choices for negative control used in COBRA as these DNA are known to lack DNA methylation. Blood DNA, though not as devoid of methylation as sperm DNA, is far easier to work with in the subsequent bisulfite conversion step and the PCR step.
Bisulfite conversion of MeDIP DNA, positive and negative DNA is achieved by using the Easy DNA Methylation kit. For most validation targets, bisulfite converted DNA derived from this straight forward approach is sufficient to yield methylation status. There are, however, regions in the genome that are less accessible to the chemicals used in the bisulfite modification reaction. This will require the more tedious protocol described elsewhere (14).
Bisulfite PCR products from the region of interest are divided into two equal portions. One portion is restricted by methylation-sensitive enzyme and the other is mock-restricted. Both portions are then purified and run out on agarose gel (PAGE gel is used if better separation is desired). This scheme provides an uncut PCR fragment with intensity approximate that of a sample devoid of DNA methylation. When a sample has methylation sites close to one end of the PCR product or just having low level of methylation, the disappearance of the unrestricted band in comparison to the mock-restricted sample will add confidence to the methylation call.
A representation of validation is depicted in Fig. 10.3. Herein we validate promoter methylation found in tamoxifen-resistant MCF-7 cells (OHT) but not in regular MCF-7 cells. In this figure, positive control was derived from genomic DNA methylated by SssI enzyme prior to bisulfite conversion to retain this methylation information. The negative control was bisulfite converted blood DNA known to lack DNA methylation. Control and sample DNA were interrogated by Bst UI to reveal methylated CpG dinucleotides within the region bracketed by the PgR promoter primers. The presence of restricted fragments with a concomitant decrease in the top unrestricted band signified the presence of methylation in the PgR promoter.
Other assays suitable for data validations include quantitative methylation-specific PCR (qMSP, 12) and the time- and resources-expensive bisulfite sequencing.
Fig. 10.3.

COBRA validation of PgR promoter methylation in OHT (tamoxifen-resistant MCF-7 cells) but not in untreated MCF-7 cells. Positive control (genomic DNA methylated with SssI) showed 100% in the interrogated region. Negative control (blood DNA) should little to no restricted bands by BstUI (not methylated in this region). The parental MCF-7 cells harbored no methylation in this region whereas tamoxifen-resistant OHT cells showed signs of methylation in the PgR promoter, potentially signifying the onset of gene silencing.
Footnotes
Different researchers have suggested widely varying values from 200 to 1000 bp. For example, a study reported a fragment size of 600 bp (20) whereas another reported a lower fragment size of 300–600 bp (21) in their MeDIP protocol. In the protocol listed by a commercial company (NimbleGen System, Inc) a much wider fragment size range (200–1000 bp) is cited. The protocols of chromatin immunoprecipitation (ChIP) are very similar to MeDIP. The average fragment size after sonication is around 200–600 bp (21, 22).
The amount of immunoprecipitated DNA is typically very small. One solution is to perform multiple pull down experiments. If source material is limited, one can amplify the pull down DNA prior to labeling and microarray analysis. There are several approaches described in the literature for this step, they include whole genome amplification using the Sigma WGA2 kit, ligation-mediated PCR (LM-PCR; http://www.chiponchip.org/protocol_itm3.html) or T7 amplication (23). After amplifying MeDIP and input samples, the resultant amplicons should be evaluated on an agarose gel to validate that their fragment size range matches the pull down DNA and input DNA. It is also pertinent that there should not be distinct banding pattern present in the amplified products.
| Company | Array designs | Total probes | Probe length |
|---|---|---|---|
| NimbleGen | Whole-Genome | 2.1 million | 50– mer |
| CpG island-Plus-Promoter | 385,000 | 50–75 mer | |
| Agilent Technologies | Human CpG island | 237,220 | 95 bp |
| Mouse CpG island | 97,652 | 95 bp | |
| Phalanx Biotech | Human One Array | 32050 | 60 mer |
| Mouse One Array | 31802 | 71 Mer |
There are several genomic DNA isolation kits commercial available now, you can choose one of them. Purity is determined by calculating the ratio of absorbance at 260 nm to absorbance at 280 nm. Pure DNA has an A260/A280 ratio of 1.8–2.0 indicating the absence of protein and an A260/230 ratio of >2.0, indicating the absence of other organic compounds such as ethanol. Impure DNA will lead to nonspecific binding and affect MeDIP pull down.
After isolation of genomic DNA from your samples, run a 1.5% agarose gel to check the quality of the DNA and make sure there is no contamination with RNA, since the antibody also can recognize 5-methylcytidine in RNA. If there is a contamination with RNA, you may see smear RNA located in the front of the gel.
Wide range of genomic DNA can be successfully fragmented by sonication (from breast progenitor cells to breast cancer cell lines). However, the efficiency of sonication varies with DNA concentration, and machine itself (probe-type sonicator produces different outcomes in comparison to Bioruptor). Therefore, it is very important to systematically check the size of the fragmented DNA. Some of the factors that will alter the fragment size such as water temperature, ice/water ratio in the sonication vessel, DNA dissolved in the water or IP buffer, duration of reset between each energy pulse, DNA concentration and volume of DNA per tube, and batch of the eppendorf tubes, etc.
MeDIP-chip is only limited to identify CpG islands that are highly methylated in genomes. This is because the antibody used in the assay can only bind to >4 nearby methylated CpG sites. Therefore, low-density methylation of CpG islands is likely not detectable by MeDIP-chip. To improve the methylation coverage by MeDIP-chip, methyl-CpG immunoprecipitation (MCIp, 24) assay is one of the alternative methods.
| Clone | Host Species | Type | Company | Quantity |
|---|---|---|---|---|
| 33D3 | Mouse | Monoclonal Ab | GenWay | 1 mg/ml |
| 33D3 | Mouse | Monoclonal Ab | Eurogentec | 1 mg/ml |
| 33D3 | Mouse | Monoclonal Ab | ProSci | 0.05 mg |
| 33D3 | Mouse | Monoclonal Ab | Calbiochem | |
| 33D3 | Mouse | Monoclonal Ab | Epigentek | 1 mg/ml |
| 33D3 | Mouse | Monoclonal Ab | Affinity BioReagents | 100 μg |
| 33D3 | Mouse | Monoclonal Ab | Santa Cruze | 50 μg/0.5 ml |
| 33D3 | Mouse | Monoclonal Ab | AbCam | 50 μg |
| 33D3 | Mouse | Monoclonal Ab | AbD SeroTec | 0.1 mg |
NanoDrop ND-3300 Fluorospectrometer uses PicoGreen dye to stain nucleic acid for quantitating double-stranded DNA (dsDNA). The PicoGreen assay provides a highly sensitive means of dsDNA quantiation with minimal consumption of sample. The ND-3300 fluorospectrometer has demonstrated a detection range for dsDNA bound with PicoGreen reagent of 1 –1000 pg/μml. Compared to the NanoDrop ND-1000, ND-3300 provides more accurate readout especially for the immunoprecipitation DNA samples.
In order to evaluate the enrichment of methylated DNA after MeDIP, we use quantitative PCR to measure the change of Ct between immunoprecipitation DNA and input DNA. We usually can have 40–200-fold increase after MeDIP based on different target genes.
References
- 1.Zilberman D, Gehring M, Tran RK, Ballinger T, Henikoff S. Genome-wide analysis of Arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription. Nat Genet. 2007;39:61–69. doi: 10.1038/ng1929. [DOI] [PubMed] [Google Scholar]
- 2.Rauch T, Wang Z, Zhang X, Zhong X, Wu X, Lau SK, Kernstine KH, Riggs AD, Pfeifer GP. Homeobox gene methylation in lung cancer studied by genome-wide analysis with a microarray-based methylated CpG island recovery assay. Proc Natl Acad Sci. 2007;104:5527–5532. doi: 10.1073/pnas.0701059104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Esteller M. Epigenetics in Cancer. N Engl J Med. 2008;358:1148–1159. doi: 10.1056/NEJMra072067. [DOI] [PubMed] [Google Scholar]
- 4.Liu K, Wang YF, Cantemir C, Muller MT. Endogenous assays of DNA methyltransferases: Evidence for differential activities of DNMT1, DNMT2, and DNMT3 in mammalian cells in vivo. Mol Cell Biol. 2003;23:2709–19. doi: 10.1128/MCB.23.8.2709-2719.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rollings RA, Haghighi F, Edwards JR, Das R, Zhang MQ, Ju J, Bestor TH. Large-scale structure of genomic methylation patterns. Genome Res. 2006;16:157–163. doi: 10.1101/gr.4362006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Laird PW. Cancer epigenetics. Hum Mol Genet. 2005;14:R65–76. doi: 10.1093/hmg/ddi113. [DOI] [PubMed] [Google Scholar]
- 7.Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- 8.Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33:245–254. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- 9.Das PM, Signal R. DNA methylation and cancer. J Clin Oncol. 2004;22:4632–4642. doi: 10.1200/JCO.2004.07.151. [DOI] [PubMed] [Google Scholar]
- 10.Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer. 2004;4:143–153. doi: 10.1038/nrc1279. [DOI] [PubMed] [Google Scholar]
- 11.Suzuki MM, Bird A. DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet. 2008;9:465–476. doi: 10.1038/nrg2341. [DOI] [PubMed] [Google Scholar]
- 12.Herman JG, Graff JR, Myohanen S, et al. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA. 1996;93:9821–9826. doi: 10.1073/pnas.93.18.9821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eads CA, Danenberg KD, Kawakami K, et al. CpG island hypermethylation in human colorectal tumors is not associated with DNA methyltransferase overexpression. Cancer Res. 1999;59:2302–2306. [PubMed] [Google Scholar]
- 14.Xion Z, Laird PW. COBRA: a sensitive and quantitative DNA methylation assay. Nucleic Acids Res. 1997;25:2532–2534. doi: 10.1093/nar/25.12.2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gonzalgo ML, Jones PA. Rapid quantitation of methylation differences at specific sites using methylation-sensitive single nucleotide primer extension (Ms-SNuPE) 1997;25:2529–2531. doi: 10.1093/nar/25.12.2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gitan RS, Shi H, Chen CM, Yan PS, Huang TH. Methylation-specific oligonucleotides microarray: a new potential for high-throughout methylation analysis. Genome Res. 2002;12:158–164. doi: 10.1101/gr.202801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yan PS, Chen CM, Shi H, Rahmatpanah F, Wei SH, Huang TH. Applications of CpG island microarrays for high-throughout analysis of DNA methylation. J Nutr. 2002;132:2430S–2434S. doi: 10.1093/jn/132.8.2430S. [DOI] [PubMed] [Google Scholar]
- 18.Weber M, Davies JJ, Wittig D, Oakeley EL, Haase M, Lam WL, Schübeler D. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat Genet. 2005;37:853–862. doi: 10.1038/ng1598. [DOI] [PubMed] [Google Scholar]
- 19.Lee TI, Johnstone SE, Young RA. Chromatin immunoprecipitation and microarray-based analysis of protein location. Nature Protocols. 2006;1:729–748. doi: 10.1038/nprot.2006.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Down TA, Rakyan VK, Turner DJ, Flicek P, Li H, Kulesha E, Graf S, Johnson N, Herrero J, Tomazou EM, Thorne NP, Backdahl L, Herberth M, Howe KL, Jackson DK, Miretti MM, Marioni JC, Birney E, Hubbard TJP, Durbin R, Tavare S, Beck S. A Bayesian deconvolution strategy for immunoprecipitation-based DNA methylome analysis. Nat Biotech. 2008;26:779–785. doi: 10.1038/nbt1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jacinto FV, Ballestar E, Esteller M. Methyl-DNA immunoprecipitation (MeDIP): hunting down the DNA methylation. Biotechniques. 2008;44:35–43. doi: 10.2144/000112708. [DOI] [PubMed] [Google Scholar]
- 22.Kininis M, Chen BS, Diehl AG, Isaacs GD, Zhang T, Siepel AC, Clark AG, Kraus WL. Genomic analyses of transcription factor binding, histone acetylation, and gene expression reveal mechanistically distinct classes of estrogen-regulated promoters. Mol Cell Biol. 2007;27:5090–5104. doi: 10.1128/MCB.00083-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu CL, Schreiber SL, Bernstein BE. Development and validation of a T7 based linear amplification for genomic DNA. BMC Genomics. 2003;4:19. doi: 10.1186/1471-2164-4-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schilling E, Rehli M. Global, comparative analysis of tissue-specific promoter CpG methylation. Genomics. 2007;90:314–323. doi: 10.1016/j.ygeno.2007.04.011. [DOI] [PubMed] [Google Scholar]