

Abstract
Importance of the field
Although significant progress has been made in delivering therapeutic agents through micro and nanocarriers, precise control over in vivo biodistribution and disease-responsive drug release has been difficult to achieve. This is critical for the success of next generation drug delivery devices, since newer drugs, designed to interfere with cellular functions, must be efficiently and specifically delivered to diseased cells. The major constraint in achieving this has been our limited repertoire of particle synthesis methods, especially at the nanoscale. Recent developments in generating shape-specific nanocarriers and the potential to combine stimuli-responsive release with nanoscale delivery devices show great promise in overcoming these limitations.
Areas covered in this review
Here we discuss how recent advancements in fabrication technology allow synthesis of highly monodisperse, stimuli-responsive, drug-carrying nanoparticles of precise geometries. We also review how particle properties, specifically shape and stimuli responsiveness, affect biodistribution, cellular uptake, and drug release.
What the reader will gain
The reader is introduced to recent developments in intelligent drug nanocarriers and new nanofabrication approaches that can be combined with disease-responsive biomaterials. This will provide insight into the importance of controlling particle geometry and incorporating stimuli responsive materials into drug delivery.
Keywords: Controlled drug delivery, nanoparticles, nanoimprint lithography, triggered release, shape and size specific nanoparticles
1. Introduction
The goals associated with designing an ideal drug carrier have evolved significantly over the past decades. Initial research efforts were focused on encapsulating therapeutic agents to protect them from degradation and to control drug release kinetics. More recently, the challenge has been to improve the effectiveness and efficiency of drug delivery, and therefore reduce side effects, such that drug carriers are preferentially releasing therapeutic agents at the site of “injury” (i.e., diseased or damaged tissues). Ultimately, the goal is to have precise control over (a) drug biodistribution, (b) cell targeting, (c) in vivo drug stability, (d) circulation kinetics, and (e) drug release mechanisms, such that therapeutic agents are released primarily in response to a disease-specific or physiologically relevant signal.
Targeting drugs to specific diseased cells and tissues following systemic delivery requires nanoscale drug carriers. Nanocarriers can enhance the efficacy of therapeutic treatments because they have the ability to rapidly distribute to pathophysiological sites in the body and can deliver drugs intracellularly. It is well established that nanoparticles can passively accumulate in tumors due to the enhanced permeability of tumor capillaries and retention in tumor tissues (EPR effect) and are thus well studied for systemic delivery of anti-cancer drugs.
Classically, drug nanocarriers have been synthesized using self-assembly or emulsion-based methods. These efforts have led to the development of several nanoparticle formulations, such as Doxil and Abraxane, which have been approved for clinical treatment of cancer [1]. A large number of polymer and lipid-based nanocarrier formulations produced using such “bottom-up” synthesis methods have been widely reported in the literature [2–8]. However, these bottom-up processes produce particles (or nanocomplexes) that are primarily spherical or near spherical in shape and are mostly polydisperse. Almost all of our current knowledge regarding the in vivo performance of drug nanocarriers is based on these spherical particles of varying size and compositions. It is widely accepted in the fluid dynamics community that shape (geometry and aspect ratio) plays a major role in how particles are transported through a fluid, especially within narrow tubes (as would be the case for drug carriers in blood capillaries or lymphatics) [9–12]. However, the inability to precisely control geometry and polydispersity of nano-sized particles has prevented experimental verification of this shape effects at the nanoscale. New methods that allow for finer control over nano and microparticle fabrication now allow drug delivery researchers to explore how precisely controlled particle shape and size affect particle transport (i.e., biodistribution, organ and cellular targeting, circulation dynamics, intracellular uptake etc.) and drug release kinetics in vitro and in vivo. We believe that our ability to finely tune particle geometry could provide a critical step in developing the next generation of drug carriers.
Triggered drug release is another major effort in current drug delivery research. To date, most sub-micron to nanoscale drug delivery carriers that have been reported employ diffusion and/or hydrolysis controlled released systems, primarily due to our long standing focus on particle synthesis using lipids and hydrolytically degradable polymers. Diffusion and hydrolysis-controlled systems have the potential to release encapsulated agents outside the target tissue, for instance in systemic circulation, thus exacerbating side effects. It is also difficult to predict or study in vivo drug release kinetics from these systems, especially from the routine in vitro assays described in most reports. Despite progress in surface ligand-based targeting strategies, it is still conceivable that a significant amount of drug carriers will reach physiologically healthy tissues, and in the absence of a second release control signal, a large dose of drugs could be released into these tissues.
Since diseased cells and tissues might express or over-express unique surface markers and proteins it is often possible to selectively target them either through specific ligand interactions or stimuli-based drug release mechanisms. Stimuli responsive carriers have been engineered to release their payload in response to local (i.e., physiological or pathophysiological) cues, including pH and enzymes, or in response to external stimuli, such as ultrasound, magnetic fields, electromagnetic radiation, and temperature. This approach is a powerful means for the development of “intelligent therapeutics”. A wide variety of physiologically sensitive hydrogel-based materials have been developed [13]. However, only a few of these materials (e.g., pH-sensitive materials) have been reported for use in nanoscale carriers. We believe that the integration of particle geometry and disease-triggered drug release concepts into our existing knowledge of surface ligand-based targeting would result in a quantum change in the efficacy of the next generation drug carriers.
In the following sections we discuss in detail (a) recent advances in fabricating shape-specific, polymer based nano- and microcarriers that could be utilized for drug delivery, (b) how particle shape and stimuli-triggered release could affect the performance of drug nanocarriers, and (c) current progress in triggered-release nanoparticles and how to incorporate triggered release properties into shape-specific nanoparticles. Because of the vast number of reviews already published that discuss drug carrier development [2–4, 14–18] our review focuses specifically on the emerging field of nano- and microfabricated polymer particles for targeted drug delivery. Finally, we present our thoughts on how these new directions in drug delivery could allow for significant improvements in disease outcome.
2. Shape and Size Specific Nanocarriers
Besides disease-responsive release, the two key properties of an effective nanocarrier are (a) efficient targeting to specific tissues and cells [5] and (b) avoiding rapid clearance (i.e., remaining in circulation) for a significant amount of time to increase particle accumulation in target tissues [1]. Circulation time, targeting, and the ability to overcome biological barriers could depend on the shape (e.g., aspect ratio) and size of a particle since these properties are likely to influence particle transport behavior in the blood, especially in small capillaries and tumor vasculature, as well as how cells sense and respond to the particle for endocytosis [9–12, 19–21]. In addition, geometry affects surface to volume ratio and hence is likely to affect degradation and drug release kinetics [22, 23]. Before discussing how size and shape affect drug carrier efficacy it is first necessary to elaborate on recently developed fabrication methods that permit control over particle geometry.
2.1 Fabrication of Shape and Size Specific Nanocarriers
Over the past several decades, a variety of nanoparticle carriers, synthesized using mostly “bottom-up” approaches, such as liposomal nanocarriers [7, 8], micellar formulations [6], solid polymeric nanoparticles [5], and polymer-drug conjugates [24], have shown promising results as potential drug delivery vehicles. Recently, advancements in “top-down” approaches, including micro and nano electromechanical system (MEMS and NEMS)-based fabrication processes have generated the potential for producing uniform micro- and nanosized particles of precise shape and size [25, 26]. Such top-down fabrication methods could provide precise, pre-designed control over particle geometry (shape, aspect ratio) and composition, which is often difficult to achieve using bottom-up approaches that tend to rely on emulsions or self-assembly. In this section, we will focus on advancements in fabrication methods for shape-specific nano- and microscale drug carriers and discuss their advantages and disadvantages.
2.1.1 Bottom-up synthesis
Extensive research has been devoted to the development of micro- and nanoscale drug carriers composed of a wide variety of organic and inorganic materials. These carriers include liposomes, nanoemulsions, solid lipid nanoparticles, micelles, dendrimers, biodegradable and non-degradable polymeric carriers, as well as inorganic semiconducting or magnetic nanoparticles [5–8, 24, 27–31]. Most of these carriers are colloidal molecular assemblies that are largely driven by hydrophobic-hydrophilic interactions, van der Waals forces, hydrogen bonding, or ionic interactions, which produces spherical or near spherical particles, often with high polydispersity. The physicochemical characteristics, in vivo drug release profiles, degradation kinetics, and biotransport properties of these carriers are variable and difficult to evaluate and reproduce, especially at clinically relevant pharmaceutical scales. In addition, combining multiple functionalities (i.e., both targeting as well as various stimuli-sensitive properties like enzyme responsiveness) in a controlled and reproducible manner could be difficult in self-assembled carrier systems. A rich literature already exists on these types of carriers and the details of their formulation are beyond the scope of this review and therefore will not be discussed further.
2.1.2 Top-down synthesis
Recent advancements in micro- and nanofabrication techniques have made them suitable for use with biomedical materials thereby allowing applications in micro- and nanoscale drug delivery systems and tissue engineering [25, 26]. Numerous micro- and nanoimprint lithography processes, including soft lithography [32], thermal embossing [33–37], step and flash lithography [33, 38, 39], and UV embossing [40–42] have been reported with nanometer resolution [26]. At the micron scale, Desai et al. explored the use of microfabrication techniques for synthesizing biocapsules for immunoisolation of “drug” releasing cell transplants [43, 44]. They also studied photolithography for fabricating shape-specific drug delivery micro-devices and particles [45–48]. For instance, the group demonstrated the fabrication of multilayered microparticles that can be released from the substrate by selective etching of a sacrificial layer [46, 48, 49]. Doyle et al. have also developed a unique use of microfabrication techniques, in which they combine flow-through microfluidic channels with a microscopic projection photolithography to fabricate shape-specific microparticles and two-faced Janus particles [50–52]. This method can produce approximately 100 identical, shape-specific particles in less than 0.1 seconds. Recently, Champion et al.[53] have reviewed fabrication of shape and size specific microparticles. Hence, this will not be discussed in much detail here. We will, instead, focus on the fabrication of drug delivery particles with nanoscale features.
Out of the wide variety of nanofabrication processes, nanoimprint lithography (NIL), including step and flash imprint lithography (S-FIL) [54], particle replication in non-wetting templates (PRINT) [55–60], and solvent molding-based fabrication [61, 62], have been shown to be powerful methods for fabricating polymer nanocarriers of specific shape, size, and aspect ratio. The following sections discuss these methods in detail.
2.1.2.1 Step and Flash Imprint Lithography (S-FIL)
Using a modified S-FIL process (Figure 1A) [33], Roy and colleagues have developed triggered release nanocarriers of specific shape, size, and aspect ratio [54]. S-FIL is a UV-NIL method that has numerous advantages over other nanofabrication methods. In the S-FIL process, a photo-polymerizable polymer, like poly(ethylene glycol) diacrylate (PEGDA), is dispensed in droplets as small as picoliter onto a substrate. This is done in a specific, computer-controlled pattern that minimizes the amount of drug-polymer solution required [63], thereby reducing fabrication cost over traditional spin coating methods. A pre-patterned transparent quartz template is pressed into the polymer droplets, causing it to spread and fill the nanofeatures patterned on the template. The template-substrate assembly is then exposed to a short UV pulse to cure the polymer. The quartz mold is then removed revealing the desired nanostructures on the surface of the substrate (silicon wafers). Not only are these patterned quartz templates re-usable, but they also enable sub-100 nm alignment of nano-features with pre-existing patterns on a substrate. In order to release the imprinted nanoparticles from the substrate the residual layer in between the particles is first removed using a short, low power, oxygen plasma etch. This is followed by dissolving the “release layer” (coated onto the substrate prior to imprinting) using a mild, one-step aqueous process [54]. This particle harvest method avoids physical force-based removal (e.g., scraping), which has the potential to damage nanoparticle shape. Figure 2B shows well preserved 400 nm particles with a square cross-section following release in an aqueous solution.
Figure 1. Schematics of shape-specific nano and microcarrier fabrication methods.

(A) Modified Step and Flash Imprint Lithography (S-FIL) method [54]; (B) Particle Replication In Non-wetting Templates process (PRINT)[64]; (C) Solvent molding-based manipulation of spherical particles into non-spherical geometries [61].
Figure 2. Various shape and size specific nanoparticles fabricated using S-FIL, PRINT and Solvent Molding.

(A–C) Modified Step and Flash Imprint Lithography (S-FIL) fabricated nanoparticles composed of 75%(v) PEG-diacrylate.[54]: (A) 50 nm × 50 nm × 100 nm pillars, (scale bar = 100 nm), (B) 400 nm × 400 nm × 525 nm particles released from the substrate (scale bar = 2μm), (C) 200 nm × 200nm × 525 nm pillars (scale bar = 500 nm); (D–E) Particle replication in non-wetting templates (PRINT) process: (D) 2 μm wide pillar PRINT particles that are composed of insulin (25%wt) (scale bar = 5 μm) [65], (E) 1 μm diameter by ~500 nm long cylindrical particles composed of 67 wt% trimethylolpropane ethoxylate triacrylate, 20 wt% PEG monomethylether monometharcylate, 10 wt% aminoethyl methacrylate hydrochloride, 2 wt% fluorescein-o-acrylate, and 1 wt% 2,2-diethoxyacetophenone (scale bar = 5 μm) [124]; (F–H) Non-spherical particle geometries formed by solvent molding-based manipulation of spherical polystyrene particles: (F) Elliptical disks (scale bar = 2 μm) [61], (G) “UFOs” (scale bar = 2 μm) [61], and (H) Barrels (scale bar = 2 μm) [61].
S-FIL is an extremely flexible method for forming imprints with a wide range of shapes, sizes, and material compositions. The composition of these nanostructures is only constrained by the viscosity, wettability, and photocrosslinking nature of the monomers. Drug molecules can be simply mixed with the monomer or macromer solution prior to imprinting in order to encapsulate them within the nanoimprinted particles. In addition, since S-FIL does not use reduction lenses like conventional photolithography methods, its resolution is not limited by optical diffraction as it is with other methods that are based on far field optics. Consequently, imprint patterns are defined by the quartz template features [33], the resolution of which is controlled by the resolution of electron-beam lithography, which is routinely sub-25 nm. Poly(ethylene glycol)-based particle structures fabricated using S-FIL and ranging from 800 nm to 50 nm has already been reported (Figure 2A–C) [54]. In addition, using SFIL, a variety of biological agents like proteins, peptides, DNA, and RNA have been encapsulated within these shape-specific nanoparticles.
Since S-FIL is a commercially available process used in the semiconductor industry, with appropriate development, this technique could provide a high-throughput, commercially and clinically viable particle fabrication method to generate shape-specific nanocarriers. It also avoids shear forces (such as those found in emulsion based systems), as well as high temperature or organic solvents, which are often necessary for in situ polymerization and liposome formation.
2.1.2.2 Particle Replication in Non-wetting Templates (PRINT)
DeSimone et al. recently demonstrated the ability to form nano-size particles from various biocompatible polymers as well as proteins, including insulin and albumin, using a PRINT fabrication method (Figure 1B) [60, 64]. The PRINT process utilizes a non-wetting elastomeric fluoropolymer-based flexible mold to form micro- to nanoscale features without a residual layer. Using a lamination based process the mold is filled with a monomer solution by placing it between the patterned mold and another film. The mold, solution, and counter sheet are then passed through a roller and as the mold passes under the roller, the counter sheet is peeled away, leaving the patterns in the mold filled with the solution. The filled mold is then dried to remove residual solvent in the molding solution or, depending on the molding solution, the mold could also be exposed to UV irradiation to photopolymerize the solution. Although a residual layer is avoided, unlike with S-FIL, the particles are not readily released from the surface. Rather, they are harvested from the substrate using physical scraping [57]. Similar to S-FIL, monodisperse particles (Figure 2D–E) can be achieved using the PRINT method, making it a highly versatile process for fabrication of micro- and nanoparticles for drug delivery applications [57, 65].
2.1.2.3 Solvent Molding Methods
Mitragotri and colleagues have reported unique solvent-based methods (Figure 1C) to generate polystyrene micro- and nanoparticles of various shapes, such as elliptical and circular discs and barrels, as shown in Figure 2F–H [61]. To fabricate these particles, commercially available spherical polystyrene nano- and microparticles of various sizes are suspended in an aqueous solution of polyvinyl alcohol and cast into patterned films. These films are then manipulated in different ways (e.g., stretching) to generate particles of various shapes and dimensions (submicron to 16 μm). Although this represents a simple, easily adaptable method, it is likely to be difficult to achieve size ranges and particle dimensions similar to those achieved by the PRINT and S-FIL methods.
In one approach (top panel of Figure 1C), the particles are exposed to an organic solvent or heat to liquefy the polystyrene and then the film is stretched in one or two dimensions depending on the desired particle shape. Alternatively, the films are stretched first and then the polystyrene is liquefied, filling the voids created during stretching (bottom panel of Figure 1C). In both approaches, the particles are re-solidified into their new shape by extracting the solvent or cooling below the glass transition temperature. The film is then dissolved and the particles collected [61]. Although this technique is limited to materials that can easily undergo phase changes or be solubilized, a wide variety of micron to sub-micron scale particles of very specific and unique shapes can be fabricated.
2.2 Effects of Shape and Size on the Fluid Dynamics of Nanocarriers
Ultimately, for a nanocarrier to be effective in delivering drugs to a diseased tissue, it should be able to efficiently interact with the capillary wall and “migrate” to the target tissue (e.g., tumors) before being cleared away by the reticuloendothelial system or being filtered by the lungs, liver, and spleen. It has been reported that microparticles larger than 5 μm are trapped in the capillary beds of the liver, whereas microparticles of ~1–5 μm localize within the liver where they are phagocytosed by Kupffer cells [20, 21]. Nanoparticles that are less than 1 μm and larger than 200 nm are easily filtered out in the spleen while those smaller than 100 nm remain in blood vessels within fenestrae of the endothelial lining [12, 20, 21].
The geometry of nanocarriers (e.g., shape, aspect ratio, and ratio of particle dimensions to vessel diameter) directly affects their margination dynamics, i.e., the lateral drift of particles towards the blood vessel wall [1, 66]. Ferrari and colleagues have shown, through theoretical modeling, that size can significantly affect how particles interact with tumor capillaries during transport [67–69]. Recent reports also suggest a significant role for particle shape in the in vivo performance of delivery vehicles [61, 70]. Specifically, shape and shape-related form factors like aspect ratio or edge geometry affects particle transport characteristics, influences cell-particle interactions, and alters drug release kinetics [53, 71].
Particles flowing in the blood stream experience a number of forces, including hemodynamic forces, buoyancy, as well as electrostatic forces [72]. These forces govern particle trajectory and are due to van der Waals interactions, electrical double layers (EDL), steric interactions, and solvation. As discussed by Decuzzi and colleagues, the force exerted on a particle is affected by the geometry of the particle [69]. By adjusting the material properties, geometry, and surface charge of the particles, these governing forces can be tuned, thus permitting control over particle motion in vivo.
Hemodynamic forces are fluid resistant forces opposing a particle in motion. For example, drag forces on the particle are proportional to its shape in a fluid at moderate to high Reynolds number. Goldmann et al. have demonstrated that hydrodynamic forces increase as the radius of a spherical particle increase and no lateral drift is observed unless an external force (e.g., van der Waals) or electrostatic interactions, are present [73]. However, non-spherical particles could also experience tumbling and rolling due to unequal moments created from hemodynamic forces. For instance, cuboid shaped particles would have a larger drag force due to a normal stagnation plane facing the flow and the possible flow separation at the corners, resulting in a large pressure drag compared to spherical particles. This phenomenon can be used to control margination dynamics. Decuzzi et al. demonstrated that ~3.5 μm ellipsoidal particles with an aspect ratio of two (length to width) drift laterally towards capillary walls due to rotating and tumbling in comparison to spherical particles, which remain centered in the flow channel. Recently, Discher and co-workers reported that self-assembled, filomicelles (i.e., filamentous polymeric micelles) with very high aspect ratios (22–60 nm in diameter, and 2–8 μm in length) remain in the blood stream ten times longer than spherical particles [70, 71]. Another interesting finding was the significant localization of these filomicelles in lung tissue [70]. Unfortunately, the micron-size dimension of these particles is not suitable for intracellular delivery of drugs.
Along with hemodynamic forces, particles in flow also experience buoyancy forces that are proportional to the particle volume, density differences between the particle and fluid, and non-specific electrostatic and van der Waals forces. Through theoretical modeling of spherical particles on the micron to nanometer scale, Decuzzi et al. demonstrated that buoyancy forces dominate when the particle radius is sufficiently large (10000 nm or larger), whereas van der Waals forces between the particle and the blood vessel wall dominate when the particle radius is sufficiently small (1000 nm-50 nm) [66]. As the radius of the particle decreases, the time needed for the particle to reach the endothelium wall increases up to a maximum “critical” radius (around 150 nm); as the radius decreases past this point, the time to reach the wall decreases. Based on these results, particles intended for long circulation times within the blood stream should be close to this critical radius [66]. Due to the balances of the forces acting on the nanocarrier, including van der Waals forces and the hydrodynamic drag, it was concluded that nanocarriers in the 100 nm size range demonstrate a propensity to stay away from the endothelium, and therefore may not be optimal for drug delivery [1, 66, 74]. Particles smaller or larger than 100 nm tend to drift to the edge of the blood stream, making them more advantageous to deliver therapeutic agents to endothelial cells and tumor sites because these sizes are more likely to be taken up by cells [1]. Although fluid mechanics concepts and governing equations indicate that particle geometry should significantly affect their flow characteristics in blood vessels, especially in small capillaries, it is necessary to study, using both computational and experimental models, how shape, size, and aspect ratio of non-spherical nanoscale particles affect in vivo circulation time and accumulation in diseased tissues (e.g., tumors).
2.3 Effects of Shape and Size on Particle Internalization
Particle geometry can also play a role in intracellular uptake of nanocarriers. Regardless of the circulation dynamics associated with any given particle (discussed in section 2.2), particles that are 500 nm and larger are generally only successfully phagocytosed by macrophages and antigen presenting cells. On the other hand, other somatic cells are capable of endocytosing sub-500 nm particles [61]. For drug applications, endocytosis of nanocarriers is often necessary for drugs that target intracellular molecules. It is also imperative to avoid nanocarrier phagocytosis by macrophages, which leads to rapid clearance of particles from the body. It is generally accepted that if particle surface properties are favorable, smaller particles get internalized more efficiently by non-phagocytic cells. Surface curvature, which is dependent on both particle size and shape, could affect interactions between cell and particle surfaces and thus internalization kinetics and efficacy. In this section we will highlight recent research that supports the hypothesis that particle geometry, and more specifically surface curvature, influences cellular internalization.
Chan and colleagues reported that spherical gold nanoparticles have a higher propensity to be internalized in vitro by HeLa cells compared to rod-shaped particles of similar dimensions [30, 31]. Spherical nanoparticles with diameters of 14 or 75 nm were taken up by cells 375–500% more compared to 74 × 14 nm rod-shaped particles. One potential reason for this disparity in in vitro cell uptake could be the difference in particle curvature, which affects the particle contact area with the cell membrane. The Chan group has also shown that cellular uptake is size dependent [30, 31]. Further, Jiang et al. showed that gold and silver nanoparticles with 2–100 nm size range could modify essential cell signaling processes with, 40 and 50 nm particles having the most impact [31]. The affects of shape and size of metallic particles on cellular internalization were also reported by Xu et al. using layered double hydroxide Mg6Al2 nanoparticles with two distinct particle morphologies: 1) rods that were 30–60 nm in width and 100–200 nm in length, and 2) hexagonal sheets that were 50–150 nm wide (laterally) and 10–20 nm thick [75, 76]. The group found that both shapes are quickly taken up by CHO-K1, NIH 3T3, and HEK 293T cell and that rod-like particles specifically target the nucleus while the sheet-like particles are retained in the cytoplasm [75, 76]. To evaluate the cellular mechanisms that controlled internalization of these nanoparticles, a series of uptake inhibitor studies were conducted which showed that internalization of both shapes was mediated by clathrin-medicated endocytosis [75, 76].
Mitragotri et al have also demonstrated that size and curvature affects how a particle “interacts” with the cell and consequently how they are internalized. They demonstrated that in vitro internalization of microscale, solvent-molded non-spherical particles (as described above), is a function of particle orientation with respect to the cell membrane (Figure 3J–L). Intracellular uptake by macrophages was possible if the cell interacted with the particle along the “narrower” dimension as opposed to the “longer” dimension (Figure 3J, K) [61]. These results clearly support the idea that particle curvature affects cellular internalization, but the mechanism of differential uptake should be investigated further.
Figure 3. Effects of particle shape and size on cellular internalization.

(A) Internalization profile of PRINT fabricated particles in HeLa cells over 4 hours. The legend indiates particle diameter and volume [57]. (B–I) SEM images of PRINT particles (all composed of 67 wt% trimethylolpropane ethoxylate triacrylate, 20 wt% PEG monomethylether monometharcylate, 10 wt% aminoethyl methacrylate hydrochloride, 2 wt% fluorescein-o-acrylate, and 1 wt% 2,2-diethoxyacetophenone) evaluated in the internalization study shown in panel A (scale bars = 1μm) [57]: (B) 150 by 450 nm particles, (C) 100 by 300 nm particles, (D) 200 by 200 nm particles; (E–F) Cylindrical microparticles that are 1 μm tall but vary in diameter (scale bars = 1 μm): (E) 0.5 μm and (F) 1 μm; (G–I) Cubic microparticles of varying widths (scale bars = 20 μm): (G) 2 μm, (H) 3 μm, and (I) 5 μm; (J–L) Colored SEM images of aveolar marcophages (brown) interacting with different shape polystyrene particles (purple) [62]: (J) cell membrane engulfing the particle; (scale bar = 10μm) (K) cell attaching itself to the flat side of the elliptical particle (scale bar = 5 μm), and (L) the cell membrane progressing around a spherical particle (scale bar = 5 μm).
DeSimone et al. have also reported preliminary results on the effects of PRINT-fabricated nanoparticle shape and size on in vitro cellular uptake [57]. Internalization of particles of different sizes and aspect ratios, ranging from 5 μm square (cross section) to 100 × 300 nm rods, were evaluated in HeLa cells. The nanoscale cylindrical particles had the highest percentage of cellular internalization over time (Figure 3A). Specifically, nanoparticles with 150 nm diameter and 450 nm height (Figure 4B) exhibited the highest up-take percentage and were internalized four times faster than symmetrical particles (aspect ratio of 1, 200 × 200 nm) (Figure 3D). These findings suggest that particle aspect ratio could play an important role in cellular uptake. However, in the same experiment, 100 nm diameter particles (Figure 3C) with an aspect ratio of 3 had a lower degree of internalization compared to 150 nm particles with the same aspect ratio. This would indicate that the uptake kinetics is likely a function of both size and shape. The cylindrical shaped particles (Figure 3E–F) with 500 nm or 1 μm diameters and 1 μm height had reduced internalization in comparison to smaller particles but showed higher uptake than micron-sized square cross-section particles. The internalization profiles of the cylindrical particles were similar to the profile associated with 2 μm cubic particles (Figure 3G). Larger (5 μm and 3 μm cubic particles) (Figure 3H–I) did not show significant uptake by HeLa cells (Figure 3B–D).
Figure 4. Examples of responsive nanocarriers for triggered drug release.
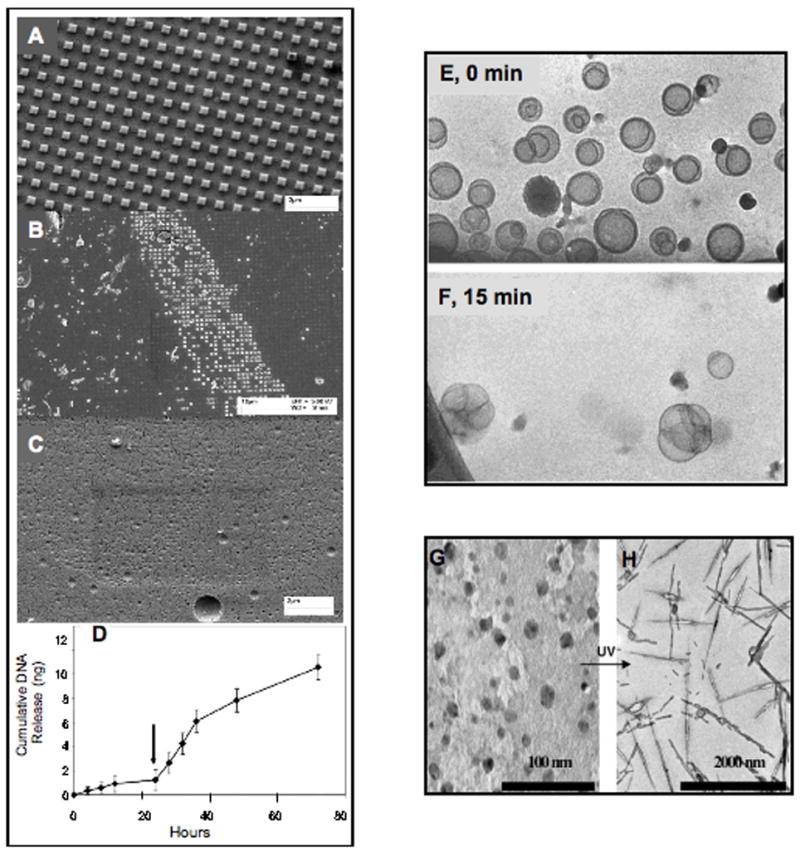
(A–C) SEM images demonstrating the enzymatic degradation of imprinted 75% (v) PEGDA-GFLGK-diacrylate nanocarriers: (A) Control particles after 48 h in PBS (i.e., no Cathepsin B) (scale bar = 2 μm), (B) Particles after 12 h in Cathepsin B (25 U/mL) (scale bar = 10 μm), (C) Particles after 48 h in Cathepsin B (25 U/mL) (scale bar = 2 μm). (D) Release profile of stimuli-responsive imprinted PEG-DAGFLGK-DA particles encapsulating 0.16% (w/w) plasmid DNA in response to 20 U/mL Cathepsin B[54]. (E, F) Time-dependent cryogenic transmission electron microscopy of 3:97 (R)-1,2-di-O-(1′Z,9′Z-octadecadienyl)-glyceryl-3-(ω-methoxy-poly(ethylene glycolate), MW5000)/fusogenic lipid 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (BVEP/DOPE) unilamellar liposomes at pH 4.5. Liposomes undergo fusing and drug release over time[17]. (G, H) Photoinduced changes in the aggregation of light sensitive nanoparticles. TEM images for 1.4 wt % 1:3 cetyltrimethylamonium bromide/sodium 4-hexylphenylazosulfonate (CTAB/C6PAS) systems in D2O (G) before and (H) after irradiation [18].
The results summarized above, demonstrate that particle aspect ratio, shape, and volume all affect cellular internalization of nanoparticles. A more comprehensive evaluation of uptake as a function of each of these particle characteristics needs to be performed. It is critical that these experiments are conducted while attempting to hold other particle properties (i.e., volume, surface area, material composition) constant. Nevertheless, the theoretical modeling and in vitro studies taken together provide significant evidence to support the idea that nanoparticle-based drug delivery to target cells can be “controlled” through rational design of particle geometry.
3. Responsive Nanocarriers for Triggered Drug Release
Although particle geometry can be designed to optimize drug carrier localization in vivo, it is also critical that additional control on drug release is incorporated within the carrier design. It is well known that particle composition can be tuned to control the kinetics and mechanism of payload release. The following section provides an overview of various triggered release mechanisms used in drug carrier design.
Nanocarrier systems in which drug release is triggered by physiological or pathological cues are generally prepared with materials that are responsive to pH, oxidation potential, or enzymes [77]. In all cases the physiological stimulus causes some physical or chemical change to the carrier material, such that the nanocarriers collapse or degrade, releasing drug either intracellularly or extracellularly.
The earliest attempts to direct drug delivery to specific tissues were primarily relevant to cancer therapy because there is a clear physiological difference between cancerous tissue and healthy tissue. Many delivery methods rely on the enhanced permeability and retention (EPR) effect associated with tumors (particle size effects) as well as the differential pH of the tumor microenvironment (pH sensitive carriers) [29, 77–85]. These have been reviewed by Peer et al. [2]. For pH sensitive carriers, anionic or cationic materials are often incorporated into block copolymers with one or two other hydrophobic (e.g., poly(lactic acid)) or hydrophilic (e.g., PEG) polymers. Generally, the key to designing these heteromaterials is that one block must be protonatable at a pH less than physiological pH (7.4), such that in the presence of the more acidic extracellular environment (pH 6.5–7), associated with tumors and other diseased tissues, sections of these materials can undergo a change in hydrophobicity. This results in swelling, shrinking, or collapse of the nanocarrier resulting in drug release. Alternatively, materials can be selected that degrade (i.e., hydrolyze) rapidly with pH change [17, 77, 78, 84]. Following hydrolysis of acid-labile subunits the hydrophobic portion of the nanocarrier is either exposed to the aqueous environment or degraded into hydrophilic products resulting in drug release (Figure 4E, F) [17, 77].
Other pH sensitive nanocarriers have been developed that can also be classified as ligand-triggered drug release systems. These nanocarriers respond to pH similarly to those systems described above. However, drug release is triggered by a more dramatic pH decrease that results from ligand binding to, and breakdown by, nanoparticle-immobilized enzyme. This has been applied to insulin delivery [82, 86]. A significant limitation of pH triggered release, as a means of targeted drug delivery, is that it is not specific to diseased tissues, since there are healthy tissues and cells that could also have an acidic extracellular environment. For physiological triggered release to be effective, a highly specific disease-related trigger needs to be used. Enzyme sensitive biopolymers could allow such disease-induced drug delivery.
In most pathophysiological conditions, there are specific enzymes and proteins that are up regulated to meet the needs of the diseased tissue. For instance, enzymes and proteins usually associated with rapidly growing cells are up-regulated in cancers. Development of biomaterials that are sensitive to these specific enzymes or proteins allows for fabrication of highly specific triggered release systems. These delivery systems are designed to be crosslinked by peptide sequences or other ligands that are recognized and broken down by a particular enzyme that is expressed in a specific diseased environment or cell. Therefore, in the presence of the appropriate enzyme the ligand linkers are cleaved and the nanocarriers’ contents are released. Although enzyme-responsive drug delivery and tissue engineering platforms have been widely explored in the macroscale [87, 88], there have been few reports on the development of enzyme-degradable nanocarriers. Examples of enzyme-triggered drug releasing nanoparticles include those responsive to elastase [89, 90], trypsin [91], (phospho)lipase [92, 93], metalloproteinase [94–96], proteinase K [97], pronase E [98], and quinone reductases.[99]
Nanoparticles that are sensitive to changes in oxidation potential have also been developed. These take advantage of the more reductive intracellular environment as compared to the extracellular fluid. There are two typical approaches to synthesize these nanoparticles, both involving disulfide linkages. In one approach, lipid-polymer conjugates are created by tethering a hydrophobic lipid to a hydrophilic polymer via disulfide bonds [83, 100, 101]. The hydrophilic shell stabilizes the liposomes in the extracellular environment, but once within a cell the shell is lost because the disulfide bonds are susceptible to cleavage by reductive substances, such as glutathione. The loss of the hydrophilic coating destabilizes the liposomes and results in drug release. A similar destabilization of nanocarrier integrity is achieved in the cellular milieu when disulfide linkers are used to crosslink the core of polyplex-micelles [102].
Although nanocarriers that are sensitive to internal triggers require no additional external regulation or guidance, most of the internal triggers that these systems rely on are hardly unique to diseased tissues. If nanoparticles are non-specifically taken up by healthy tissues the acidic pH associated with endosomes and lysozomes and the reductive intracellular environment will trigger drug release from pH-sensitive and redox-sensitive drug carriers, respectively. Therefore, tissue-specific delivery is not optimal. While the EPR effect will bias nanocarrier accumulation to most diseased tissues, uptake by healthy tissues is usually unavoidable because the aforementioned nanoparticles are not actually targeted to any particular tissue. Also, the EPR effect is not necessarily a feature of all diseased or damaged tissues. Enzyme sensitive nanocarrier systems are, however, considered advantageous compared to redox- and pH-sensitive systems because drug release depends on the presence of a particular enzyme, which if only present in the diseased cells will limit drug release to the target cells. Regardless, during circulation and once near or inside healthy tissues these types of nanocarriers, like most drug carriers sensitive to physiological stimuli, are still prone to drug release (even if it is at a slower rate). This is due to the fact that even healthy cells express, to some extent, most of the enzymes that are over-expressed in diseased tissues. It will be critical for future research in this area to “tune” the enzyme-triggered degradation to the local concentration of the enzyme (i.e., incorporate threshold-based degradation).
In addition to physiological or pathophysiological triggers, there are a vast array of nanocarrier systems that are responsive to external physical cues [16], such as light [18, 103–105], temperature [106–110], ultrasound [15, 111–114], and magnetic fields [14]. In all these cases, time must be allowed for nanoparticle uptake into diseased tissues prior to the application of external stimuli. Nanocarriers that are designed to respond to these external triggers, in theory only release carrier contents at the location of the stimulus application. Most light responsive nanoparticles conceptually function like many pH- and redox-sensitive carriers, in that the destabilization of nanocarriers is achieved upon irreversible photo-degradation of an outer hydrophilic layer (Figure 4G, H) [18] or cleavage (photolysis in this case) of labile polar head groups from hydrophobic “tails” [103]. On the other hand, temperature sensitive nanocarriers are designed from polymers that have a phase transition [106, 107] or a positive swelling transition [109] at biologically acceptable temperatures (35–42°C). The change in physical properties (e.g., liquification, swelling) that accompanies such material transitions causes drug release from the carriers. For further information on these types of nanoparticles the reader is referred to a review written by Needham and Dewhirst on the application of hyperthermia-responsive liposomes for drug delivery to solid tumors [115]. Alternatively, ultrasound responsive nanocarriers, including pluronic micelles [15, 27, 28, 111, 112, 116, 117] and DNA-polycation polyplexes [118–121] have been shown to give rise to improved delivery in the presence of ultrasonic irradiation.
Despite the fact that externally triggered drug release from nanocarrier systems generally permits improved site-specific drug delivery, and therefore, requires lower drug doses compared to systems that are designed to respond to physiological stimuli, the cost of treatment would be higher and patient compliance would likely be reduced. In addition, majority of the external trigger is restricted to superficial tissues and is not applicable to internal organs or tumors.
4. Incorporation of Triggered-release into Shape-specific Drug Nanocarriers
In the prior sections of this review we have presented the advantages of fabricating shape and size specific nanoparticles, as well as the advantages of disease-responsive nanoparticles. Incorporating enzymatically degradable biomaterials into shape and size specific nanoparticles combines both these concepts and could provide an interesting approach for the development of “intelligent” drug nanocarriers. Particles fabricated using the modified S-FIL process have been designed to have well controlled geometry, as well as enzyme-triggered release mechanism. These PEG-based particles were nanoimprinted using a penta-peptide crosslinker (GFLGK) [54]. This peptide linker is degraded by the lysosomal enzyme Cathepsin B, which is present intracellularly and is also highly up regulated in certain tumors (present at high levels in the tumor microenvironment). Enzyme-triggered degradation of imprints containing the GFLGK peptide was evaluated using SEM (Figure 4A–C). The nanoparticles began to degrade within 30 minutes and between 24 and 48 hours all particles were fully degraded. On the other hand, particles exposed to aqueous solution without Cathepsin B remained intact during this period. Stimuli-responsive release of encapsulated antibodies (Alexa Fluor 594 labeled goat anti mouse IgG, Invitrogen) and plasmid DNA from imprinted PEGDA-GFLGK-DA (75% v) nanoparticles were also evaluated. The amount of released agents over time before and after addition of Cathepsin B was measured and as shown in Figure 4D, efficient enzyme triggered release was achieved. These results demonstrate that it is feasible to incorporate both geometrical (i.e., size, shape, and aspect ratio) parameters and disease-triggered drug release mechanisms in a single nanocarrier.
5. Expert Opinion
The use of nanofabrication methods that are compatible with biological reagents permit, for the first time, the evaluation of particle geometry as a tunable parameter in nanoscale drug delivery systems. Despite the promise of such methods in reducing drug-related side effects and increasing efficacy, there remain significant challenges. A major concern is the number (and hence mass) of nanoparticles that can be readily synthesized using these sophisticated methods. Both the S-FIL and PRINT methods are yet to demonstrate (a) large, pharmaceutical scale synthesis of nanoparticles and (b) fabrication of particles with high drug loading efficacy. Also, the versatility of the solvent molding methods, in terms of material composition, is yet to be demonstrated. The combination of manipulating nanoscale geometry and incorporating triggered release within the same nanocarrier design opens up new directions in drug delivery. It would be beneficial if both the PRINT and solvent molding processes could incorporate disease-sensitive release triggers within the synthesized nanocarriers. But above all, it remains to be demonstrated conclusively whether size, shape, and triggered release have the proposed benefits in vivo or not, either in anti-cancer therapy or even in other disease models. It is encouraging that more studies are being reported that address the effects of shape and size on particle biodistribution through both theoretical and experimental approaches [122, 123]. However, extensive in vivo studies are necessary to prove that these parameters matter and that they indeed alter capillary transport, cell uptake, and drug release kinetics, especially for nanoparticles. It is also imperative that more in-depth theoretical modeling studies along with in vitro experiments using micro- and nanofluidics be conducted to better describe and quantitatively assess how particle geometry and triggered release affect the intrinsic behavior of nanoparticles. In parallel, development of strategies for large scale manufacturing that includes (a) high-throughput production of enough particles to perform extensive animal studies and (b) minimal “waste” of expensive biomolecules and drugs, is needed. The promise of such systems can only be achieved if the cost benefit ratio is attractive to biotech and pharmaceutical companies. Therefore, inter-disciplinary research involving biomedical, chemical, and mechanical/manufacturing engineers along with chemists and biologists is necessary to move the field forward.
Article highlights box
Integration of particle geometry and disease-triggered drug release concepts into our existing knowledge of surface ligand-based targeting would result in a quantum change in the efficacy of the next generation drug carriers.
Top-down fabrication methods could provide precise, pre-designed control over particle geometry (shape, aspect ratio) and composition, which is often difficult to achieve using bottom-up approaches that tend to rely on emulsions or self-assembly.
The geometry of nanocarriers (e.g., shape, aspect ratio, and ratio of particle dimensions to vessel diameter) directly affects their margination dynamics
Particle aspect ratio, shape, and volume all affect cellular internalization of nanoparticles
Although particle geometry can be designed to optimize drug carrier localization in vivo, it is also critical that additional control on drug release is incorporated within the carrier design. It is well known that particle composition can be tuned to control the kinetics and mechanism of payload release
Extensive in vivo studies are necessary to prove that these parameters matter and that they indeed alter capillary transport, cell uptake, and drug release kinetics, especially for nanoparticles. It is also imperative that more in-depth theoretical modeling studies along with in vitro experiments using micro- and nanofluidics be conducted to better describe and quantitatively assess how particle geometry and triggered release affect the intrinsic behavior of nanoparticles
Acknowledgments
The authors would like to acknowledge partial funding from the National Science Foundation and the National Institutes of Health for their work on shape-specific nanoparticles.
Footnotes
6. Conflict of interest: The authors do not have any conflict of interest to declare.
References
- 1.Ferrari M. Cancer nanotechnology: opportunities and challenges. Nature Reviews Cancer. 2005;5(3):161–71. doi: 10.1038/nrc1566. [DOI] [PubMed] [Google Scholar]
- 2.Peer D, Karp JM, Hong S, et al. Nanocarriers as an emerging platform for cancer therapy. Nature Nanotechnology. 2007;2(12):751–60. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]
- 3.Zhang SF, Uludag H. Nanoparticulate Systems for Growth Factor Delivery. Pharmaceutical Research. 2009;26(7):1561–80. doi: 10.1007/s11095-009-9897-z. [DOI] [PubMed] [Google Scholar]
- 4.Fenske DB, Chonn A, Cullis PR. Liposomal Nanomedicines: An Emerging Field. Toxicologic Pathology. 2008;36(1):21–9. doi: 10.1177/0192623307310960. [DOI] [PubMed] [Google Scholar]
- 5.Duncan R. The dawning era of polymer therapeutics.(Report) Nature Reviews Drug Discovery. 2003;2(5):347. doi: 10.1038/nrd1088. 14. [DOI] [PubMed] [Google Scholar]
- 6.Jeong JH, Kim SW, Park TG. Molecular design of functional polymers for gene therapy. Progress in Polymer Science. 2007;32(11):1239–74. [Google Scholar]
- 7.Samad A, Sultana Y, Aqil M. Liposomal Drug Delivery Systems: An Update Review. Current Drug Delivery. 2007;4(4):297–305. doi: 10.2174/156720107782151269. [DOI] [PubMed] [Google Scholar]
- 8.Torchilin VP. Recent advances with liposomes as pharmaceutical carriers.(Report) Nature Reviews Drug Discovery. 2005;4(2):145. doi: 10.1038/nrd1632. 16. [DOI] [PubMed] [Google Scholar]
- 9.Goldsmith HL, VTT Rheological aspects of thrombosis and hemostasis - basic principles and applications - Icth-Report - Subcommittee on Rheology of the International Committee on Thrombosis and Hemostasis. Thromb Haemost. 1986;55 (3):415–35. [PubMed] [Google Scholar]
- 10.Patil VRS, Campbell CJ, Yun YH, et al. Particle Diameter Influences Adhesion under Flow. Biophysical Journal. 2001;80(4):1733–43. doi: 10.1016/s0006-3495(01)76144-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lamprecht A, Schäfer U, Lehr C-M. Size-Dependent Bioadhesion of Micro- and Nanoparticulate Carriers to the Inflamed Colonic Mucosa. Pharmaceutical Research. 2001;18(6):788–93. doi: 10.1023/a:1011032328064. [DOI] [PubMed] [Google Scholar]
- 12.Stolnik S, Illum L, Davis SS. Long circulating microparticulate drug carriers. Advanced Drug Delivery Reviews. 1995;16(2–3):195–214. [Google Scholar]
- 13.Robert Langer NAP. Advances in biomaterials, drug delivery, and bionanotechnology. AIChE Journal. 2003;49(12):2990–3006. [Google Scholar]
- 14.Brazel CS. Magnetothermally-responsive Nanomaterials: Combining Magnetic Nanostructures and Thermally-Sensitive Polymers for Triggered Drug Release. Pharmaceutical Research. 2009;26(3):644–56. doi: 10.1007/s11095-008-9773-2. [DOI] [PubMed] [Google Scholar]
- 15.Husseini GA, Pitt WG. Ultrasonic-Activated Micellar Drug Delivery for Cancer Treatment. Journal of Pharmaceutical Sciences. 2009;98(3):795–811. doi: 10.1002/jps.21444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meng F, Zhong Z, Feijen J. Stimuli-Responsive Polymersomes for Programmed Drug Delivery. Biomacromolec. 2009;10(2):197–209. doi: 10.1021/bm801127d. [DOI] [PubMed] [Google Scholar]
- 17.Boomer JA, Inerowicz HD, Zhang ZY, et al. Acid-triggered release from sterically stabilized fusogenic liposomes via a hydrolytic DePEGylation strategy. Langmuir. 2003;19(16):6408–15. [Google Scholar]
- 18.Eastoe J, Vesperinas A, Donnewirth AC, et al. Photodestructible vesicles. Langmuir. 2006;22(3):851–3. doi: 10.1021/la052882r. [DOI] [PubMed] [Google Scholar]
- 19.Moghimi SM, Hunter AC, Murray JC. Long-circulating and target-specific nanoparticles: theory to practice. Pharm Rev. 2001;53(2):283–318. [PubMed] [Google Scholar]
- 20.Illum L, Davis SS, Wilson CG, et al. Blood clearance and organ deposition of intravenously administered colloidal particles - the effects of particle-size, nature and shape. 1982;12:2–3. (Int. J. Pharm.):135–46. [Google Scholar]
- 21.Tabata Y, Ikada Y. Phagocytosis of polymer microspheres by macrophages. New Polymer Materials. 1990:107–41. [Google Scholar]
- 22.Panyam J, Dali MM, Sahoo SK, et al. Polymer degradation and in vitro release of a model protein from poly(,-lactide-co-glycolide) nano- and microparticles. Journal of Controlled Release. 2003;92(1–2):173–87. doi: 10.1016/s0168-3659(03)00328-6. [DOI] [PubMed] [Google Scholar]
- 23.Dunne M, Corrigan OI, Ramtoola Z. Influence of particle size and dissolution conditions on the degradation properties of polylactide-co-glycolide particles. Biomaterials. 2000;21(16):1659–68. doi: 10.1016/s0142-9612(00)00040-5. [DOI] [PubMed] [Google Scholar]
- 24.Pasut G, Veronese FM. Polymer-drug conjugation, recent achievements and general strategies. Progress in Polymer Science. 2007;32(8–9):933–61. [Google Scholar]
- 25.Ainslie KM, Desai TA. Microfabricated implants for applications in therapeutic delivery, tissue engineering, and biosensing. Lab on a Chip. 2008;8(11):1864–78. doi: 10.1039/b806446f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gates BD, Xu Q, Stewart M, et al. New Approaches to Nanofabrication: Molding, Printing, and Other Techniques. Chemical Reviews. 2005;105(4):1171–96. doi: 10.1021/cr030076o. [DOI] [PubMed] [Google Scholar]
- 27.Gao Z, Fain HD, Rapoport N. Ultrasound-enhanced tumor targeting of polymeric micellar drug carriers. Molecular Pharmaceutics. 2004;1(4):317–30. doi: 10.1021/mp049958h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gao ZG, Fain HD, Rapoport N. Controlled and targeted tumor chemotherapy by micellar-encapsulated drug and ultrasound. Journal of Controlled Release. 2005;102(1):203–22. doi: 10.1016/j.jconrel.2004.09.021. [DOI] [PubMed] [Google Scholar]
- 29.Gao ZG, Lee DH, Kim DI, et al. Doxorubicin loaded pH-sensitive micelle targeting acidic extracellular pH of human ovarian A2780 tumor in mice. Journal of Drug Targeting. 2005;13(7):391–7. doi: 10.1080/10611860500376741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chithrani BD, Chan WCW. Elucidating the Mechanism of Cellular Uptake and Removal of Protein-Coated Gold Nanoparticles of Different Sizes and Shapes. Nano Lett. 2007;7(6):1542–50. doi: 10.1021/nl070363y. [DOI] [PubMed] [Google Scholar]
- 31.Jiang W, KimBetty YS, Rutka JT, et al. Nanoparticle-mediated cellular response is size-dependent. Nat Nano. 2008;3(3):145–50. doi: 10.1038/nnano.2008.30. [DOI] [PubMed] [Google Scholar]
- 32.Xia Y, Whitesides GM. Soft Lithography. Angew Chem, Int Ed. 1998;37(5):550–75. doi: 10.1002/(SICI)1521-3773(19980316)37:5<550::AID-ANIE550>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 33.Resnick DJ, Sreenivasan SV, Wilson CG. Step and flash imprint lithography. materials today. 2005:34–42. [Google Scholar]
- 34.Koh WG, Revzin A, Pishko MV. Poly(ethylene glycol) Hydrogel Microstructures Encapsulating Living Cells. Langmuir. 2002;18(7):2459–62. doi: 10.1021/la0115740. [DOI] [PubMed] [Google Scholar]
- 35.Stephen YC, Peter RK, Zhang W, et al. Sub - 10 nm imprint lithography and applications. Journal of Vacuum Science & Technology B: Microelectronics and Nanometer Structures. 1997;15(6):2897–904. [Google Scholar]
- 36.Chou SY, Krauss PR, Renstrom PJ. Imprint Lithography with 25-Nanometer Resolution. Science. 1996;272(5258):85–7. [Google Scholar]
- 37.Bacher W, Bade K, Matthis B, et al. Fabrication of LIGA mold inserts. Microsystem Technologies. 1998;4(3):117–9. [Google Scholar]
- 38.Matthew C, Annette G, Byung Jin C, et al. Patterning nonflat substrates with a low pressure, room temperature, imprint lithography process. Journal of Vacuum Science & Technology B: Microelectronics and Nanometer Structures. 2001;19(6):2162–72. [Google Scholar]
- 39.Jan H, Martin V, Kees van den H, et al. Mold-assisted nanolithography: A process for reliable pattern replication. AVS; 1996. pp. 4124–8. [Google Scholar]
- 40.Bender M, Otto M, Hadam B, et al. Fabrication of nanostructures using a UV-based imprint technique. Microelectronic Engineering. 2000;53(1–4):233–6. [Google Scholar]
- 41.Gale MT. Replication techniques for diffractive optical elements. Microelectronic Engineering. 1997;34(3–4):321–39. [Google Scholar]
- 42.Shvartsman FP. Holographic optical elements by dry photopolymer embossing. SPIE. 1991;1461(5):313–20. [Google Scholar]
- 43.Desai TA, Chu WH, Tu JK, et al. Microfabricated immunoisolating biocapsules. Biotechnology and Bioengineering. 1998;57(1):118–20. doi: 10.1002/(sici)1097-0290(19980105)57:1<118::aid-bit14>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 44.Desai TA, Hansford DJ, Kulinsky L, et al. Nanopore Technology for Biomedical Applications. Biomedical Microdevices. 1999;2(1):11–40. [Google Scholar]
- 45.Tao SL, Desai TA. Micromachined polymeric devices for applications in targeted drug delivery. Journal of the Association for Laboratory Automation. 2004;9(3):155–8. [Google Scholar]
- 46.Tao SL, Popat K, Desai TA. Off-wafer fabrication and surface modification of asymmetric 3D SU-8 microparticles. Nat Protocols. 2007;1(6):3153–8. doi: 10.1038/nprot.2006.451. [DOI] [PubMed] [Google Scholar]
- 47.Tao SL, Desai TA. Microfabricated drug delivery systems: from particles to pores. Advanced Drug Delivery Reviews. 2003;55(3):315–28. doi: 10.1016/s0169-409x(02)00227-2. [DOI] [PubMed] [Google Scholar]
- 48.Ahmed A, Bonner C, Desai TA. Bioadhesive Microdevices for Drug Delivery: A Feasibility Study. Biomedical Microdevices. 2001;3(2):89–96. [Google Scholar]
- 49.Tao SL, Desai TA. Microfabrication of Multilayer, Asymmetric, Polymeric Devices for Drug Delivery. Advanced Materials. 2005;17(13):1625–30. [Google Scholar]
- 50.Dendukuri D, Hatton TA, Doyle PS. Synthesis and Self-Assembly of Amphiphilic Polymeric Microparticles. Langmuir. 2006;23(8):4669–74. doi: 10.1021/la062512i. [DOI] [PubMed] [Google Scholar]
- 51.Dendukuri D, Pregibon DC, Collins J, et al. Continuous-flow lithography for high-throughput microparticle synthesis. Nat Mater. 2006;5(5):365–9. doi: 10.1038/nmat1617. [DOI] [PubMed] [Google Scholar]
- 52.Dendukuri D, Tsoi K, Hatton TA, et al. Controlled Synthesis of Nonspherical Microparticles Using Microfluidics. Langmuir. 2005;21(6):2113–6. doi: 10.1021/la047368k. [DOI] [PubMed] [Google Scholar]
- 53.Champion JA, Katare YK, Mitragotri S. Particle shape: A new design parameter for micro- and nanoscale drug delivery carriers. Journal of Controlled Release. 2007;121(1–2):3–9. doi: 10.1016/j.jconrel.2007.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54**.Glangchai LC, Caldorera-Moore M, Shi L, et al. Nanoimprint lithography based fabrication of shape-specific, enzymatically-triggered smart nanoparticles. Journal of Controlled Release. 2008;125(3):263–72. doi: 10.1016/j.jconrel.2007.10.021. (Paper describing the SFIL technology for nanoaprticle syntehsis) [DOI] [PubMed] [Google Scholar]
- 55.Euliss LE, Welch CM, Maynor BW, et al. Advances in Resist Technology and Processing XXIII 2006. San Jose, CA, USA: SPIE; 2006. Monodisperse nanocarriers: novel fabrication of polymeric nanoparticles for bio-nanotechnology; p. 61534A-8. [Google Scholar]
- 56.Gratton SEA, Pohlhaus PD, Lee J, et al. Nanofabricated particles for engineered drug therapies: A preliminary biodistribution study of PRINT^(TM) nanoparticles. Journal of Controlled Release. 2007;121(1):10–8. doi: 10.1016/j.jconrel.2007.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gratton SEA, Ropp PA, Pohlhaus PD, et al. The effect of particle design on cellular internalization pathways. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(33):11613–8. doi: 10.1073/pnas.0801763105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kenton BW, Natasha SW, Kevin PH, et al. In: Soft lithography using perfluorinated polyether molds and PRINT technology for fabrication of 3-D arrays on glass substrates. Michael JL, editor. SPIE; 2006. p. 61513F. [Google Scholar]
- 59.Pandya AA, Maynor BW, Gratton SEA, et al. Emerging Lithographic Technologies X. San Jose, CA, USA: SPIE; 2006. Fabrication of organic nanoparticles by PRINT: master generation using lithographic and RIE techniques; p. 61513C-6. [Google Scholar]
- 60**.Rolland JP, Maynor BW, Euliss LE, et al. Direct Fabrication and Harvesting of Monodisperse, Shape-Specific Nanobiomaterials. J Am Chem Soc. 2005;127(28):10096–100. doi: 10.1021/ja051977c. First paper describing the PRINT method for nanoparticle synthesis. [DOI] [PubMed] [Google Scholar]
- 61**.Champion JA, Katare YK, Mitragotri S. Making polymeric micro- and nanoparticles of complex shapes. Proceedings Of The National Academy Of Sciences Of The United States Of America. 2007;104(29):11901–4. doi: 10.1073/pnas.0705326104. Paper describing the solvent-molding method for particle synthesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Champion JA, Mitragotri S. Role of target geometry in phagocytosis. Proc Natl Acad Sci U S A. 2006;103(13):4930–4. doi: 10.1073/pnas.0600997103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Truskett VN, Watts MPC. Trends in imprint lithography for biological applications. Trends in Biotechnology. 2006;24(7):312–7. doi: 10.1016/j.tibtech.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 64.Dorian AC, Kevin PH, Joseph MD. Top-down particle fabrication: control of size and shape for diagnostic imaging and drug delivery. Wiley Interdisciplinary Reviews: Nanomedicine and Nanobiotechnology. 2009;1(4):391–404. doi: 10.1002/wnan.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kelly JY, DeSimone JM. Shape-Specific, Monodisperse Nano-Molding of Protein Particles. Journal of the American Chemical Society. 2008;130(16):5438–9. doi: 10.1021/ja8014428. [DOI] [PubMed] [Google Scholar]
- 66**.Decuzzi P, Lee S, Bhushan B, et al. A Theoretical Model for the Margination of Particles within Blood Vessels. Annals of Biomedical Engineering. 2005;33(2):179–90. doi: 10.1007/s10439-005-8976-5. Paper describing margination dynamics of shape-specific particles. [DOI] [PubMed] [Google Scholar]
- 67.Decuzzi P, Ferrari M. The adhesive strength of non-spherical particles mediated by specific interactions. Biomaterials. 2006;27(30):5307–14. doi: 10.1016/j.biomaterials.2006.05.024. [DOI] [PubMed] [Google Scholar]
- 68.Decuzzi P, Causa F, Ferrari M, et al. The effective dispersion of nanovectors within the tumor microvasculature. Ann Biomed Eng. 2006;34(4):633–41. doi: 10.1007/s10439-005-9072-6. [DOI] [PubMed] [Google Scholar]
- 69.Decuzzi P, Pasqualini R, Arap W, et al. Intravascular Delivery of Particulate Systems: Does Geometry Really Matter? Pharmaceutical Research. 2009 Jan;26(1):235–43. doi: 10.1007/s11095-008-9697-x. [DOI] [PubMed] [Google Scholar]
- 70.Geng Y, Dalhaimer P, Cai S, et al. Shape effects of filaments versus spherical particles in flow and drug delivery. Nat Nano. 2007;2(4):249–55. doi: 10.1038/nnano.2007.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nishiyama N. Nanomedicine: Nanocarriers shape up for long life. Nat Nano. 2007;2(4):203–4. doi: 10.1038/nnano.2007.88. [DOI] [PubMed] [Google Scholar]
- 72.Furlani EP, Ng KC. Analytical model of magnetic nanoparticle transport and capture in the microvasculature. Physical Review E (Statistical, Nonlinear, and Soft Matter Physics) 2006;73(6):061919. doi: 10.1103/PhysRevE.73.061919. [DOI] [PubMed] [Google Scholar]
- 73.Goldman AJ, Cox RG, Brenner H. Slow viscous motion of a sphere parallel to a plane wall--II Couette flow. Chemical Engineering Science. 1967;22(4):653–60. [Google Scholar]
- 74.Liotta LA, Ferrari M, Petricoin E. Clinical proteomics: Written in blood. Nature. 2003;425(6961):905. doi: 10.1038/425905a. [DOI] [PubMed] [Google Scholar]
- 75.Ladewig K, Xu ZP, Lu GQ. Layered double hydroxide nanoparticles in gene and drug delivery. Expert Opinion on Drug Delivery. 2009;6(9):907–22. doi: 10.1517/17425240903130585. [DOI] [PubMed] [Google Scholar]
- 76.Xu ZP, Niebert M, Porazik K, et al. Subcellular compartment targeting of layered double hydroxide nanoparticles. Journal of Controlled Release. 2008;130(1):86–94. doi: 10.1016/j.jconrel.2008.05.021. [DOI] [PubMed] [Google Scholar]
- 77.Chen W, Meng F, Li F, et al. pH-Responsive Biodegradable Micelles Based on Acid-Labile Polycarbonate Hydrophobe: Synthesis and Triggered Drug Release. Biomacromolecules. 2009;10:1727–35. doi: 10.1021/bm900074d. [DOI] [PubMed] [Google Scholar]
- 78.Oh KT, Kim D, You HH, et al. pH-sensitive properties of surface charge-switched multifunctional polymeric micelle. International Journal of Pharmaceutics. 2009;376(1–2):134–40. doi: 10.1016/j.ijpharm.2009.04.021. [DOI] [PubMed] [Google Scholar]
- 79.Lee ES, Oh KT, Kim D, et al. Tumor pH-responsive flower-like micelles of poly(L-lactic acid)-b-poly (ethylene glycol)-b-poly(L-histidine) Journal of Controlled Release. 2007;123(1):19–26. doi: 10.1016/j.jconrel.2007.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bae Y, Jang WD, Nishiyama N, et al. Multifunctional polymeric micelles with folate-mediated cancer cell targeting and pH-triggered drug releasing properties for active intracellular drug delivery. Molecular Biosystems. 2005;1(3):242–50. doi: 10.1039/b500266d. [DOI] [PubMed] [Google Scholar]
- 81.Oh KT, Oh YT, Oh NM, et al. A smart flower-like polymeric micelle for pH-triggered anticancer drug release. International Journal of Pharmaceutics. 2009;375(1–2):163–9. doi: 10.1016/j.ijpharm.2009.04.005. [DOI] [PubMed] [Google Scholar]
- 82.Jo SM, Kim JC. Glucose-triggered release from liposomes incorporating poly(N-isopropylacrylamide-co-methacrylic acid-co-octadecylacrylate) and glucose oxidase. Colloid and Polymer Science. 2009;287(4):379–84. [Google Scholar]
- 83.Zalipsky S, Qazen M, Walker JA, et al. New detachable poly(ethylene glycol) conjugates: Cysteine-cleavable lipopolymers regenerating natural phospholipid, diacyl phosphatidylethanolamine. Bioconjugate Chemistry. 1999;10(5):703–7. doi: 10.1021/bc990031n. [DOI] [PubMed] [Google Scholar]
- 84.Guo X, Szoka FC. Steric stabilization of fusogenic liposomes by a low-pH sensitive PEG-diortho ester-lipid conjugate. Bioconjugate Chemistry. 2001;12(2):291–300. doi: 10.1021/bc000110v. [DOI] [PubMed] [Google Scholar]
- 85.Shenoy D, Little S, Langer R, et al. Poly(ethylene oxide)-modified poly(beta-amino ester) nanoparticles as a pH-sensitive system for tumor-targeted delivery of hydrophobic drugs: Part 2. In vivo distribution and tumor localization studies. Pharmaceutical Research. 2005;22(12):2107–14. doi: 10.1007/s11095-005-8343-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jain SK, Amit KC, Chalasani KB, et al. Enzyme triggered pH sensitive liposomes for insulin delivery. Journal of Drug Delivery Science and Technology. 2007;17(6):399–405. [Google Scholar]
- 87.Kim S, Chung EH, Gilbert M, et al. Synthetic MMP-13 degradable ECMs based on poly(N-isopropylacrylamide-co-acrylic acid) semi-interpenetrating polymer networks. I. Degradation and cell migration. Journal of Biomedical Materials Research Part A. 2005;75A(1):73–88. doi: 10.1002/jbm.a.30375. [DOI] [PubMed] [Google Scholar]
- 88.Kim S, Healy KE. Synthesis and characterization of injectable poly(N-isopropylacrylamide-co-acrylic acid) hydrogels with proteolytically degradable cross-links. Biomacromolecules. 2003;4(5):1214–23. doi: 10.1021/bm0340467. [DOI] [PubMed] [Google Scholar]
- 89.Pak CC, Ali S, Janoff AS, et al. Triggerable liposomal fusion by enzyme cleavage of a novel peptide-lipid conjugate. Biochimica Et Biophysica Acta-Biomembranes. 1998;1372(1):13–27. doi: 10.1016/s0005-2736(98)00041-8. [DOI] [PubMed] [Google Scholar]
- 90.Pak CC, Erukulla RK, Ahl PL, et al. Elastase activated liposomal delivery to nucleated cells. Biochimica Et Biophysica Acta-Biomembranes. 1999;1419(2):111–26. doi: 10.1016/s0005-2736(98)00256-9. [DOI] [PubMed] [Google Scholar]
- 91.Hu LR, Ho RJY, Huang L. Trypsin Induced Destabilization of Liposomes Composed of Dioleoylphosphatidylethanolamine and Glyophorin. Biochemical and Biophysical Research Communications. 1986;141(3):973–8. doi: 10.1016/s0006-291x(86)80139-5. [DOI] [PubMed] [Google Scholar]
- 92.Davidsen J, Jorgensen K, Andresen TL, et al. Secreted phospholipase A(2) as a new enzymatic trigger mechanism for localised liposomal drug release and absorption in diseased tissue. Biochimica Et Biophysica Acta-Biomembranes. 2003;1609(1):95–101. doi: 10.1016/s0005-2736(02)00659-4. [DOI] [PubMed] [Google Scholar]
- 93.Sahu A, Bora U, Kasoju N, et al. Synthesis of novel biodegradable and self-assembling methoxy poly(ethylene glycol)-palmitate nanocarrier for curcumin delivery to cancer cells. Acta Biomaterialia. 2008;4(6):1752–61. doi: 10.1016/j.actbio.2008.04.021. [DOI] [PubMed] [Google Scholar]
- 94.Elegbede AI, Banerjee J, Hanson AJ, et al. Mechanistic studies of the triggered release of liposomal contents by matrix metalloproteinase-9. Journal of the American Chemical Society. 2008;130(32):10633–42. doi: 10.1021/ja801548g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Peng KW, Morling FJ, Cosset FL, et al. A gene delivery system activatable by disease-associated matrix metalloproteinases. Human Gene Therapy. 1997;8(6):729–38. doi: 10.1089/hum.1997.8.6-729. [DOI] [PubMed] [Google Scholar]
- 96.Sarkar N, Banerjee J, Hanson AJ, et al. Matrix metalloproteinase-assisted triggered release of liposomal contents. Bioconjugate Chemistry. 2008;19(1):57–64. doi: 10.1021/bc070081p. [DOI] [PubMed] [Google Scholar]
- 97.Xiong XY, Tam KC, Gan LH. Effect of enzymatic degradation on the release kinetics of model drug from Pluronic F127/poly(lactic acid) nano-particles. Journal of Controlled Release. 2005;108(2–3):263–70. doi: 10.1016/j.jconrel.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 98.Romberg B, Flesch FM, Hennink WE, et al. Enzyme-induced shedding of a poly(amino acid)-coating triggers contents release from dioleoyl phosphatidylethanolamine liposomes. International Journal of Pharmaceutics. 2008;355(1–2):108–13. doi: 10.1016/j.ijpharm.2007.11.055. [DOI] [PubMed] [Google Scholar]
- 99.Ong W, Yang YM, Cruciano AC, et al. Redox-Triggered Contents Release from Liposomes. Journal of the American Chemical Society. 2008;130(44):14739–44. doi: 10.1021/ja8050469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kirpotin D, Hong KL, Mullah N, et al. Liposomes with detachable polymer coating: Destabilization and fusion of dioleoylphosphatidylethanolamine vesicles triggered by cleavage of surface-grafted poly(ethylene glycol) Febs Letters. 1996;388(2–3):115–8. doi: 10.1016/0014-5793(96)00521-2. [DOI] [PubMed] [Google Scholar]
- 101.Tang FX, Hughes JA. Introduction of a disulfide bond into a cationic lipid enhances transgene expression of plasmid DNA. Biochemical and Biophysical Research Communications. 1998;242(1):141–5. doi: 10.1006/bbrc.1997.7923. [DOI] [PubMed] [Google Scholar]
- 102.Matsumoto S, Christie RJ, Nishiyama N, et al. Environment-Responsive Block Copolymer Micelles with a Disulfide Cross-Linked Core for Enhanced siRNA Delivery. Biomacromolecules. 2009;10(1):119–27. doi: 10.1021/bm800985e. [DOI] [PubMed] [Google Scholar]
- 103.Chandra B, Mallik S, Srivastava DK. Design of photocleavable lipids and their application in liposomal “uncorking”. Chemical Communications. 2005;24:3021–3. doi: 10.1039/b503423j. [DOI] [PubMed] [Google Scholar]
- 104.Chen K, Preuss A, Hackbarth S, et al. Novel photosensitizer-protein nanoparticles for Photodynamic therapy: Photophysical characterization and in vitro investigations. Journal of Photochemistry and Photobiology B-Biology. 2009;96(1):66–74. doi: 10.1016/j.jphotobiol.2009.04.006. [DOI] [PubMed] [Google Scholar]
- 105.Zhang ZY, Smith BD. Synthesis and characterization of NVOC-DOPE, a caged photoactivatable derivative of dioleoylphosphatidylethanolamine. Bioconjugate Chemistry. 1999;10(6):1150–2. doi: 10.1021/bc990087h. [DOI] [PubMed] [Google Scholar]
- 106.Chiu GNC, Abraham SA, Ickenstein LM, et al. Encapsulation of doxorubicin into thermosensitive liposomes via complexation with the transition metal manganese. Journal of Controlled Release. 2005;104(2):271–88. doi: 10.1016/j.jconrel.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 107.Kono K, Murakami T, Yoshida T, et al. Temperature Sensitization of Liposomes by use of thermosensitive block copolymers synthesized by living cationic polymerization: Effect of copolymer chain length. Bioconjugate Chemistry. 2005;16(6):1367–74. doi: 10.1021/bc050004z. [DOI] [PubMed] [Google Scholar]
- 108.Owens DE, Eby JK, Jian Y, et al. Temperature-responsive polymer-gold nanocomposites as intelligent therapeutic systems. Journal of Biomedical Materials Research Part A. 2007;83A(3):692–5. doi: 10.1002/jbm.a.31284. [DOI] [PubMed] [Google Scholar]
- 109.Owens DE, Jian YC, Fang JE, et al. Thermally responsive swelling properties of polyacrylamide/poly(acrylic acid) interpenetrating polymer network nanoparticles. Macromolecules. 2007;40(20):7306–10. [Google Scholar]
- 110.Yuan Q, Venkatasubramanian R, Hein S, et al. A stimulus-responsive magnetic nanoparticle drug carrier: Magnetite encapsulated by chitosan-grafted-copolymer. Acta Biomaterialia. 2008;4(4):1024–37. doi: 10.1016/j.actbio.2008.02.002. [DOI] [PubMed] [Google Scholar]
- 111.Husseini GA, Myrup GD, Pitt WG, et al. Factors affecting acoustically triggered release of drugs from polymeric micelles. Journal of Controlled Release. 2000;69(1):43–52. doi: 10.1016/s0168-3659(00)00278-9. [DOI] [PubMed] [Google Scholar]
- 112.Marin A, Muniruzzaman M, Rapoport N. Acoustic activation of drug delivery from polymeric micelles: effect of pulsed ultrasound. Journal of Controlled Release. 2001;71(3):239–49. doi: 10.1016/s0168-3659(01)00216-4. [DOI] [PubMed] [Google Scholar]
- 113.O’Neill BE, Vo H, Angstadt M, et al. Pulsed High Intensity Focused Ultrasound Mediated Nanoparticle Delivery: Mechanisms and Efficacy in Murine Muscle. Ultrasound in Medicine and Biology. 2009;35(3):416–24. doi: 10.1016/j.ultrasmedbio.2008.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Pitt WG, Husseini GA. Ultrasound in drug and gene delivery - Preface. Advanced Drug Delivery Reviews. 2008;60(10):1095–6. doi: 10.1016/j.addr.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 115.Needham D, Dewhirst MW. The development and testing of a new temperature-sensitive drug delivery system for the treatment of solid tumors. Advanced Drug Delivery Reviews. 2001;53(3):285–305. doi: 10.1016/s0169-409x(01)00233-2. [DOI] [PubMed] [Google Scholar]
- 116.Husseini GA, Pitt WG. Micelles and nanoparticles for ultrasonic drug and gene delivery. Advanced Drug Delivery Reviews. 2008;60(10):1137–52. doi: 10.1016/j.addr.2008.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Nelson JL, Roeder BL, Carmen JC, et al. Ultrasonically activated chemotherapeutic drug delivery in a rat model. Cancer Research. 2002;62(24):7280–3. [PubMed] [Google Scholar]
- 118.Hosseinkhani H, Kushibiki T, Matsumoto K, et al. Enhanced suppression of tumor growth using a combination of NK4 plasmid DNA-PEG engrafted cationized dextran complex and ultrasound irradiation. Cancer Gene Therapy. 2006;13(5):479–89. doi: 10.1038/sj.cgt.7700918. [DOI] [PubMed] [Google Scholar]
- 119.Hosseinkhani H, Tabata Y. Ultrasound enhances in vivo tumor expression of plasmid DNA by PEG-introduced cationized dextran. Journal of Controlled Release. 2005;108(2–3):540–56. doi: 10.1016/j.jconrel.2005.08.027. [DOI] [PubMed] [Google Scholar]
- 120.Aoyama T, Hosseinkhani H, Yamamoto S, et al. Enhanced expression of plasmid DNA-cationized gelatin complex by ultrasound in murine muscle. Journal of Controlled Release. 2002;80(1–3):345–56. doi: 10.1016/s0168-3659(02)00029-9. [DOI] [PubMed] [Google Scholar]
- 121.Chumakova OV, Liopo AV, Andreev VG, et al. Composition of PLGA and PEI/DNA nanoparticles improves ultrasound-mediated gene delivery in solid tumors in vivo. Cancer Letters. 2008;261(2):215–25. doi: 10.1016/j.canlet.2007.11.023. [DOI] [PubMed] [Google Scholar]
- 122.Sei-Young L, Mauro F, Paolo D. Shaping nano-/micro-particles for enhanced vascular interaction in laminar flows. Nanotechnology. 2009;(49):495101. doi: 10.1088/0957-4484/20/49/495101. [DOI] [PubMed] [Google Scholar]
- 123**.Decuzzi P, Godin B, Tanaka T, et al. Size and shape effects in the biodistribution of intravascularly injected particles. Journal of Controlled Release. 2009 Oct 27; doi: 10.1016/j.jconrel.2009.10.014. [Epub ahead of print] In-vivo results describing biodistribution of shape-specific particles. [DOI] [PubMed] [Google Scholar]
- 124.Gratton S, Napier M, Ropp P, et al. Microfabricated Particles for Engineered Drug Therapies: Elucidation into the Mechanisms of Cellular Internalization of PRINT Particles. Pharmaceutical Research. 2008;25(12):2845–52. doi: 10.1007/s11095-008-9654-8. [DOI] [PMC free article] [PubMed] [Google Scholar]