

Abstract
The central cavity of K+-selective ion channels is lined by four S6 transmembrane α-helices. An Ala residue is located near the midpoint of each S6 and marks the narrowest point of the central cavity. In hERG1 channels, we determined the functional consequences of substituting this conserved Ala (Ala653) with other hydrophobic or charged amino acids. Mutant channels were expressed in Xenopus oocytes and ionic currents measured by using the two-microelectrode voltage clamp technique. Substitution of Ala653 with bulkier hydrophobic residues (Val, Leu, Ile, Met, Phe, Trp) did not prevent ion conduction, but the mutant channels activated at more negative potentials compared to wild-type channels. The half-point for voltage dependent activation was shifted by −54 mV for the most conservative hydrophobic mutation, A653V. Oxidation of A653C hERG1 channels induced a maintained current at negative membrane potentials. This effect was not reversible with dithiothreitol, indicating that the sulfhydryl side-chains of Cys653 were oxidized to a negatively charged sulfinic or sulfonic acid. Substitution of Ala653 with acidic (Asp, Glu) or basic (Arg, Lys) residues prevented channel deactivation. Thus, an Ala at position 653 in hERG1 is required for normal voltage dependence of channel gating and a charged residue in this position prevents channel closure.
Keywords: potassium channel, gating, voltage clamp, Xenopus oocyte
Introduction
High resolution x-ray crystal structures of several bacterial K+ channels (KcsA, MthK, KvAP) [1–4] and mammalian Kv1.2 [5] have provided unique and detailed insights into the structural basis of ion permeation and gating of ion channels. KcsA was crystallized in the closed state, whereas the MthK and Kv1.2 structures represent the open state. In the closed KcsA channel, four straight α-helices line the central cavity and form a right-handed bundle crossing whose narrow aperture (~4 Å) and hydrophobic nature is incompatible for the passage of hydrated K+ ions [1]. In the open MthK channel [2,3], these inner helices are bent near their midpoint at Gly83 and splay away from the center, creating a wider aperture that permits the passage of K+. The narrowest constriction of the pore in MthK is 12 Å in diameter, and is located five residues C-terminal to the gating hinge at Ala88 [2].
Site-directed mutagenesis and voltage clamp analysis of mutant ion channels provides a complementary, function-based approach to study the structural components that mediate channel gating. Voltage-gated (Kv) channels such as Shaker, Kv1.2 and hERG1 are formed by coassembly of four identical subunits, each containing six transmembrane domains (S1-S6). The S6 domains of Kv channel subunits are homologous to the innermost helices of the bacterial channels and include residues equivalent to Gly83 and Ala88 of MthK (Fig. 1A).
Fig. 1.

Location of conserved Ala in the inner helix (S6) of hERG1 subunit. (A) Sequence comparison of the inner helix/S6 domain for several K+-selective channel subunits. The highly conserved Ala residue is highlighted, and is located 5 residues C-terminal to the Gly hinge (outlined by box). (B) Homology model for hERG1 based on MthK structure [3], showing three S6 and three S5 subunits. Ala653 is shown as van der Waals spheres. (C) Pore domain of hERG1 (four S5 and S6 domains) viewed from the intracellular side.
Because Ala88 is positioned near the narrowest constriction of the inner pore (Fig. 1B and C), it was proposed that a residue larger than Ala might occlude the open channel and reduce or block K+ ion conductance [2], a notion supported by the finding that mutation of the equivalent Ala to a Val in Shaker [6] or Kv1.5 [7] renders the channel nonfunctional. However, mutation of the conserved Ala to a much larger residue (Trp or Asp) in Shaker generates functional channels with relatively normal biophysical properties [8]. The importance of this conserved Ala in other Kv channels has not been explored.
hERG1 (human ether-a-go-go related gene type 1) is highly expressed in the heart where they conduct IKr, the rapid delayed rectifier K+ current [9,10]. Loss of function mutations or pharmacological block of these channels prolongs cardiac repolarization and increases the risk of life-threatening ventricular fibrillation [11]. Here we determined the functional consequences of mutating the conserved S6 Ala residue (Ala653) in hERG1. Substitution of Ala653 with larger hydrophobic residues did not prevent ion conduction and in fact, stabilized the open state of the channel as measured by a negative shift in the voltage dependence of channel activation. Substitution of Ala653 in hERG1 with a basic or acidic amino acid locked channels in an open state, most likely by charge repulsion.
Materials and Methods
Site-directed mutagenesis and expression of hERG1
Wild-type (WT) and mutant HERG1 (aka KCNH2 or Kv11.2) cDNAs were subcloned into the pSP64 oocyte expression vector (Promega, Madison, WI). Mutation of WT HERG1 cDNA was performed using the Quickchange site-directed mutagenesis kit (Stratagene, La Jolla, CA). Mutation constructs were confirmed by restriction enzyme and DNA sequence analyses. Complementary RNAs (cRNA) for injection into oocytes were prepared with either SP6 Cap-Scribe (Roche, Indianapolis, IN) or mMessage mMachine SP6 (Ambion, Austin, TX), following linearization of the expression construct with EcoR1. Isolation, culture and injection of Xenopus laevis oocytes were performed as described previously [12].
Isolation and voltage clamp of oocytes
Stage IV or V oocytes were collected from Xenopus laevis frogs anaesthetized with tricaine (0.17%). After surgery, frogs were allowed to recover for at least a month before being used again for oocyte isolation. After the final oocyte collection, frogs were killed humanely as approved by the University of Utah IACUC committee. Individual oocytes were isolated by treatment with collagenase, injected with ~50 nl of cRNA and then incubated at 18°C until studied as described previously [13].
Whole cell currents were recorded at room temperature in oocytes 2–8 days after cRNA injection by using standard two-electrode voltage clamp techniques [13], a GeneClamp 500 amplifier and a Digidata 1200 digitizer (Molecular Devices, Union City, CA) that was interfaced with a Pentium computer. Ionic currents were on-line filtered at 500 Hz and digitized at 2 kHz. pClamp9 software (Molecular Devices) was used to generate voltage clamp command potentials and acquire membrane currents.
Oxidation of hERG1 channels
Tert-butyl hydroperoxide (tbHO2) was applied to oocytes to determine the effects of oxidation on A653C hERG1 channels. Oocytes were placed in a 0.2 ml recording chamber and exposed to an extracellular solution containing 2 mM tbHO2 for 8 min followed by 12 min of washout. The flow rate of solution was held constant at 0.75 ml/min during which time a 2-s test pulse to +40 mV was applied every 15 s from a holding potential of −65 mV. Each test pulse was followed by a 3-s pulse to −100 mV to activate tail currents. Current-voltage (I-V) relationships were recorded before tbHO2 application and again after washout. The I-V protocol consisted of 2-s pulses to potentials ranging from −130 mV to +40 mV (applied in 10-mV increments) followed by a 3-s pulse to −110 mV. For oocytes expressing C555A/A653C or C566A/A653C hERG1 channels, the holding potential during the I-V protocol was −100 mV and test potentials ranged from −120 mV to +30 mV.
In some experiments, we attempted to reverse the effects of tbHO2 by subsequent treatment of oocytes with 20 mM dithiothreitol (DTT). After tbHO2 washout, DTT was applied for 9 min followed by 9 min of washout. I-V relationships were recorded before the start of tbHO2, at the end of tbHO2 washout, and at the end of DTT washout. Washout of tbHO2 and DTT was necessary to reverse channel block caused by these compounds.
Solutions
The extracellular solutions for most experiments contained the following: 96 mM NaCl, 4 mM KCl, 1 mM CaCl2, 1 mM MgCl2 and 5 mM HEPES; pH adjusted to 7.6 with NaOH. A653R(K, E, D) hERG1 channels expressed poorly. Therefore, currents for these four mutant channels were recorded in oocytes bathed in a high K external (96K) solution that contained the following: 96 mM KCl, 4 mM NaCl, 1 mM CaCl2, 1 mM MgCl2 and 5 mM HEPES; pH adjusted to 7.6 with KOH.
Data analysis
The voltage-dependence of current activation was determined by analysis of tail currents (Itail) measured after 2-s test pulses to a variable test potential (Vt). Normalized conductance values (G/Gmax) were plotted versus Vt and the relationship fitted to the Boltzmann function (Eqn. 1), where V1/2 is the potential at which the current is half-activated and k is the slope factor. The maximum value of Itail used to estimate Gmax was determined for individual oocytes by extrapolation of the fitted Boltzmann function to more positive potentials.
| Eqn. 1 |
Fully-activated I-V relationships were determined by a two pulse protocol. The first pulse was to +40 mV to fully activate channels and was followed by a test pulse to a potential that was varied from −140 to 0 mV. The maximum slope conductance was calculated from a linear fit of peak Itail measured in response to a Vt applied in 10-mV increments and ranging from −140 to −100 mV.
pCLAMP9 and Origin 7.5 (OriginLab, Northhampton, MA) software were used for off-line data analysis and preparation of figures. Data are presented as mean ± SEM (n = number of oocytes). Statistical significance was determined by t-test.
Results
Substitution of Ala653 with hydrophobic residues shifts voltage-dependence of hERG1 channel activation
To determine if substitution of Ala653 with other amino acids would prevent K+ conduction through the pore or alter channel gating, Ala653 was mutated to Gly and 7 larger hydrophobic residues (Met, Val, Cys, Ile, Leu, Phe, Trp).
Trp has the largest side-chain of the natural amino acids, so we first compared the maximum currents conducted by oocytes expressing WT or A653W hERG1 channels. Oocytes from the same isolation batch were injected with 8 ng of either WT or A653W hERG1 cRNA. Three days later, fully-activated I-V relationships were determined from currents recorded from both groups of oocytes. The maximum slope conductance was 55.3 ± 8.6 μS for WT (n = 11) and 25.5 ± 2.1 μS for A653W hERG1 (n = 10) channels. Thus, mutation of Ala653 to Trp reduced maximum current ~2-fold. In addition to a reduced current magnitude, A653W channels also activated at more negative membrane potentials and deactivated slower compared to WT channels (Table 1).
Table 1.
Biophysical parameters for WT and mutant hERG1 channels.
| Ala653 substitution | V1/2 | ΔV1/2 | k | ΔG0 | ΔΔG0 | n | τslow deactivation | τfast deactivation | Arel | n |
|---|---|---|---|---|---|---|---|---|---|---|
| mV | mV | mV | kcal/mol | kcal/mol | ms | ms | ||||
| (WT) | −29±0.7 | n.a. | 7.4±0.2 | −2.4±0.1 | n.a | 11 | 296±19 | 32±2 | 0.87 | 11 |
| Gly | −34±0.6* | −5 | 9.1±0.3* | −2.3±0.1 | −0.09 | 17 | 1014±49* | 137±12* | 0.57 | 11 |
| Met | −44±0.5* | −15 | 8.4±0.2* | −3.2±0.1* | −0.8 | 19 | 328±15 | 17±1* | 0.93 | 10 |
| Cys | −59±0.7* | −29 | 8.5±0.2* | −4.1±0.1* | −1.76 | 23 | 696±66* | 107±14* | 0.78 | 14 |
| Trp | −58±0.5* | −29 | 8.3±0.3† | −4.2±0.2* | −1.86 | 8 | 1175±52* | 49±4* | 0.33 | 11 |
| Phe | −60±1.2* | −31 | 7.6±0.2 | −4.7±0.2* | −2.38 | 6 | 701±23* | 61±3* | 0.68 | 10 |
| Ile | −66±0.7* | −37 | 8.9±0.3* | −4.5±0.2* | −2.09 | 8 | 393±12* | 31±2 | 0.84 | 8 |
| Leu | −68±0.5* | −39 | 8.9±0.2* | −4.5±0.1* | −2.16 | 8 | 410±7* | 35±2 | 0.78 | 11 |
| Val | −83±1.3* | −54 | 8.2±0.2 | −6.1±0.2* | −3.7 | 14 | 787±23* | 78±7* | 0.56 | 10 |
V1/2 and k were determined from fitting G-V relationships to a Boltzmann function; τSlow and τFast: slow and fast deactivation time constants measured at −130 mV; Arel: agnitude of the fast component of deactivation/magnitude Itail, measured at −130 mV; n.a., not applicable.
P<0.01;
P<0.05
Substitution of Ala653 with residues other than Trp also shifted the voltage dependence of hERG1 channels to more negative potentials and/or altered the rate of current deactivation. As examples, currents recorded at a Vt of −80, −40 and −10 mV from cells expressing WT, A653G, A653I or A653V hERG1 channels are shown in Fig. 2A. WT hERG1 channels were closed at −80 mV and only partially activated at −40 mV. Upon repolarization to −130 mV, WT hERG1 Itail deactivated rapidly with a fast time constant of ~30 ms. The voltage dependence of A653G channel activation was similar to WT, but these mutant channels deactivated much slower. A653I channels were partially activated at −80 and fully activated at −40 mV. A653V channels were half-activated at −80 and also fully activated at −40 mV. The V1/2 for activation of A653I and A653V channels was −66 mV and −83 mV, representing a shift of −37 and −54 mV when compared to WT channels (Fig. 2B). The magnitude of the shift in the activation curve varied with the amino acid present at position 653 as follows: Val > Leu = Ile > Cys = Phe > Trp > Met > Gly (Fig. 2B). The V1/2 and k values for the conductance-voltage (G-V) relationship and the kinetics of deactivation determined at −130 mV for all the mutant channels are summarized in Table 1.
Fig. 2.

Mutation of Ala653 alters voltage dependence of hERG1 channel activation. (A) Representative current traces recorded at indicated Vts (in mV) for WT channels and for three mutant channels harboring indicated amino acid substitution for Ala653. Itail was measured at −130 mV. (B) G-V relationships for WT and Ala653 mutant hERG1 channels expressed in Xenopus oocytes. The labels refer to residue substituted for Ala653. The smooth curves are fits to a Boltzmann function and the fitted parameters V1/2 and k are presented in Table 1.
A653V/F656V double mutant channels
A653V induced the largest shift in the half-point of hERG1 channel activation. Presumably, this mutation introduced a new interaction, or altered an existing one that stabilized the open state of hERG1. Homology models for hERG1 based on the pore region of Kv1.2 and MthK in their putative open states were used to identify nearby residues that could potentially interact with Val653. In the homology model based on Kv1.2 [5], Val653 is located ~3.6 Å away from Phe656 on the same subunit and ~4.1 Å from Phe656 in an adjacent subunit. In the homology model based on MthK [2], the distance between Val653 and Phe656 on adjacent subunits is only ~2.9 Å (Fig. 3). These models suggest that a hydrophobic interaction between Val653 and Phe656 in A653V channels might have caused the negative shift in activation. To test this idea, Phe656 was mutated to a smaller hydrophobic residue (Val) in A653V hERG1. In the homology model based on MthK, the distance between Val653 and Val656 on adjacent subunits in the double mutant was predicted to increase to ~5.5 Å. We hypothesized that the two introduced Val residues would not interact and therefore, the V1/2 for activation of the double mutant channel would be similar to the V1/2 for WT or F656V hERG1. Currents recorded at a Vt of −80, −40 and −10 mV from WT, F656V, F656V/A653V, and A653V hERG1 channels are shown in Fig. 4A. A653V hERG1 channels were fully activated at a potential of −40 mV. In contrast, F656V and F656V/A653V channels were similar to WT channels and activated at more positive potentials and required a pulse to −10 mV to attain full activation. The G-V relationship and Boltzmann fits for multiple cells are summarized in Fig. 4B. Thus, a second mutation (F656V) prevented the −50 mV shift in the half-point of channel activation induced by A653V.
Fig. 3.
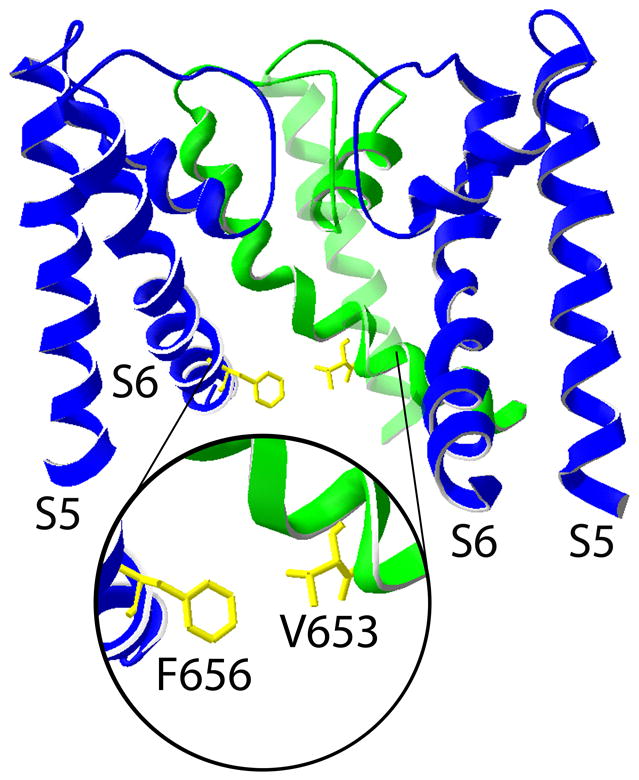
Phe656 is located in close proximity to Val653 on an adjacent subunit of the hERG1 channel. Structural model for the hERG1 pore based on the MthK channel [3]. The S5-S6 regions of three subunits are shown. The inset provides an expanded view of the proximity and orientation of one Phe656 residue relative to one Val653 residue located on an adjacent subunit.
Fig. 4.

Phe656V restores normal voltage dependence of activation altered by A653V mutation. (A) Representative current traces recorded at indicated Vts (in mV) for oocytes expressing WT, F656V, A653V/F656V or A653V hERG1 channels. (B) G-V relationship for WT and three mutant hERG1 channels. Currents were activated with 2-s test pulses. V1/2 values determined from fitting relationship to a Boltzmann function were −23.7 ± 0.6 mV for WT (n = 8), −83 ± 1.3 mV for A653V (n = 14), −25.6 ± 0.2 mV for F656V (n = 16), and −27.5 ± 0.4 mV for A653V/F656V channels (n = 10). k values were 7.7 ± 0.6 mV for WT, 8.6 ± 0.2 mV for A653V, 8.7 ± 0.2 mV for F656V, and 7.3 ± 0.4 mV for A653V/F656V channels.
Oxidization of A653C hERG1 disrupts channel deactivation
Our findings suggested that the side-chains of amino acids in position 653 of hERG1 do not face directly towards the central cavity, but are instead oriented in such a way as to favor contact with residues on adjacent subunits (e.g., the Val653-Phe656 interaction proposed above). To further probe the side-chain orientation, we determined if Cys residues introduced at position 653 could be cross-linked by oxidation. If a disulfide bridge was formed between Cys653 residues located on diagonal subunits, we would expect the channel to be non-conducting. We applied tbHO2, a membrane permeable oxidizing agent, to cells expressing WT or A653C hERG1 channels. Following a test pulse to +20 mV, the peak Itail at −110 mV was −6.5 ± 1.5 μA (n = 8) before oxidation and −7.3 ± 1.8 μA after oxidation for WT hERG1 channels, an 8 ± 3% increase (NS). For A653C channels, peak Itail was −2.7 ± 1.3 μA (n = 9) before oxidation and −1.4 ± 0.7 μA after oxidation, a 48 ± 7% decrease (P<0.01). Application and subsequent washout of tbHO2 also greatly increased the maintained (non-deactivating) component of Itail for A653C (Fig. 5A and B). At a potential of −130 mV, the relative amplitude of non-deactivating Itail for A653C channels increased from 0.07 to 0.53 after cells were oxidized with tbHO2 (Fig. 5C). In contrast, WT channels were unaffected by identical tbHO2 treatment (Fig. 5C). Thus, oxidation of A653C hERG1 prevented the channels from fully closing at negative potentials.
Fig. 5.

Oxidizing conditions alter gating of A653C hERG1 channels. (A and B) Current traces recorded from A653C channels before (A) and after (B) application of 2 mM tbHO2. Oxidized channels did not fully close at negative potentials. Inset above panel A shows voltage pulse protocol. (C) The voltage dependence of activation for WT and A653C channels before and after application of tbHO2. The G-V relationship for WT channels was not appreciably altered by oxidation. Control (■): V1/2 = −25.0 ± 0.3 mV, k = 8.4 ± 0.2 mV; after tbHO2 (□): V1/2 = −28.0 ± 0.2 mV, k = 8.5 ± 0.2 mV (n = 8). Oxidation of A653C channel currents caused a negative shift in V1/2 and an increase in the minimum value for G/Gmax. Control (●): V1/2 = −77.4 ± 1.5 mV, k = 14.7 ± 1.4 mV; after tbHO2 (○): V1/2 = −84.5 ± 2.5 mV, k = 24.1 ± 2.2 mV (n = 9).
If the altered gating of oxidized A653C hERG1 channels resulted from formation of a disulfide bond, then the effect should be reversible by application of the reducing agent DTT. However, DTT (20 mM) did not reverse the effects of tbHO2 on the I-V (Fig. 6A) or G-V (Fig. 6B) relationships for A653C hERG1 channels. For these experiments, the holding potential was −90 mV. At this holding potential, the effect of channel oxidation was less than that achieved with a holding potential of −65 mV (Fig. 5). This suggests that tbHO2 only has easy access to Cys653 when the channels are in the open state.
Fig. 6.

DTT does not reverse the effects of oxidation on A653C hERG1 channels. (A) I-V relationships for A653C channels before and after oxidation with 2 mM tbHO2 and subsequent treatment with 20 mM DTT (n = 10). The holding potential was −90 mV. Only mutant channels conduct inward currents at a Vt negative to −80 mV. Currents were normalized to the peak of the outward current (1.18 ± 0.24 μA at −30 mV) under control conditions. (B) G-V relationships for A653C channels under control conditions (V1/2 = −60.8 ± 0.5 mV, k = 9.6 ± 0.4 mV, n = 10) and after oxidation with tbHO2 (V1/2 = −67.6 ± 0.6 mV, k = 10.5 ± 0.5 mV, n = 10). Subsequent application of DTT (V1/2 = −59.7 ± 0.5 mV, k = 11.6 ± 0.5 mV, n = 10) did not reverse the effects of tbHO2 on minimum G/Gmax.
A653C does not interact with Cys residues in the S5 domain of hERG1
The inability of DTT to reverse the effects of oxidation on A653C hERG1 makes it extremely unlikely that a disulfide bond can form between Cys653 residues of adjacent subunits or between residues located on opposite sides of the tetrameric channel complex. However, the possibility remained that Cys653 could form a disulfide bond with a native Cys residue in hERG1 and that this bond was not accessible by DTT.
There are two native Cys residues located in the S5 domain (Cys555 and Cys566) that could conceivably interact with introduced Cys653 in the S6 domain. Mutation of the native Cys residues in S5 to Ala did not alter the response of A653C hERG1 to oxidation by tbHO2. The relative amplitude of non-deactivating Itail for C555A/A653C hERG1 increased from 0.07 to 0.3 after application of tbHO2 (Fig. 7A). A similar result was obtained from cells expressing C566A/A653C hERG1 channels (Fig. 7B). Thus, the altered channel gating caused by oxidation of Cys653 hERG1 was not caused by formation of a disulfide between two introduced Cys653 residues or between Cys653 and a native Cys in the S5 domain.
Fig. 7.

Mutations of Cys residues in the S5 domain do not prevent oxidation-induced alteration of A653C hERG1 channel gating. (A) G-V relationship for C555A/A653C mutant channels before (V1/2 = −62.6 ± 0.2 mV, k = 9.9 ± 0.2 mV, n = 8) and after 2 mM tbHO2 (V1/2 = −68.1 ± 0.1 mV, k = 10.0 ± 0.1 mV, n = 8). (B) G-V relationship for C566A/A653C channels before (V1/2 = −59.9 ± 0.9 mV, k = 11.2 ± 0.8 mV, n = 5) and after 2 mM tbHO2 (V1/2 = −72.2 ± 2.0 mV, k = 20.6 ± 1.7 mV, n = 5). For both mutant channels, currents shown were elicited at a Vt of −30 mV and Itail was measured at −120 mV. The holding potential was −100 mV.
Oxidation of the thiol group of Cys produces an unstable intermediate, sulfenic acid [14,15]. This unstable intermediate can either form a disulfide bond with another thiol group in close proximity, or it can be further oxidized to the more stable sulfinic or sulfonic acid. We suggest that tbHO2 oxidized the thiol of A653C to either sulfinic or sulfonic acid and that the resulting negative charged side chains of multiple Cys653 residues prevented channels from closing due to like-charge repulsion.
Substitution of Ala653 with a charged residue results in channels that are constitutively open
To further test our charge repulsion hypothesis suggested by the oxidation experiments, we mutated Ala653 to a residue with a positive (Arg, Lys) or a negative (Glu, Asp) charged side chain. For experiments with these mutant channels, oocytes were bathed in 96K extracellular solution and channels were activated by a pulse to +40 mV. Itail was recorded upon repolarization to a potential of −120 mV. Whereas WT hERG1 current deactivated rapidly at −120 mV (Fig. 8, middle trace), A653R hERG1 channels failed to deactivate, and instead conducted a maintained inward current at −120 mV (Fig. 8, bottom trace). Similar results were obtained for cells expressing the other three mutant channels containing a charged amino acid at position 653. I-V and normalized G-V relationships for these mutant channels are summarized in Figs. 9A and 9B. Together, these findings strongly suggest that substitution of Ala653 with a charged residue disrupts voltage-dependent closure of hERG1 channels by a charge repulsion mechanism.
Fig. 8.

A653R hERG1 channels are constitutively open. Top panel illustrates the pulse protocol used to activate currents, shown in lower three panels, that were recorded from an uninjected oocyte or oocytes injected with WT or A653R hERG1 cRNA. Unlike WT hERG1 currents, A653R channels do not deactivate. Oocytes were bathed in 96 K external solution.
Fig. 9.

Substitution of Ala653 with charged amino acids prevents channel closure. (A) I-V relationships for WT (n = 10), A653R (n = 17), A653K (n = 17), A653E (n = 15), and A653D (n = 7) hERG1 channel currents. At negative potentials, WT channels are closed, whereas the mutant channels conduct large currents. Oocytes were bathed in 96 K external solution. (B) Channel activation was voltage-dependent for WT (V1/2 = −27.6 ± 0.3 mV, k = 9.0 ± 0.2 mV, n = 10), but not for mutant hERG1 channels.
Discussion
The S6 domains of Kv channels contain a highly conserved Ala residue (Ala653 in hERG1), located five residues towards the C-terminus from the putative Gly hinge (Gly648 in hERG). The crystal structure of MthK in the open state revealed that the equivalent Ala in this bacterial channel was positioned near the narrowest constriction of the inner pore with its side chain projected towards the center of the channel pore [2]. This orientation suggested that substitution of Ala with a bulkier residue might block ion conduction. A “hydrophobic seal” can be formed by inter-subunit interaction of activation gate residues located near the C-terminus of S6 in Shaker [16] or the TM2 domain of KcsA channels [17]. To test the hypothesis that a similar “seal” might form near the midpoint of the S6 domains, we substituted Ala653 with several other hydrophobic residues, including Val, Leu, Ile, Met, Phe and Trp. All of these mutations stabilized the open state of hERG1, measured as negative shifts in the voltage dependence of channel activation. In addition, the substitution that produced the bulkiest side-chain (A653W) decreased current magnitude, an effect that could result from a reduced single channel conductance, decreased open probability or reduced trafficking. Together our results suggest that the side chains of residues at position 653 do not face directly towards the center of the pore, or that the distance between them is too great to cause significant occlusion of the pore or impede ion conductance.
To further test whether the side chains of 653 residues were directed towards the center axis of the pore, we determined if oxidation of Cys653 hERG1 channels could cross-link a pair of Cys residues and occlude the pore. A disulfide bond between two Cys residues can be readily formed only when they are orientated orthogonally and their β-carbons are located within 5 Å of each other [18]. Although tbHO2 altered the biophysical properties of A653C hERG1 channels, the effects were not reversed by treatment with DTT. Therefore, the spatial arrangements of the Cys653 residues were unfavorable for disulfide bond formation, either between Cys residues on adjacent or opposite subunits. It seems more likely that the sulfhydryl groups of Cys653 residues were oxidized to sulfinic or sulfonic acid and that charge repulsion between adjacent or diagonal subunits prevented the channels from closing normally. The gating phenotype displayed by replacing Ala653 with charged amino acids (Arg, Lys, Glu, Asp) supports this interpretation. Charged residue substitutions strongly bias the channel towards the open state, preventing any measurable deactivation, even at potential as negative as −140 mV.
Oxidation of A653C channels also decreased Itail amplitude. Because this effect was not reversed with DTT application, it is unlikely that oxidized Cys653 residues form a disulfide bridge. Instead, we suggest that the modified sulfhydryl groups reduce the conductance, or perhaps mean open time of single channels.
We found that the large negative shift in the voltage dependence of channel activation induced by the A653V mutation could be rescued by an additional mutation (F656V). It was previously shown that mutation of Phe656 to other hydrophobic residues (Trp, Tyr, Met, Leu, Ile) had relatively little effect on channel gating [19]. However, substitution of Phe656 with charged, or polar residues (Arg, Glu, Ser) disrupted hERG1 deactivation [19]. Thus, hydrophobic interactions between Phe656, located in a region of each S6 domain presumed to form the inner bundle crossing, is required for normal channel closure. A pair of hydrophobic interactions between Val653 and Phe656 on an adjacent subunit may favor the open state of the channel by hindering intra-subunit interactions between Phe656 residues or by stabilizing the paired S6 subunits in a splayed outward conformation.
Ala471 in Shaker is equivalent to Ala653 in hERG1. A471W and A471D Shaker channels retain relatively normal function [8]; however, A471V Shaker channels do not functionally express [6]. Presumably, A471V Shaker subunits fold and co-assemble to form channels that are non-conducting. Because charged substitutions of the conserved Ala in S6 have very different functional consequences in hERG1 and Shaker, it seems likely that the orientation of the S6 domain for these two channels differs significantly from that predicted simply by sequence alignment based on the location of the conserved Ala and Gly hinge. Most importantly, the residues located C-terminal of Ala653 in the S6 of hERG1 (Ser-Ile-Phe-Gly) are Leu-Pro-Val-Pro in most other well-characterized Kv channels. S6 bends at the Pro-Val-Pro motif in the open state of Kv1.2 [5] and is proposed to have an important role in the gating of Shaker and related channels [20,21].
In summary, hydrophobic substitutions of Ala653 significantly alter the voltage dependence of hERG1 channels. Mutation to a charged residue stabilizes the channel in a constitutively open state. The contrast between the functional consequences of mutations in this conserved Ala between hERG1 and Shaker indicate an important difference in the orientation of the S6 domains in these channels.
Acknowledgments
We thank Kam Hoe Ng and Wes Perrine for technical assistance. This work was supported by National Institutes of Health Grants HL065299 and HL55236 (to M.C.S.).
References
- 1.Doyle DA, Morais Cabral J, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, MacKinnon R. The structure of the potassium channel: Molecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- 2.Jiang Y, Lee A, Cadene M, Chait BT, MacKinnon R. The open pore conformation of potassium channels. Nature. 2002;417:523–526. doi: 10.1038/417523a. [DOI] [PubMed] [Google Scholar]
- 3.Jiang Y, Lee A, Chen J, Cadene M, Chait BT, MacKinnon R. Crystal structure and mechanism of a calcium-gated potassium channel. Nature. 2002;417:515–522. doi: 10.1038/417515a. [DOI] [PubMed] [Google Scholar]
- 4.Jiang Y, Lee A, Chen J, Ruta V, Cadene M, Chait BT, MacKinnon R. X-ray structure of a voltage-dependent K+ channel. Nature. 2003;423:33–41. doi: 10.1038/nature01580. [DOI] [PubMed] [Google Scholar]
- 5.Long SB, Campbell EB, Mackinnon R. Crystal structure of a mammalian voltage-dependent shaker family K+ channel. Science. 2005;309:897–903. doi: 10.1126/science.1116269. [DOI] [PubMed] [Google Scholar]
- 6.Yifrach O, MacKinnon R. Energetics of pore opening in a voltage-gated K+ channel. Cell. 2002;111:231–239. doi: 10.1016/s0092-8674(02)01013-9. [DOI] [PubMed] [Google Scholar]
- 7.Decher N, Kumar P, Gonzalez T, Renigunta V, Sanguinetti MC. Structural basis for competition between drug binding and Kvbeta 1.3 accessory subunit-induced N-type inactivation of Kv1.5 channels. Mol Pharmacol. 2005;68:995–1005. doi: 10.1124/mol.105.011668. [DOI] [PubMed] [Google Scholar]
- 8.Hackos DH, Chang TH, Swartz KJ. Scanning the intracellular S6 activation gate in the shaker K+ channel. J Gen Physiol. 2002;119:521–532. doi: 10.1085/jgp.20028569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sanguinetti MC, Jiang C, Curran ME, Keating MT. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell. 1995;81:299–307. doi: 10.1016/0092-8674(95)90340-2. [DOI] [PubMed] [Google Scholar]
- 10.Trudeau M, Warmke JW, Ganetzky B, Robertson GA. HERG, a human inward rectifier in the voltage-gated potassium channel family. Science. 1995;269:92–95. doi: 10.1126/science.7604285. [DOI] [PubMed] [Google Scholar]
- 11.Sanguinetti MC, Tristani-Firouzi M. HERG potassium channels and cardiac arrhythmia. Nature. 2006;440:463–469. doi: 10.1038/nature04710. [DOI] [PubMed] [Google Scholar]
- 12.Shih TM, Smith RD, Toro L, Goldin AL. High-level expression and detection of ion channels in xenopus oocytes. Methods Enzymol. 1998;293:529–556. doi: 10.1016/s0076-6879(98)93032-4. [DOI] [PubMed] [Google Scholar]
- 13.Stühmer W. Electrophysiological recording from Xenopus oocytes. Methods Enzymol. 1992;207:319–339. doi: 10.1016/0076-6879(92)07021-f. [DOI] [PubMed] [Google Scholar]
- 14.Claiborne A, Mallett TC, Yeh JI, Luba J, Parsonage D. Structural, redox, and mechanistic parameters for cysteine-sulfenic acid function in catalysis and regulation. Adv Protein Chem. 2001;58:215–276. doi: 10.1016/s0065-3233(01)58006-7. [DOI] [PubMed] [Google Scholar]
- 15.Poole LB, Karplus PA, Claiborne A. Protein sulfenic acids in redox signaling. Annu Rev Pharmacol Toxicol. 2004;44:325–347. doi: 10.1146/annurev.pharmtox.44.101802.121735. [DOI] [PubMed] [Google Scholar]
- 16.Kitaguchi T, Sukhareva M, Swartz KJ. Stabilizing the closed s6 gate in the shaker Kv channel through modification of a hydrophobic seal. J Gen Physiol. 2004;124:319–332. doi: 10.1085/jgp.200409098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Armstrong CM. Voltage-gated k channels. Science STKE. 2003:re10. doi: 10.1126/stke.2003.188.re10. [DOI] [PubMed] [Google Scholar]
- 18.Careaga CL, Falke JJ. Thermal motions of surface alpha-helices in the D-galactose chemosensory receptor. Detection by disulfide trapping. J Mol Biol. 1992;226:1219–1235. doi: 10.1016/0022-2836(92)91063-u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fernandez D, Ghanta A, Kauffman GW, Sanguinetti MC. Physicochemical features of the hERG channel drug binding site. J Biol Chem. 2004;279:10120–10127. doi: 10.1074/jbc.M310683200. [DOI] [PubMed] [Google Scholar]
- 20.del Camino D, Holmgren M, Liu Y, Yellen G. Blocker protection in the pore of a voltage-gated K+ channel and its structural implications. Nature. 2000;403:321–325. doi: 10.1038/35002099. [DOI] [PubMed] [Google Scholar]
- 21.Webster SM, Del Camino D, Dekker JP, Yellen G. Intracellular gate opening in shaker K+ channels defined by high-affinity metal bridges. Nature. 2004;428:864–868. doi: 10.1038/nature02468. [DOI] [PubMed] [Google Scholar]