

Abstract
In recent years, investigators have carried out several studies designed to evaluate whether human tumor-associated antigens might be exploited as targets for active specific immunotherapy, specifically human cancer vaccines. Not too long ago such an approach would have been met with considerable skepticism because the immune system was believed to be a rigid discriminator between self and non-self which, in turn, protected the host from a variety of pathogens. That viewpoint has been challenged in recent years by a series of studies indicating that antigenic determinants of self have not induced absolute host immune tolerance. Moreover, under specific conditions that evoke danger signals, peptides from self-antigen can be processed by the antigen-presenting cellular machinery, loaded onto the major histocompatibility antigen groove to serve as targets for immune intervention. Those findings provide the rationale to investigate a wide range of tumor-associated antigens, including differentiation antigens, oncogenes, and tumor suppressor genes as possible immune-based targets. One of those tumor-associated antigens is the carcinoembryonic antigen (CEA). Described almost 40 years ago, CEA is a Mr 180–200,000 oncofetal antigen that is one of the more widely studied human tumor-associated antigens. This review will provide: (i) a brief overview of the CEA gene family, (ii) a summary of early preclinical findings on overcoming immune tolerance to CEA, and (iii) the rationale to develop mouse models which spontaneously develop gastrointestinal tumors and express the CEA transgene. Those models have been used extensively in the study of overcoming host immune tolerance to CEA, a self, tumor-associated antigen, and the experimental findings have served as the rationale for the design of early clinical trials to evaluate CEA-based cancer vaccines.
Keywords: Carcinoembryonic antigen, Poxvirus cancer vaccines, Gastrointestinal cancer
1. Introduction
CEA expression was first characterized in gastrointestinal (G.I.) cancers and circulating CEA is recognized as a valuable adjunct to postoperative surveillance and preoperative prognosis of patients diagnosed with colorectal and other types of carcinomas [1]. The gene for CEA was cloned in 1987 [2,3] and the translated protein was shown to be a part of a subgroup of the immunoglobulin (Ig) super family (reviewed in Ref. [4]). More than 30 other genes/pseudogenes related to CEA have been identified and are clustered on the long arm of human chromosome 19q13.2 [5]. Although a murine CEA gene family has been identified, no direct counterpart to human CEA has been described and probably does not exist. With the emergence of this large family of CEA-like molecules, investigators decided to adopt a nomenclature that would identify CEA and the CEA-like molecules as members of the same family [6]. The family was divided into two branches—CEA-related cell adhesion molecules, abbreviated CEACAM (pronounced c-cam), and pregnancy-specific glycoproteins (PSG). Each CEACAM consists of an Ig variable region-like amino-terminal domain followed by up to six Ig constant region-like domains and each is anchored to the cell membrane via either a (i) glycosylphosphatidylinositol (GPI) moiety or (ii) a proteinaceous transmembrane and cytoplasm domain. Of the different functions attributed to the CEACAMs, most are associated with their acting as homophilic and heterophilic intercellular adhesion molecules (reviewed in Ref. [7]). For example, there are eight described human CEACAMs—CEACAM1 (BGP), CEACAM3 (CGM1), CEACAM4 (CGM7), CEACAM5 (CEA, CD66e), CEACAM6 (NCA), CEACAM7 (CGM2), CEACAM8 (CGM6), and CEACAM21 (note: old names in parentheses). CEACAM1, CEACAM3, CEA, and CEACAM6 serve as receptors for Neisseria gonorrhea and Neisseria meningitis (reviewed in Ref. [8]). CEACAM1, CEA, and CEACAM6 may serve as receptors for diffusely adhering Escherichia coli and may influence their pathogenicity [9–11]. CEA has also been implicated in the development of hepatic metastases [1,12]. The number of human, mouse, rat, guinea pig, and non-human primates is constantly being expanded and the most current information can be found at the following website: http://cea.klinikum.uni-muenchen.de/human/ceacam/cea_fam_hum_ceacam.htm.
2. Preclinical mouse models expressing human CEA as a transgene
Because CEA, the prototype CEACAM, is anchored on the plasma membrane via a GPI linkage and is overexpressed in a wide range of epithelial tumors [1,7,13], it has been a popular target for novel cancer therapies, including cellular immunotherapy, radioimmunotherapy, antibody therapy, and cancer vaccines. In recent years, cancer vaccines that elicit the host immune system to target CEA have renewed interest in designing experimental models for preclinical vaccine studies. However, until approximately 10 years ago, using experimental murine models to study different therapeutic interventions directed at CEA had been impossible since no CEA homologue had been identified in either mice or rats. Since then several murine models expressing human CEA as a transgene have been developed and used extensively to study the generation of CEA-specific host immunity and immune-mediated antitumor responses. To date, at least four different mice expressing human CEA as a transgene have been developed [14–17]. The initial CEA transgenic mouse line used the full-length CEA cDNA containing the SV40 early promoter and CEA was expressed in all tissues [14]. The most recent model was developed using a 187-kb human bacterial artificial chromosome that contained part of the human CEA family gene cluster, including complete CEA, CEACAM3, CEACAM6, and CEACAM7 sequences [15]. That model should provide the opportunity to target other CEACAMs in vaccine-based protocols and better determine whether autoreactive T cells may mediate autoimmunity directed at normal tissues that expressed the different CEACAMs. The two remaining murine models used the complete CEA gene, including the flanking regulatory elements (approximately 33 kb), to generate CEA transgenic (CEA.Tg) mice with tissue-specific CEA expression [18], which closely recapitulated that seen in humans [16,17]. While all founder mice showed a restricted tissue pattern of CEA expression, there were significant differences between the CEA.Tg mice generated at those laboratories. The CEA.Tg generated at the University of Freiburg, Germany had ectopic CEA expression in the esophagus, small intestine, trachea, and lung [16]. Tissue CEA levels were considerably higher than those measured in the same human tissues and CEA was present in both feces and serum of the CEA.Tg mice [16,19,20]. The differences in CEA expression levels in those two CEA.Tg mouse colonies provided the opportunity for researchers to investigate whether the degree of immune tolerance might impact the ability of CEA-based vaccines to generate CEA-specific immune responses. Furthermore, the CEA.Tg [16,19] mouse strain provided a preclinical model in which to study peripheral immune tolerance and one that more approximates the CEA expression levels in late-stage cancer patients.
2.1. Initial studies on immune tolerance to CEA in CEA.Tg murine models
Using the CEA.Tg [16,19] mouse, initial studies were carried out by this laboratory to examine whether the immune system in those mice was tolerant to CEA. CEA.Tg mice were immunized twice with either whole CEA or ovalbumin (OVA) protein emulsified in adjuvant, representing self and foreign antigens, respectively. CEA.Tg mice developed strong anti-OVA serum Ig titers, whereas those same mice developed only weak IgM and no measurable IgG serum titers against CEA, underscoring the ability of the CEA.Tg mice to mount an immune response to OVA, a foreign antigen, but remaining immunologically silent to CEA, a self-antigen.
Recombinant poxviruses have been engineered to express a wide variety of foreign antigens and have been studied by numerous laboratories as candidates for active immunization. Vaccinia virus, a poxvirus, is a particularly attractive platform for a vaccine because it accepts large inserts of foreign genes, has a wide host range, and the co-presentation of a weak immunogen with the highly immunogenic vaccinia proteins can boost the host immune response to the inserted gene product. Indeed, initial studies from our laboratory demonstrated that vaccination of CEA.Tg mice with recombinant vaccinia virus engineered to express human CEA (rV-CEA) overcame host immune tolerance as evidenced by the induction of: (i) high serum titers of anti-CEA Igs, (ii) Ig class switching, and (iii) anti-CEA cell-mediated immunity [20]. Table 1 summarizes the various vaccine platforms that have been successful in overcoming immune tolerance and inducing CEA-specific immunity with accompanying antitumor responses.
Table 1.
Generation of CEA-specific host and antitumor immunity in CEA transgenic mice
| Immunogen | Vaccine platform | Anti-CEA host immunity | Antitumor response | Reference | |
|---|---|---|---|---|---|
| Humoral | Cellular | ||||
| rV-CEA | Poxvirus | + | CD4 and CD8 | +/− | [20] |
| CEA-transfected cells | Fibroblasts | + | CD8 | + | [121] |
| anti-CEA/IL-2 single chain antibody | Fusion protein | NT | NT | + | [122] |
| pW-CEA/pCD40LT-CEA and CEA-IL-4/IL-12 | DNA | NT | CD8 | + | [123–126] |
| ALVAC-CEA | Avian poxvirus | + | CD4 and CD8 | + | [82] |
| rV-CEA-TRICOM | Poxvirus + costimulation | + | CD4 and CD8 | + | [37,127,128] |
| pHI-691 | CEA minigene | NT | CD8 | + | [129] |
NT, not tested.
2.2. Transplantable CEA-expressing tumor models: limitations and future studies
The studies summarized in Table 1 have provided critical insights into the ability of different vaccine platforms to overcome immune tolerance to a tissue-specific, self-antigen. Furthermore, it is interesting that presenting CEA to the host immune system in a variety of vaccine platforms, such as recombinant poxviruses, transfected fibroblasts, and naked DNA, induced CEA-specific humoral and cell-mediated responses, and, in most cases, regression of transplantable CEA-expressing tumors. While those studies have made important contributions, the use of transplantable CEA-expressing tumors to assess vaccine-mediated antitumor immunity offers some crucial limitations which include: (i) tumor cells in which the CEA gene was inserted via a retroviral vector absent of any of the CEA regulatory elements, (ii) the growth of transplantable tumors at sites which are not authentic to G.I. cancer, (iii) the rapid proliferation of transplantable tumors which does not recapitulate the growth characteristics of carcinomas and requires a compressed time schedule for vaccine administration, and (iv) the presence of large tumors within 4–6 weeks following transplant requires that most experimental studies be completed within a time frame which may not allow for sufficient evaluation of whether autoreactive T cells elicit unwanted autoimmunity directed at CEA-expressing normal tissues. We believe that the preclinical evaluation of cancer vaccines would be better served in experimental models that develop spontaneously arising tumors over an extended time period. Those models would provide the tissue authenticity for tumor development as well as permit the evaluation of cancer vaccines over a prolonged experimental treatment period. Research efforts have focused on the development of murine models in which gastrointestinal tumors arise spontaneously and the following sections will examine those models for their ability to replicate the human disease state and serve as platforms to evaluate vaccine strategies targeted against human tumor-associated antigens.
3. Murine models of gastrointestinal cancer
3.1. Murine models of colorectal cancer
Research elucidating the molecular pathogenesis of CRC has identified inactivating germline mutations of the tumor suppressor gene, adenomatous polyposis coli (APC), and the DNA mismatch repair (MMR) genes MSH2, MSH6, MLH1, PMS1, and PMS2 in patients that are genetically predisposed to intestinal neoplasia. The APC gene was initially cloned and identified as the genetic mutation associated with familial adenomatous polyposis coli [21], an autosomal dominant form of inherited CRC [21,22]. A role for mutations in APC and β-catenin, both intermediates in the Wnt signal transduction pathway, has been characterized in cell lines and animal models of intestinal tumorigenesis. Hereditary nonpolyposis colorectal cancer (HNPCC), also known as Lynch Syndrome, is caused by mutations in one or more of the human MMR genes MSH2, MSH6, MLH1, PMS1, or PMS2 resulting in increased microsatellite instability [23,24]. Additional research implicates the SMAD proteins associated with transforming growth factor beta (TGF-β) signaling in the development of intestinal neoplasia in both mice and humans (reviewed in Ref. [25]). Murine models of intestinal neoplasia can be distinguished by the molecular mechanisms that are modulated in order to mimic human CRC: (i) Wnt/APC/β-catenin signaling pathway, (ii) DNA MMR deficiency, (iii) TGF-β signaling pathway, and (iv) combinations of APC mutant mice crossed with MMR- or SMAD-deficient mice (Table 2).
Table 2.
Murine models of colorectal cancer
| Name of model | Background strain | Primary site | Primary histology | Average # of tumors/mouse | Age (months) | Metastasis | References |
|---|---|---|---|---|---|---|---|
| Wnt/APC/β-catenin signaling pathway | |||||||
| APCMin/+ | C57BL/6 | Small intestine | Adenoma | 30 | 4 | No | [26,29,30] |
| APCMin/+/CEA.Tg | C57BL/6 | Small intestine | Adenoma | 30–50 | 4–5 | No | [37,38] |
| APC1638N/+ | C57BL/6 | Small intestine | Adenoma/carcinoma | 4 | 10–12 | Yes | [31,32,35,36,45] |
| APCΔ716/+ | C57BL/6 | Small intestine | Adenoma | 300 | 4 | No | [33] |
| APC1309/+ | N.R. | Small intestine | Adenoma | 34 | 3 | No | [34] |
| DN131-β-catenin | C57BL/6 X DBA | Small intestine | Adenoma | 1 | 1 | No | [39] |
| DN89-β-catenin | C57BL/6 X ROSA26 X 129/Sv | N.R. | N.R. | 0 | 10 | No | [40] |
| Catnb+/lox(ex3):Krt1-19+/cre | C57BL/6 | Small intestine | Adenoma | 3000 | 3 wks. | No | [41] |
| DNA mismatch repair (MMR) deficiency | |||||||
| Mlh1−/− | C57BL/6 X 129/Ola | Small intestine | Adenoma/carcinoma | 2 | 7 | No | [36] |
| Mlh1−/−Apc1638N/+ | C57BL/6 X 129/Ola | Small intestine | Adenoma/carcinoma | 45 | 3 | No | [36] |
| Mlh1−/−ApcMin/+ | C57BL/6 | Small intestine | Adenoma | 139 | 2 | No | [44] |
| Msh2−/− | C57BL/6 X 129/Ola | Small intestine | Adenoma/carcinoma | 1–2 | 6–10.5 | No | [42] |
| Msh3−/− | C57BL/6 X 129/Sv X SJL/J | Small intestine | Adenoma/carcinoma | <1 | 26 | No | [43] |
| Msh3−/−APC1638N/+ | C57BL/6 | Small/large intestine | Adenoma/carcinoma | 5 | 9 | No | [45] |
| Msh6−/− | C57BL/6 X 129/Sv X SJL/J | Small intestine | Adenoma/carcinoma | <1 | 11–12 | No | [43] |
| Msh6−/−APC1638N/+ | C57BL/6 | Small/large intestine | Adenoma/carcinoma | 26 | 4.5 | No | [45] |
| Msh3−/−Msh6−/− | C57BL/6 X 129/Sv X SJL/J | Small intestine | Adenoma/carcinoma | 2 | 7–8 | No | [43] |
| Msh3−/−Msh6−/−APC1638N/+ | C57BL/6 | Small/large intestine | Adenoma/carcinoma | 39 | 2.5 | No | [45] |
| Transforming growth factor-beta (TGF-β) signaling pathway | |||||||
| Tgfb1−/−Rag2−/− | 129S6 X CF1 | Cecum/colon | Mucinous carcinoma | 2 | 2–6 | No | [51,52] |
| Smad3−/− | 129/Sv | Colon | Mucinous carcinoma | 4 | 5–6 | Yes | [57] |
| Smad4+/− | C57BL/6 | Stomach/duodenum | Hamartomatous polyps | 2 | 24 | No | [58] |
| Smad4+/−APCΔ716/+ | C57BL/6 | Small intestine | Carcinoma | 300 | 4 | No | [58] |
N.R., not reported.
3.1.1. Wnt/APC/β-catenin signaling pathway
The APCMin/+ (Multiple Intestinal Neoplasia) mouse was developed by using the carcinogen ethylnitrosourea to introduce a germline mutation in codon 850 of the APC tumor suppressor gene [26]. The functional APC protein regulates the transcriptional coactivator, β-catenin, through a process of ubiquitination, and proteolytic degradation in the proteosome. The mutated APC gene expresses a prematurely truncated form of the APC protein that allows β-catenin to accumulate in the nucleus where it drives the transcription of genes involved in apoptosis, cellular proliferation, and cell cycle progression [27,28]. Homozygous loss of APC expression is lethal during embryonic development; however, heterozygous APCMin/+ mice develop an average of 30 adenomas per mouse primarily in the small intestines by 4 months of age [29,30]. Other APC mutant mouse models, including APC1638N/+ [31,32], APCΔ716/+ [33], and APC1309/+ [34], develop small intestinal adenomas but differ with respect to tumor burden and the age of onset (reviewed in Ref. [25]). The APC1638N/+ mouse is unique among the APC mutant mice for its ability to progressively develop aberrant crypt foci, colonic polyps, small intestinal adenomas, and carcinomas with metastatic potential [35,36]. In the absence of mutations in the Ras and P53 genes, the APC1638N/+ mice develop 5–6 adenomas and adenocarcinomas per mouse with over 75% of these lesions testing positive for a loss of the wild type APC allele and the entire mouse chromosome 18 [32]. When APCMin/+ mice are crossed with CEA transgenic mice, the resulting hybrid develops CEA-expressing intestinal adenomas [19]. The APCMin/+/CEA mice are immunocompetent and their use as an experimental model to study CEA-directed vaccine strategies against colorectal cancer will be discussed later [37,38].
Of the three transgenic animal models overexpressing a stable and mutated form of β-catenin [39–41], the most successful model developed by Harada et al. [41] used a cre-lox conditional expression system to direct the expression of stable β-catenin to intestinal polyps and the colonic mucosa. Each β-catenin mutant mice developed approximately 3000 intestinal polyps with evidence of nascent microadenomas in the colonic mucosa by 3 weeks of age [41]. Studies of the APCMin/+ and β-catenin mutant mice provide compelling evidence that genetic alterations in the Wnt signaling pathway play a causal role in intestinal and colonic tumorigenesis in mice.
3.1.2. DNA mismatch repair (MMR) deficiency
Mouse models of HNPCC have been developed through the homozygous deletion of the DNA mismatch repair genes: Mlh1−/− [36], Msh2−/− [42], Msh3−/− [43], and Msh6−/− [43]. Many of these mice succumb to lymphomas and skin tumors; however, the mice that survive develop small intestinal adenomas and carcinomas (reviewed in Ref. [25]). While the genetic backgrounds of MMR-deficient mice yield significantly different intestinal phenotypes (Table 2), Mlh1 deficiency bred into APC mutant mice results in 4- and 10-fold increases in the number of intestinal adenomas in Mlh1−/−APCMin/+ and Mlh1−/−APC1638N/+ mice compared to APCMin/+ and APC1638N/+ mice, respectively [36,44]. Mlh1 deficiency in APCMin/+ mice increases tumor multiplicity without evidence of advances in tumor progression [44]. When an Msh6 deficiency is bred into APC1638N/+ mice, the resulting offspring (Msh6−/−APC1638N/+) experience reduced survival and a 6- to 7-fold greater intestinal tumor burden compared to APC1638N/+ mice [45]. In Msh3−/−Msh6−/− mice, the Msh3, and Msh6 genes have been shown to cooperate in the modulation of tumor suppression in the intestine [43]. When Msh3−/−Msh6−/− mice are crossed with APC1638N/+ mice, survival is further reduced with concomitant increases in intestinal tumor burden in the Msh3−/−Msh6−/−APC1638N/+ offspring compared to Msh6−/−APC1638N/+ mice [45]. Thus, when MMR-deficient mice carrying APC mutations are compared to APC1638N/+ mice, the MMR-deficient phenotype appears to promote tumor initiation but not progression as evidenced by increases in tumor burden in the absence of changes in carcinoma incidence.
3.1.3. Transforming growth factor-beta (TGF-β) signaling pathway
TGF-β possesses both immunosuppressant and tumor suppressor activity; however, many epithelial carcinomas are resistant to the growth inhibitory effects of TGF-β due to mutations in TGF-β cell surface receptors. In fact, mutations in the type II TGF-β receptor (TGF-βR2) have been identified in both sporadic CRC and HNPCC [46–48]. To examine the role of TGF-β signaling in intestinal tumorigenesis, Tgfb1−/− mice were crossed with immunodeficient Rag2−/− mice because homozygous deletion of the Tgfb1 gene in immunocompetent mice results in a multifocal inflammatory disease and death at approximately 3 weeks of age [49,50]. In the presence of intestinal microflora containing Helicobacter hepaticus, the Tgfb1−/−Rag2−/− mice develop mucinous carcinomas of the cecum and colon between 2 and 6 months of age [51,52]; however, the lack of a competent immune system in this model does not allow for the evaluation of protein-directed immunotherapy regimens.
The SMAD proteins are downstream mediators in the TGF-β signaling pathway. Inactivating mutations in the SMAD2 and SMAD4 genes have been identified in human CRC [53–55]. Homozygous deletion of the Smad2 gene in mice is embryonically lethal and heterozygous Smad2+/− mice fail to develop an intestinal phenotype [56]. Mice deficient in Smad3 [57] and Smad4 [58] expression develop intestinal neoplasia. Homozygous Smad3 knockout mice on a 129/Sv genetic background become moribund with colorectal adenocarcinomas between 4 and 6 months of age [57]. A unique feature of this model is the metastatic transformation of the colonic epithelium as evidenced by metastatic infiltration of the adenocarcinoma to mesenteric and thoracic lymph nodes [57].
While homozygous deletion of the Smad4 gene is embryonically lethal, heterozygous Smad4+/− mice develop gastric and duodenal polyps at 1–2 years of age [58]. When Smad4+/− mice are crossed with APCΔ716/+ mice, the Smad4+/−APCΔ716/+ double mutant offspring develop approximately 300 small intestinal carcinomas per mouse by 4 months of age [58]. While tumor multiplicity is identical in APCΔ716/+ and Smad4+/−APCΔ716/+ mice, the predominant lesions found in APCΔ716/+ mice are adenomas while carcinomas represent the predominant lesion found in Smad4+/−APCΔ716/+ mice. Moreover, when the polyp size distribution of these mice was evaluated between 6 and 20 weeks of age, the Smad4+/−APCΔ716/+ mice were shown to have significantly larger polyps than APCΔ716/+ mice. These findings suggest that introducing a Smad4 deficiency into the APCΔ716/+ mouse plays a significant role in the malignant progression but not initiation of colorectal tumors in C57BL/6 mice [58].
3.2. Murine models of pancreatic cancer
The pancreas possesses two anatomical entities with distinct physiological functions. Over 80% of all pancreatic cancers arise from the exocrine pancreas with morphological features that are consistent with pancreatic ductal adenocarcinoma [59]. With the characterization of premalignant lesions of the pancreas known as pancreatic intraepithelial neoplasia or PanIN [60], the process of pancreatic carcinogenesis is thought to begin with PanIN lesions that progress from low- to high-grade dysplasia and finally, infiltrating ductal adenocarcinoma [61]. Genetic mutations resulting in the activation of oncogenes, such as Kras, Her2/Neu, and AKT2 are reported to occur in 95, 90, and 10–20% of all PanIN lesions, respectively [62,63]. Inactivating mutations of the tumor-suppressor genes p16INK4a, p53, p14ARF (p19ARF in mice), DPC4 [58], BRCA2, and LKB1/STK11 occur with a frequency of 90, 75, 50, 50, 5–10, and 5%, respectively [62,63]. The most common mutations (K-ras, Her2/Neu, and p16INK4a) are early events in the pathogenesis of pancreatic ductal adenocarcinoma (reviewed in Ref. [63]). Based on our current understanding of the molecular pathogenesis of this disease, murine models of pancreatic cancer have employed a variety of strategies to replicate a pancreatic phenotype that is consistent with the clinical features of human pancreatic adenocarcinoma including: (i) Ela-1-SV40 TAg, (ii) Ela-1-c-myc, (iii) TGF-α signaling pathway, and (iv) the activated KrasG12D X Ink4a/Arf deficient mice (Table 3).
Table 3.
Murine models of pancreatic cancer
| Name of model | Background strain | Primary site | Primary histology | Average # of tumors/mouse | Age (months) | Metastasis | References |
|---|---|---|---|---|---|---|---|
| Simian Virus 40 transforming antigen (SV40 TAg) models | |||||||
| Ela-1-SV40 TAg | C57BL/6 X SJL | Exocrine pancreas | Acinar cell carcinoma | 100 | 3–7 | Yes (rare) | [64] |
| Ela-1-SV40 TAg (T1-127, ET Mice) | C57BL/6 | Exocrine pancreas | Acinar cell carcinoma | 100 | 3–7 | Yes | [65] |
| MET (ET X MUC1.Tg) | C57BL/6 | Exocrine pancreas | Acinar cell carcinoma | 100 | 4–6 | Yes | [67,68] |
| c-Myc model | |||||||
| Ela-1-c-Myc | C57BL/6 X SJL | Exocrine pancreas | Acinar/ductal adenocarcinoma | 10 | 2–7 | Yes | [69] |
| Transforming growth factor-alpha (TGF-α) models | |||||||
| MT-TGFα –hGH | C57BL/6 X SJL | Exocrine pancreas | Pseudoductular metaplasia | Multifocal | 4–7 | No | [70] |
| Ela-1-TGFα-hGH | C57BL/6 | Exocrine pancreas | Acinar-to-ductal metaplasia | 107 | >1 year | No | [71,72] |
| Ela-1-TGFα-hGH X P53+/− | C57BL/6 X Balb/c | Exocrine pancreas | Acinar-to-ductal metaplasia | 90 | 7–13 | No | [72] |
| Ela-1-TGFα-hGH X P53−/− | C57BL/6 X Balb/c | Exocrine pancreas | Acinar-to-ductal metaplasia | 22 | 2–3 | Yes | [72] |
| Activated KrasG12D X Ink4a/Arf deficient model | |||||||
| Pdx1-Cre;Ink4a/Arflox/+ | FVB/n | N.R. | N.R. | 0 | 2–6 | No | [73] |
| LSL-KrasG12D;Ink4a/Arflox/lox | FVB X 129/Sv | Exocrine pancreas | Ductal PanIN lesions | Multifocal | 2–6 | No | [73] |
| Pdx1-Cre;LSL-KrasG12D;Ink4a/Arflox/lox | FVB X 129/Sv | Exocrine pancreas | Ductal adenocarcinoma | Multifocal | 2–3 | Yes | [73] |
N.R., not reported.
3.2.1. Simian Virus 40 transforming antigen (SV40 TAg) models
The elastase I gene encodes a serine protease that is synthesized in the exocrine pancreas and secreted into the lumen of the intestine. Transgenic mice expressing activated oncogenes targeted to the pancreas were first developed by fusing the rat elastase I promoter to the early region of the SV40 genome containing the gene encoding the transforming antigen [64]. The Ela-1-SV40 TAg mice express the large T antigen in acinar cells of the exocrine pancreas thereby initiating a multi-step process of acinar cell carcinogenesis [64]. During embryonic development, Ela-1-SV40 TAg mice exhibit acinar cell hyperplasia that progresses to dysplasia at 5–16 weeks of age [64]. By 3–6 months of age, the Ela-1-SV40 TAg mice become moribund due to the high incidence of acinar cell carcinomas [64]. In a related model, a transgene producing the N-terminal amino acids 1–127 of the large T antigen and the full-length small T antigen driven by the rat elastase I promoter was sufficient to induce acinar cell carcinomas and metastases in mice with wild-type p53 status [65]. While the Ela-1-SV40 TAg mice reported by Ornitz et al. [64] and the ET mice developed by Tevethia et al. [65] exhibit multi-step pancreatic carcinogenesis, these models do not replicate the ductal histology nor do they possess activating mutations of the Kras gene observed in the vast majority of human pancreatic cancers.
The tumor-associated antigen, MUC1, is overexpressed in 90% of all human pancreatic cancers [66], and thus, MUC1 represents one target for protein-directed immunotherapy. When MUC1 transgenic mice are crossed with ET mice on a C57BL/6 genetic background, the resulting MET mice form spontaneous pancreatic acinar cell carcinomas that express the human MUC1 antigen [67]. The MET mice, while immunologically tolerant of human MUC1, generate MUC1-specific cytotoxic T lymphocytes that can be stimulated to enhance survival following vaccination with a dendritic cell-tumor cell fusion and treatment with the T cell stimulant, staphylococcal enterotoxin B [68]. Thus, the MET mouse provides a platform from which to test MUC1-directed antitumor vaccine strategies in a spontaneous model of pancreatic acinar cell carcinoma.
3.2.2. c-Myc model
When the rat elastase-1 gene is used to direct expression of the c-myc oncogene to the exocrine pancreas, the Ela-1-c-Myc mice develop acinar and acinar/ductal adenocarcinomas of the pancreas between 2 and 7 months of age [69]. These findings suggest that some pancreatic ductal adenocarcinomas may originate from acinar cells that undergo acinar-to-ductal transdifferentiation, also known as acinar-to-ductal metaplasia (Table 3).
3.2.3. Transforming growth factor-alpha (TGF-α) models
TGF-α is a member of the epidermal growth factor family of proteins, a ligand for the EGF receptor (EGFR), and an activator of the EGFR-mediated signaling pathway. Overexpression of TGF-α in transgenic mice promotes epithelial hyperplasia and, in the pancreas, TGF-α promotes the proliferation of acinar cells and fibroblasts, and altered acinar cell differentiation resulting in pseudoductular acinar metaplasia [70]. When a zinc-inducible metallothionein promoter is used to regulate TGF-α overexpression in MT-TGFα mice, the magnitude of the acinar cell proliferative response to TGF-α is proportional to the level of tissue-specific TGF-α expression [70]. Additional support for an acinar cell origin of pancreatic ductal adenocarcinoma is found in Ela-1-TGFα-hGH transgenic mice overexpressing TGF-α under the control of the rat elastase I promoter. The Ela-1-TGFα-hGH mice exhibit acinar-to-ductal metaplasia, the formation of ductal tubular complexes with duct-specific expression of carbonic anhydrase and cytokeratins 8 and 18, and in animals over 1 year of age, dysplasia and malignant transformation are noted in the ductal tubular complexes [70,71]. While Ela-1-TGFα-hGH mice lack Kras mutations, EGFR overexpression is correlated with the transition from the acinar to ductal phenotype and the malignant tumors arising from the ductal tubular complexes display increased immunoreactivity for nuclear p53 [71]. These findings suggest a role for TGFα signaling in pancreatic carcinogenesis and the acinar-ductal-carcinoma sequence.
By introducing a p53-deficiency into Ela-1-TGFα-hGH mice, Wagner et al. [72] dramatically accelerated the growth of pancreatic tumors in a p53-dependent manner such that 77% of TGFα/p53+/− mice developed tumors with a mean tumor-free survival period of 220 days while 100% of TGFα/p53−/− mice developed tumors with a mean tumor-free survival period of 120 days after birth [72]. In p53-deficient Ela-1-TGFα-hGH mice, homozygous deletion of the Ink4a/Arf locus was identified in 5 of 15 tumors and loss of heterozygosity of the Smad4 locus was identified in 4 of 15 tumors [72]. In addition, the Ras/Erk signaling cascade was selectively activated in the ductal tubular complexes of these mice indicating that p53-deficient Ela-1-TGFα-hGH mice recapitulate many of the molecular and genetic features of human pancreatic adenocarcinoma [72].
3.2.4. Activated KrasG12D X Ink4a/Arf deficient model
Using a cre-lox conditional expression system, Aguirre et al. [73] engineered mice on a mixed FVB X 129/Sv genetic background that exhibit pancreas-specific expression of an activating mutation of the KrasG12D allele and a deletion of the Ink4a/Arf allele encoding the p16Ink4a and p19Arf tumor suppressor genes. The Cre-mediated gene expression profile of LSL-Kras;Ink4a/Arflox/lox mice is directed to the pancreas through the use of a Pdx1-Cre transgene that efficiently deletes loxP-containing alleles in pancreatic cells of endocrine, exocrine, and ductal lineages (reviewed in Refs. [63,74]). The resulting Pdx1-Cre;LSL-Kras;Ink4a/Arflox/lox mice develop low- to high-grade PanIN ductal lesions at 3–4 weeks of age and small multifocal pancreatic adenocarcinomas at 5–6 weeks of age [73]. The pancreatic adenocarcinomas progress rapidly, become highly invasive, and develop metastases that result in the death of all Pdx1-Cre;LSL-Kras;Ink4a/Arflox/lox mice by 11 weeks of age [73]. In this model, activation of the mutant KrasG12D allele in mice with normal Ink4a/Arf tumor suppressor activity results in Pdx1-Cre;LSL-Kras mice that develop premalignant ductal PanIN lesions that fail to progress to neoplasia [73]. These findings suggest that inactivation of the Ink4a/Arf tumor suppressor allele appears to be necessary for malignant progression to pancreatic ductal adenocarcinoma.
A molecular analysis of the pancreatic adenocarcinomas in Pdx1-Cre;LSL-Kras-Ink4a/Arflox/lox mice did not detect the presence of Smad4 or p53 mutations; however, there is amplification of the mutant KrasG12D gene transcript and evidence that activated Kras signaling contributes to malignant progression in the pancreas [73]. Consistent with the selective overexpression of EGFR and Her2/Neu in early human PanIN lesions, both EGFR and Her2/Neu are expressed in the glandular component but not the undifferentiated sarcomatoid regions of pancreatic tumors in Pdx1-Cre;LSL-Kras;Ink4a/Arflox/lox mice [73]. The Pdx1-Cre;LSL-Kras;Ink4a/Arflox/lox mouse is an ideal model for the evaluation of both prevention and therapeutic intervention strategies because it possesses the two most commonly mutated genes in human pancreatic cancer (Kras and Ink4a/Arf), overexpresses EGFR and Her2/Neu in preneoplastic but not advanced pancreatic lesions, and faithfully replicates the histopathology of human pancreatic ductal adenocarcinoma.
4. Treatment of spontaneous intestinal tumors in APCMin/+/CEA mice using a CEA-based vaccine strategy
4.1. Generation of a murine model of spontaneously arising, CEA-expressing G.I. tumors
Of the murine models in which tumors arise spontaneously, our interests focused on the APCMin/+ mice that were on the same genetic background as the CEA.Tg mice (C57BL/6) and developed adenomas over a prolonged time period that would allow for the evaluation of the antitumor and autoimmune effects of the CEA-based vaccine. Female, CEA.Tg mice were bred with male, APCMin/+ mice and the APCMin/+/CEA progeny were examined for the spontaneous development of intestinal adenomas to determine whether those adenomas overexpressed CEA. APCMin/+/CEA mice developed approximately the same number of intestinal adenomas as did APCMin/+ mice—30 to 50 per mouse—and the appearance of numerous intestinal adenomas in the APCMin/+/CEA mice coincided with developing anemia and progressive weight loss due to the disruption of the intestinal architecture caused by the numerous adenomas. Untreated APCMin/+/CEA mice, like the APCMin/+ mice, have a life expectancy of 4–5 months. Analyses of isolated adenoma tissue revealed 3- to 4-fold higher levels of CEA expression than found in adjacent normal mucosa (Table 4, Fig. 1). Thus, the APCMin/+/CEA mice offer an experimental model that develops spontaneous intestinal neoplasms that overexpress CEA in an authentic tissue site.
Table 4.
CEA expression levels in normal intestinal tissue, adenomas and sera
| GENOTYPE | CEA expression | SERUM CEAd (ng/ml) | ||
|---|---|---|---|---|
| RIAa (cpm 125I-COL-1 bound/40 μg protein) | EIAb (μg CEA/mg protein) | IHCc | ||
| APCMin/+/CEA | 44,400 ± 2150 | 3.27–4.45 | ++ | 42.2 ±7.6 |
| CEA | 12,160 ± 990 | 1.35–1.80 | + | 60.8 ±14.9 |
| APCMin/+ | NEG | NEG | NEG | NEG |
| C57BL/6 | NEG | NEG | NEG | NEG |
Protein extracts were prepared and the binding of an anti-CEA monoclonal antibody (COL-1) and an irrelevant, isotype-matched antibody (BL3) was performed as a solid phase RIA. Data are the mean ± S.E.M. of the total cpm (125I-COL-1 minus 125I-BL3) bound.
CEA levels were measured using the CEA ELISA kit and presented as the range of CEA levels. NEG, <5 ng/mg protein.
Immunohistochemistry staining intensity of an anti-CEA monoclonal antibody (COL-1) in intestinal adenomas of APCMin/+/CEA and normal intestinal tissue of CEA.Tg mice. Scoring: no staining, NEG; +, weak (pale brown intensity); ++, strong (dark brown immunoprecipitate).
Sera CEA levels (mean ± S.E.M.) measured using the CEA ELISA kit; NEG, <5 ng/ml serum.
Fig. 1.

Immunohistochemical staining of an intestinal adenoma from an APCMin/+/CEA mouse using the anti-CEA monoclonal antibody (COL-1). Dark brown staining pattern illustrates CEA overexpression in the adenomatous tissue (10×).
4.2. Second generation CEA-based recombinant poxvirus vaccines
As shown previously, insertion of CEA as a transgene into the vaccinia virus genome results in a much more vigorous CEA-specific host immune response that is most likely due to the acute local pro-inflammatory reaction that accompanies vaccinia administration. The advantages of using recombinant vaccinia virus as a priming agent are also many of the same reasons that limit its repeated use. That is, the vigorous host immune response, which is important in priming the immune responses, also limits the replication of the virus and expression of the transgene during subsequent vaccinations. Studies have identified recombinant avipox vectors that can be used as potent booster vaccinations following a primary vaccination with a recombinant vaccinia vector. There are two major types of avipox vectors: fowlpox and canarypox (ALVAC). Both are derived from avian species, are replication-deficient in mammalian cells, but efficiently and stably express inserted transgenes. Those vectors can express the transgene for up to 21 days, and can be repeatedly administered without the host generating neutralizing immunity. Recombinant poxvirus-based vaccines expressing CEA have proceeded through several evolutionary phases. Initial studies utilizing rV-CEA and recombinant fowlpox-CEA (rF-CEA) demonstrated that a diversified prime/boost vaccine regimen generated a more potent host immune response to CEA when compared with homologous vaccine regimens [75]. Still, while vaccinating CEA.Tg mice bearing subcutaneous CEA-expressing tumors with rV-CEA followed by rF-CEA did induce an anti-CEA host immune response, the strength of those responses did not elicit much of an antitumor response [75,76]. As a result, subsequent efforts were focused on improving the CEA-based vaccine by re-engineering the vaccine (both rV- and rF-) to include three co-stimulatory molecules (B7.1, LFA-3, and ICAM-1, designated TRICOM) [77]. These co-stimulatory molecules were selected because each molecule has a distinct signaling pathway in immune cells. As a result, the level of T cell activation observed with the TRICOM vector has been synergistic when compared with the level of activation using any one of the co-stimulatory molecules. In addition to the newly re-engineered poxvirus-based vaccines, experiments also identified biological adjuvants that might augment host immune responses to the recombinant poxvirus-based vaccines. Granulocyte-macrophage colony-stimulating factor (GM-CSF), by virtue of its actions as a major stimulatory cytokine for Langerhans and dendritic cells [78], may play a major role as a vaccine adjuvant. GM-CSF has been administered as a recombinant protein co-injected with antigen followed by an additional 3–4 daily injections at the injection site [79]. In our initial studies, this schedule had demonstrated GM-CSF to be an effective vaccine adjuvant [80]. However, other studies have suggested that the amount of GM-CSF produced or the route of its administration may actually impede its immune adjuvant actions [81]. As a consequence of those studies, we set out to design a delivery vehicle that would allow for long-term release of GM-CSF with accompanying restriction of the cytokine to the vaccination site. Those attributes would minimize the entry of GM-CSF into the circulation and ensure that its presence would coincide with that of antigen delivered via the poxvirus-based vaccines. That same approach might avoid the need to administer multiple injections of recombinant GM-CSF protein and improve the overall performance of this immune adjuvant. Indeed, GM-CSF was engineered as a recombinant fowlpox vector (rF-GM-CSF). A single subcutaneous injection of rF-GM-CSF increased the number of mature dendritic cells within the draining regional lymph nodes, which correlated with enhanced humoral and cellular immune responses to recombinant poxvirus-based vaccines [82,130]. Armed with this new and improved CEA-based vaccine, subsequent experimental studies were designed to test the antitumor efficacy of the vaccine in a CEA-expressing spontaneous colorectal tumor model.
4.3. Antitumor effects of a CEA-based vaccine on spontaneous intestinal murine tumors
An experimental protocol was designed and consisted of groups of either APCMin/+/CEA or APCMin/+ mice that were administered the CEA-based, non-CEA-based vaccines, or vehicle control. The CEA-based vaccine was comprised of a priming vaccination with rV-CEA-TRICOM with subsequent monthly boosts with rF-CEA-TRICOM. Mice that received the non-CEA-based vaccine were immunized with rV-TRICOM with monthly injections of rF-TRICOM. All vaccinations included rF-GM-CSF. Untreated APCMin/+/CEA or APCMin/+ mice received the vehicle (HBSS) alone. Vaccinations began at approximately 30 days of age with monthly booster vaccinations given for the entire length of the study. This aggressive vaccination plan was adopted because the APCMin/+/CEA genotypes present formidable challenges to the immune system to generate, not only anti-CEA host immunity, but also to impact adenoma formation. The APC mutation is germline and tumorigenesis is believed to initiate in utero, followed by aberrant crypt foci formation (ACF) at 2 weeks of age, and the mice develop a large tumor burden as a result of numerous intestinal adenomas [83]. CEA expression in normal tissues and its presence in circulating serum present a considerable degree of peripheral tolerance for the immune system to overcome [20]. The general health status of each mouse was followed closely with weekly body weights and regular hematocrit readings since the appearance of anemia is a good indication of tumor development. An initial indication that the CEA-based vaccine was eliciting favorable results was the continued good health status of the APCMin/+/CEA mice. As expected, APCMin/+/CEA mice treated with either the non-CEA-based vaccine or the vehicle alone developed adult-onset anemia (hematocrit values <35) at 100–120 days of age followed by progressive weight loss. Overall weight gain in those mice rarely exceeded 5–6 g, while APCMin/+/CEA mice vaccinated with the CEA-based vaccine maintained normal hematocrit levels and often reached a body weight of 28–30 g (total weight gain = 10.1 ± 1.6 g) with normal hematocrit readings for most mice. These changes seemed to be associated with the ability of the CEA-based vaccine to induce CEA-specific host immune responses since vaccinating the APCMin/+ mice with the CEA-based vaccine did not alter the course of the disease.
The total number of G.I. tumors for each of the APCMin/+/CEA mice within the different treatment groups was determined at approximately 150 days of age. The average number of G.I. tumors in the untreated APCMin/+/CEA mice was 36.7 ± 6.2. Administration of the non-CEA-based vaccine did not have any impact on tumor formation, as the average number of tumors in that group was 46.8 ± 6.2. Administration of the CEA-based vaccine reduced the average number of tumors/mouse by approximately 40% to 22.4 ± 6.8 (Fig. 2). Of the 13 APCMin/+/CEA mice that received the CEA-based vaccine, five had complete responses (0–3 adenomas), four partial responses (4–25 adenomas), and four had no response to the vaccine. Additional analyses revealed that adenoma size, as measured by the number of adenomas whose diameter was >2 mm, was also reduced, but the reduction was not statistically significant (P = 0.08) in CEA.Tg/MIN mice that received the CEA-based vaccine [37]. The reduction in the number and size of the intestinal adenomas in APCMin/+/CEA mice vaccinated with the CEA-based vaccine correlated with a significant (P < 0.01) improvement of long-term survival (Fig. 3). Mice in three control groups, CEA.Tg/MIN mice vaccinated with the non-CEA-based vaccine, vehicle alone or MIN mice that received the CEA-based vaccine, were sacrificed by 25–29 weeks due to continued weight loss and severe anemia (hematocrit levels <25). In contrast, APCMin/+/CEA mice vaccinated with the CEA-based vaccines continued to either maintain or gain body weight and their hematocrit levels remained in the normal range. At 37 weeks of age, 50% of those mice were still alive. But, by 55 weeks, all mice were severely anemic with progressive weight loss that required their sacrifice (Fig. 3). Subsequent macroscopic examination of the small intestines revealed numerous adenomas. While the data suggest that the generation of anti-CEA host immunity does suppress adenoma formation, future studies are needed to investigate the events that contribute to incomplete tumor suppression as well as the inability of some mice to mount an antitumor immune response. Several possible events include: (i) loss/reduction of MHC expression, (ii) TGF-β production, (iii) CEA loss variants, and (iv) Fas-FasL interactions and/or changes within the immune system (i.e., downregulation of T cell zeta-chain, generation of regulatory T cells).
Fig. 2.

Total number of intestinal adenomas in APCMin/+/CEA mice that received the CEA-based vaccine, non-CEA-based vaccine or vehicle alone. Each dot represents a single mouse.
Fig. 3.
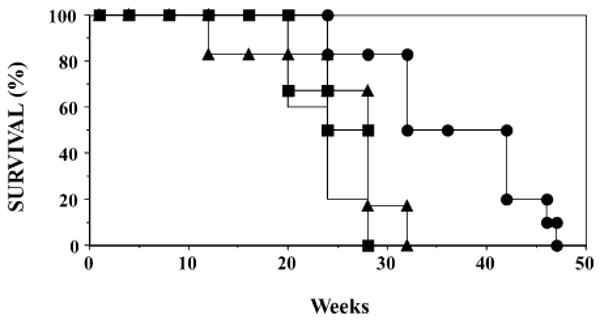
Long-term survival of vaccinated APCMin/+/CEA and APCMin/+ mice. APCMin/+/CEA mice were vaccinated with the CEA-based (n = 12, solid circles), non-CEA-based vaccine (n = 10, solid triangles) or vehicle alone (n = 12, solid line). APCMin/+ mice (n = 5, solid squares) also received the CEA-based vaccines.
One of the central arguments of using mouse models in which spontaneous tumors arise was that it would provide a longer time period for multiple vaccinations which, in turn, would be a better assessment of whether those mice develop autoimmunity as a result of an attack by the autoreactive T cells on CEA-expressing normal tissues. In the long-term survival portion of the study, approximately 50% of the APCMin/+/CEA mice were alive at 37 weeks after receiving 8–9 injections of the CEA-based vaccine. Intestinal tissues from those mice were analyzed histopathologically and by immunohistochemistry for indications of autoimmunity as well as for changes in CEA expression levels. Histopathological analyses of the intestines from APCMin/+/CEA mice which were protected from adenoma formation by administering the CEA-based vaccine revealed normal intestinal architecture (Fig. 4, panel A) and CEA expression in intestinal crypts (Fig. 4, panel B). Histological analyses of intestinal tissues isolated from APCMin/+/CEA mice that were unresponsive to the CEA-based vaccines revealed areas of epithelial cell proliferation (adenomas) (Fig. 4, panel C, arrows), ectatic crypts, and lymphocyte infiltration. Furthermore, those areas of adenoma formation also expressed higher CEA levels when compared with normal intestinal tissues (Fig. 4, panel D). In summary, immunohistochemistry of normal intestinal tissues revealed no significant differences in CEA expression levels in APCMin/+/CEA mice in which the CEA-based vaccine either reduced or had no effect on the number of intestinal adenomas.
Fig. 4.

Histopathological (A and C) and immunohistochemical (B and D) analyses of intestinal tissues of APCMin/+/CEA mice that received the CEA-based vaccine. Panel A shows an H&E staining of ileum revealing normal architecture from an APCMin/+/CEA mouse that did not develop intestinal adenomas following CEA-based vaccinations (25×). Panel B illustrates COL-1 MAb staining (30×) showing CEA expression in the crypts of the normal intestines of the APCMin/+/CEA mouse in panel A. Panel C illustrates H&E staining of an adenoma from an APCMin/+/CEA mouse in which the CEA-based vaccine did not reduce tumor formation (25×). Areas of epithelial cell proliferation (adenomas) are identified by the arrows. Panel D shows COL-1 staining of CEA expression (20×) in an adenoma from the APCMin/+/CEA mouse, which was described in panel C.
Spleen, pancreas, lung, liver, and kidneys from individual APCMin/+/CEA mice that received either the CEA-based, non-CEA-based vaccine or the vehicle alone were examined for macro- and microscopic indications of pathology. Spleens were enlarged and most other organs were pale when taken from mice that had multiple intestinal adenomas. Splenic enlargement was attributed to extramedullary hematopoiesis and the paleness of other organs associated with severe anemia since no other pathology was found. APCMin/+/CEA in which the CEA-based vaccine reduced the number of intestinal adenomas had normal sized spleens. Sporadic hydronephrosis was found in all three groups of mice. Serum samples from individual mice were analyzed for the presence of anti-DNA and anti-nuclear antibody titers and no changes were observed in any of the three groups of APCMin/+/CEA mice. In conclusion, the findings indicate that overexpression of CEA in the intestinal adenomas of APCMin/+/CEA mice can be exploited as a vaccine target. Vaccination with the CEA-based vaccine significantly reduced tumor multiplicity with a commensurate improvement in overall survival of the APCMin/+/CEA mice without any overt or microscopic indications of autoimmune complications.
5. Future directions: combination therapies: cancer vaccine and chemotherapeutic agents
As chemotherapeutic and immune-based approaches to cancer treatment become more mature, there is growing interest in their combination into chemoimmuno-based interventions for cancer therapy and/or prevention. From a cancer vaccine standpoint, those approaches might be exploited to (i) improve the potency of the vaccine by improving the immune recognition of tumor cells, thus, making them more susceptible to targeted lysis and/or (ii) combine another molecular target(s) together with the CEA-based vaccine. Both approaches are being investigated by numerous laboratories. Our interests have been to investigate whether targeting distinct molecules using chemotherapeutic agents might be compatible with the administration of the CEA-based cancer vaccine. The cyclooxygenase isoenzymes (COX) present potential targets for therapeutic and/or prevention intervention. Two COX isoenzymes have been identified: COX-1 is expressed in most tissues and necessary for healthy mucosa, kidneys and platelets [84], and COX-2, which is virtually undetectable in most tissues, but induced in response to inflammation, cytokines, growth factors and other stimuli [85,86]. Both COX isoenzymes convert arachidonic acid to PGH2, which serves as the substrate for a number of prostaglandin synthetases. Additionally, COX overexpression has been linked with resistance to apoptosis and tumor growth promotion [87]. A regular intake of NSAIDS, such as aspirin, ibuprofen, and sulindac, inhibits both COX enzymes with an associated reduction of cancer risk [88–92]. Subsequent findings reported that prolonged suppression of COX-1 activity caused unwanted side effects [93], which underscored the need to develop selective COX-2 inhibitors. COX-2 mRNA and protein are overexpressed in neoplastic epithelial cells [94–96] and COX-2 interruption by pharmacological agents or a gene knockout reduced tumor development in a variety of experimental murine models [97–99]. Celecoxib, one of the most studied COX-2 inhibitors, reduced the multiplicity and size of intestinal tumors (primarily adenomas) in the MIN mouse model [100]. In a subsequent clinical study, oral celecoxib (Celebrex®) administration significantly reduced the number of colorectal polyps in patients with FAP [101].
The APCMin/+/CEA mice offer an experimental model system in which one can investigate whether COX-2 and CEA can be simultaneously targeted in a combined chemoimmuno-based approach to cancer prevention and/or therapy. One of the first questions was whether combining a potent anti-inflammatory agent, such as celecoxib, with a pro-inflammatory vaccine might be counterproductive. Indeed, conflicting data does exist as to whether a reduction of COX-2 levels in T cells is associated with functional changes [102,103]. Therefore, prior to designing a study that incorporates a COX-2 inhibitor and the CEA-based vaccine, experiments were carried out to address whether chronic COX-2 suppression might negatively impact innate and/or adaptive host immunity. To address those questions, COX-2 expression was initially interrupted by placing groups of C57BL/6 mice on diets supplemented with 500, 1000, or 1500 ppm celecoxib. During a 3–4 month time period, the general health status of individual mice was closely monitored. Chronic exposure to celecoxib was indicated by the appearance of eosinophilia (>0.24 eosinophils per 103 cells/μl) [104] and a drop in plasma PGE2 levels [105]. Despite those changes, individual weight gain and CBC/differential counts were similar for mice whether or not they were fed a celecoxib-supplemented diet. Likewise, there were no obvious changes in splenic cell numbers or expression of lymphocyte cell surface markers. Several studies have reported that COX-2 is transcriptionally upregulated following activation of isolated human T cells in vitro, and that this upregulation was blocked by the addition of COX-2 inhibitors [102,106]. Yet, in a more recent study, downregulation of COX-2 mRNA by the in vitro addition of COX-2 antisense oligonucleotides did not impede T cell activation [103]. We decided to examine COX-2 expression levels in splenic T cells purified from mice fed either the control diet or the 1500-ppm celecoxib-supplemented diet (Fig. 5). RT-PCR-based analyses of the total RNA from splenic T cells from mice fed the control diet revealed very low COX-2 mRNA levels in unstimulated T cells which were increased approximately 9-fold by Con A stimulation (Fig. 5, lower panel, relative units). In contrast, COX-2 mRNA levels were undetectable in either resting or Con A-stimulated T cells isolated from mice fed the 1500 ppm celecoxib-supplemented diet (Fig. 5). The findings are also consistent with the hypothesis that COX-2 expression is regulated by a positive feedback loop and any disruption within the pathways would be expected to suppress COX-2 expression levels [107]. Despite the significant reduction in COX-2 mRNA levels in T cells from mice fed the diet supplemented with 1500 ppm of celecoxib, those T cells responded normally to Con A and anti-CD3 stimulation. In other experiments, mice in which COX-2 had been genetically ablated (i.e., COX-2 knockout mice) were vaccinated to examine whether the absence of COX-2 might negatively impact the generation of antigen-specific immunity. Host immune responses in vaccinated COX-2 KO mice were similar to those generated in COX-2+/+ mice.
Fig. 5.
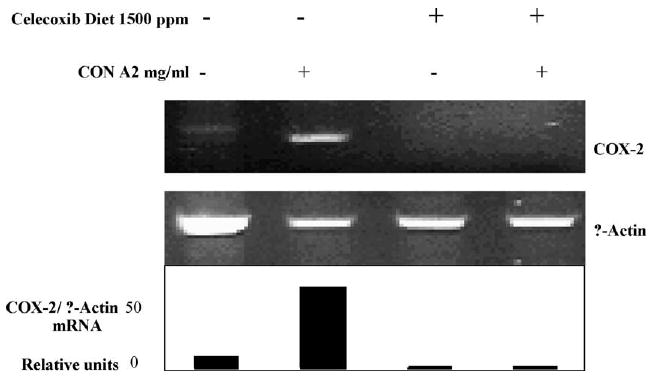
Celecoxib effects on T cell COX-2 mRNA expression levels. Splenic T cells were purified from mice fed either the control diet (no celecoxib) or the 1500 ppm celecoxib-supplemented diet and incubated in the absence or presence of 2.0 μg/ml Con A for 24 h. Total RNA was isolated and the RT-PCR amplification using primer pairs for COX-2 and β-actin was carried out. Gel images were taken and COX-2 mRNA transcript levels were quantified with normalization for β-actin expression as shown in the lower panel.
Those findings indicated that disruption of COX-2 expression with dietary celecoxib administration or ablation of the COX-2 gene did not severely impact the ability of the immune system to respond to innate stimuli or to generate adaptive host immune responses. As a result, an experimental protocol was designed to investigate whether a combined treatment of APCMin/+/CEA mice with dietary celecoxib and regularly scheduled CEA-based vaccinations might lead to a more potent antitumor response than administering either agent alone. APCMin/+/CEA mice were placed on either the control or celecoxib (1000 ppm) supplemented diet at 30 days of age at which time they received the primary vaccination comprised of rV-CEA-TRICOM or rV-TRICOM combined with rF-GM-CSF. Mice remained on the celecoxib-supplemented diet for the entire study and received monthly booster vaccinations with rF-CEA-TRICOM or rF-TRICOM with rF-GM-CSF. APCMin/+/CEA mice that were fed the control diet and remained untreated or vaccinated with the non-CEA-based vaccines TRICOM + rF-GM-CSF developed 38.8 ± 3.2 and 37.8 ± 2.1 intestinal tumors per mouse, respectively (Fig. 6). As reported earlier [20,38], administration of the CEA-based vaccine or dietary celecoxib as single agents significantly reduced the number of intestinal tumors. APCMin/+/CEA mice fed the celecoxib-supplemented diet averaged 13.4 ± 2.0 intestinal tumors per mouse (P < 0.001 versus mice fed the control diet and administered the vehicle alone, Fig. 6). The average number of intestinal tumors in APCMin/+/CEA mice vaccinated with the CEA-based vaccine (i.e., rV-rF-CEA-TRICOM + rF-GM-CSF) was 21.1 ± 2.6 (P < 0.05 versus vehicle or non-CEA-based vaccine treated APCMin/+/CEA mice) (Fig. 6). However, combining the CEA-based vaccine with dietary celecoxib substantially reduced intestinal tumor multiplicity to 2.1 ± 0.6, a reduction of 95% when compared with untreated APCMin/+/CEA mice. In fact, six of 16 APCMin/+/CEA mice administered the CEA-based vaccine and dietary celecoxib were tumor-free at 150–160 days of age. Statistical analyses using the linear regression coefficient of the combined treatment, i.e., CEA vaccine and celecoxib (β = −12.18), approximated the additive effect of each treatment alone, i.e., CEA-based vaccine (β = −8.46) and celecoxib treatment (β = −5.93). The significant reduction in intestinal tumor burden achieved by treating APCMin/+/CEA mice with dietary celecoxib combined with CEA-based vaccination was also accompanied by a profound improvement in overall survival (Fig. 7). Administration of the CEA-based vaccine alone significantly improved overall survival (P < 0.001 versus either vehicle control or non-CEA vaccine treated mice) of the APCMin/+/CEA mice, as did dietary celecoxib (P < 0.001 versus matched treatment groups of mice fed the control diet). However, all APCMin/+/CEA mice receiving either celecoxib or CEA-based vaccine eventually developed anemia with progressive weight loss with an overall survival of 40–60% by 18 months of age. For comparison, at 18 months of age 100% (12/12) of those APCMin/+/CEA mice that received both dietary celecoxib and the CEA-based vaccine were alive, a significant improvement in overall survival (X2 = 25.27; P < 0.001 versus CEA-based vaccine alone, and X2 = 7.76; P = 0.0054 versus celecoxib diet alone). The specific antitumor mechanisms of the combined treatment of CEA.Tg/MIN mice with the CEA-based vaccine and dietary celecoxib are currently under study. In APCΔ716/+ mice, COX-2 expression was found in polyp stromal cells and polyps >1 mm, but not in polyps <1 mm in diameter [108]. If a similar COX-2 tissue expression profile exists in the APCMin/+/CEA mice, then the target of early celecoxib administration would be the polyp stroma, not the epithelial cells. We know that the intestinal adenomas that appear in the APCMin/+/CEA mice overexpress CEA by approximately 3- to 4-fold when compared with normal intestinal mucosa. One possible hypothesis is that dietary celecoxib keeps the size of the tumors in check by targeting stromal COX-2 expression, thereby allowing for the CEA-based vaccine to attack the adenoma cells that emerge and overexpress CEA. That combination would be an effective approach to inhibit tumor formation at its earliest stages and account for those APCMin/+/CEA mice that remain tumor-free for up to 18 months of age. As in previous studies, extensive analyses were carried out to determine whether the combined treatment of CEA.Tg/MIN mice with dietary celecoxib and the CEA-based vaccine also induced changes in CEA expression (tongue, trachea, esophagus, stomach, intestine, cecum, and colon) associated with autoreactive T cells. No macroscopic or microscopic abnormalities were found in the tongue, trachea, esophagus, or stomach of the APCMin/+/CEA mice fed the celecoxib-supplemented diet and administered >10 injections of the CEA-based vaccine. In those CEA.Tg/MIN mice that received the combined treatment and were found to be tumor-free, histopathological analyses found the entire G.I. tract judged to be within normal histological limits. Those results provide another example of a CEA-based vaccination protocol that generates substantial antitumor immunity with little, or no, evidence of autoreactive T cells resulting in autoimmune pathology. The reasons(s) that the CEA-based vaccine elicits strong antitumor responses without accompanying autoimmune involvement remain an intriguing observation. Some possible explanations include: (i) a selective susceptibility of tumor tissue to immune attack due to a combination of the disruption of tissue architecture and CEA overexpression and (ii) a “braking” action of tolerizing APCs and/or regulatory T cells on autoreactive T cells [109].
Fig. 6.

Total number of intestinal tumors in APCMin/+/CEA mice that were fed either a control diet or a 1000 ppm celecoxib-supplemented diet. Mice also received the vehicle alone, the CEA-based vaccine or the non-CEA-based vaccine as indicated. Dots represent individual mice and horizontal lines the mean number of tumors for each treatment group. *N, total number of mice in that treatment group.
Fig. 7.

Long-term survival of APCMin/+/CEA mice fed either the control or 1000 ppm celecoxib-supplemented diets ± the indicated vaccine. APCMin/+/CEA mice fed the control diet were injected with the vehicle alone (n = 10, solid line, no symbol), the CEA-based vaccine (n = 11, open circles) or the non-CEA-based vaccine (n = 6, open triangles). Other groups of APCMin/+/CEA mice were fed the celecoxib-supplemented diet and also injected with the vehicle alone (n = 10, closed squares), the CEA-based vaccine (n = 12, closed circles) or the non-CEA-based vaccine (n = 7, closed triangles).
Finally, the data provide a strong argument for future experimental as well as clinical efforts to combine cancer vaccines with NSAIDs-targeting COX expression. A recent study reported that COX-2 inhibition enhanced the efficacy of an adenovirus-based intratumoral therapy [110]. The most immediate population that may benefit from a combined CEA-based vaccine with celecoxib is patients diagnosed with familial adenomatus polyposis (FAP). Celecoxib (Celebrex®) is being prescribed for FAP patients based on a 30% reduction in polyp burden [101]. In several early clinical studies, administration of the CEA-based vaccine has been well tolerated and able to generate anti-CEA host immune responses [111–113]. One question that remains is whether CEA overexpression is present in the polyps of the FAP patients. CEA levels, as measured by quantitative analyses, were 2- to 6-fold higher in tubulovillous adenomas and hyperplastic polyps when compared with CEA levels in normal mucosa from healthy donors [114]. A larger study is needed to not only examine CEA expression in colorectal polyps, but also determine the tissue expression of COX-2. A high percentage of other carcinomas including colorectal (FAP, HNPCC, sporadic), gastric, pancreas, breast (invasive, DCIS), non-small cell lung, cervical, and head and neck [115–120] overexpress both CEA and COX-2. Experimental studies are needed to determine the breadth of combining a cancer vaccine with targeting a COX isoenzyme. While the present results encourage the use of this combined treatment to inhibit the progression from pre-malignancy to tumors, the effectiveness of treating overt cancers with such a combined chemoimmuno-based approach requires additional research.
References
- 1.Sikorska H, Shuster J, Gold P. Clinical applications of carcinoembryonic antigen. Cancer Detect Prev. 1988;12:321–355. [PubMed] [Google Scholar]
- 2.Oikawa S, Nakazato H, Kosaki G. Primary structure of human carcinoembryonic antigen (CEA) deduced from cDNA sequence. Biochem Biophys Res Commun. 1987;142:511–518. doi: 10.1016/0006-291x(87)90304-4. [DOI] [PubMed] [Google Scholar]
- 3.Beauchemin N, Benchimol S, Cournoyer D, Fuks A, Stanners CP. Isolation and characterization of full-length functional cDNA clones for human carcinoembryonic antigen. Mol Cell Biol. 1987;7:3221–3230. doi: 10.1128/mcb.7.9.3221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thompson JA, Grunert F, Zimmermann W. Carcinoembryonic antigen gene family: molecular biology and clinical perspectives. J Clin Lab Anal. 1991;5:344–366. doi: 10.1002/jcla.1860050510. [DOI] [PubMed] [Google Scholar]
- 5.Olsen A, Teglund S, Nelson D, Gordon L, Copeland A, Georgescu A, Carrano A, Hammarstrom S. Gene organization of the pregnancy-specific glycoprotein region on human chromosome 19: assembly and analysis of a 700-kb cosmid contig spanning the region. Genomics. 1994;23:659–668. doi: 10.1006/geno.1994.1555. [DOI] [PubMed] [Google Scholar]
- 6.Beauchemin N, Draber P, Dveksler G, Gold P, Gray-Owen S, Grunert F, Hammarstrom S, Holmes KV, Karlsson A, Kuroki M, Lin SH, Lucka L, Najjar SM, Neumaier M, Obrink B, Shively JE, Skubitz KM, Stanners CP, Thomas P, Thompson JA, Virji M, von Kleist S, Wagener C, Watt S, Zimmermann W. Redefined nomenclature for members of the carcinoembryonic antigen family. Exp Cell Res. 1999;252:243–249. doi: 10.1006/excr.1999.4610. [DOI] [PubMed] [Google Scholar]
- 7.Hammarstrom S. The carcinoembryonic antigen (CEA) family: structures, suggested functions and expression in normal and malignant tissues. Semin Cancer Biol. 1999;9:67–81. doi: 10.1006/scbi.1998.0119. [DOI] [PubMed] [Google Scholar]
- 8.Dehio Christoph, Gray-Owen D Scott, Meyer TF. The role of neisserial Opa proteins in interactions with host cells. Trends Microbiol. 1998;6:489–495. doi: 10.1016/s0966-842x(98)01365-1. [DOI] [PubMed] [Google Scholar]
- 9.Benchimol S, Fuks A, Jothy S, Beauchemin N, Shirota K, Stanners CP. Carcinoembryonic antigen, a human tumor marker, functions as an intercellular adhesion molecule. Cell. 1989;57:327–334. doi: 10.1016/0092-8674(89)90970-7. [DOI] [PubMed] [Google Scholar]
- 10.Oikawa S, Inuzuka C, Kuroki M, Matsuoka Y, Kosaki G, Nakazato H. Cell adhesion activity of non-specific cross-reacting antigen (NCA) and carcinoembryonic antigen (CEA) expressed on CHO cell surface: homophilic and heterophilic adhesion. Biochem Biophys Res Commun. 1989;164:39–45. doi: 10.1016/0006-291x(89)91679-3. [DOI] [PubMed] [Google Scholar]
- 11.Berger CN, Billker O, Meyer TF, Servin AL, Kansau I. Differential recognition of members of the carcinoembryonic antigen family by Afa/Dr adhesins of diffusely adhering Escherichia coli (Afa/Dr DAEC) Mol Microbiol. 2004;52:963–983. doi: 10.1111/j.1365-2958.2004.04033.x. [DOI] [PubMed] [Google Scholar]
- 12.Hostetter RB, Augustus LB, Mankarious R, Chi KF, Fan D, Toth C, Thomas P, Jessup JM. Carcinoembryonic antigen as a selective enhancer of colorectal cancer metastasis. J Natl Cancer Inst. 1990;82:380–385. doi: 10.1093/jnci/82.5.380. [DOI] [PubMed] [Google Scholar]
- 13.Chevinsky AH. CEA in tumors of other than colorectal origin. Semin Surg Oncol. 1991;7:162–166. doi: 10.1002/ssu.2980070309. [DOI] [PubMed] [Google Scholar]
- 14.Hasegawa T, Isobe K, Tsuchiya Y, Oikawa S, Nakazato H, Ikezawa H, Nakashima I, Shimokata K. Establishment and characterisation of human carcinoembryonic antigen transgenic mice. Br J Cancer. 1991;64:710–714. doi: 10.1038/bjc.1991.386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chan CH, Stanners CP. Novel mouse model for carcinoembryonic antigen-based therapy. Mol Ther. 2004;9:775–785. doi: 10.1016/j.ymthe.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 16.Eades-Perner AM, van der Putten H, Hirth A, Thompson J, Neumaier M, von Kleist S, Zimmermann W. Mice transgenic for the human carcinoembryonic antigen gene maintain its spatiotemporal expression pattern. Cancer Res. 1994;54:4169–4176. [PubMed] [Google Scholar]
- 17.Clarke P, Mann J, Simpson JF, Rickard-Dickson K, Primus FJ. Mice transgenic for human carcinoembryonic antigen as a model for immunotherapy. Cancer Res. 1998;58:1469–1477. [PubMed] [Google Scholar]
- 18.Hefta LJ, Schrewe H, Thompson JA, Oikawa S, Nakazato H, Shively JE. Expression of complementary DNA and genomic clones for carcinoembryonic antigen and nonspecific cross-reacting antigen in Chinese hamster ovary and mouse fibroblast cells and characterization of the membrane-expressed products. Cancer Res. 1990;50:2397–2403. [PubMed] [Google Scholar]
- 19.Thompson JA, Eades-Perner AM, Ditter M, Muller WJ, Zimmermann W. Expression of transgenic carcinoembryonic antigen (CEA) in tumor-prone mice: an animal model for CEA-directed tumor immunotherapy. Int J Cancer. 1997;72:197–202. doi: 10.1002/(sici)1097-0215(19970703)72:1<197::aid-ijc28>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 20.Kass E, Schlom J, Thompson J, Guadagni F, Graziano P, Greiner JW. Induction of protective host immunity to carcinoembryonic antigen (CEA), a self-antigen in CEA transgenic mice, by immunizing with a recombinant vaccinia-CEA virus. Cancer Res. 1999;59:676–683. [PubMed] [Google Scholar]
- 21.Kinzler KW, Nilbert MC, Su LK, Vogelstein B, Bryan TM, Levy DB, Smith KJ, Preisinger AC, Hedge P, McKechnie D. Identification of FAP locus genes from chromosome 5q21. Science. 1991;253:661–665. doi: 10.1126/science.1651562. [DOI] [PubMed] [Google Scholar]
- 22.Groden J, Thliveris A, Samowitz W, Carlson M, Gelbert L, Albertsen H, Joslyn G, Stevens J, Spirio L, Robertson M. Identification and characterization of the familial adenomatous polyposis coli gene. Cell. 1991;66:589–600. doi: 10.1016/0092-8674(81)90021-0. [DOI] [PubMed] [Google Scholar]
- 23.Peltomaki P. Deficient DNA mismatch repair: a common etiologic factor for colon cancer. Hum Mol Genet. 2001;10:735–740. doi: 10.1093/hmg/10.7.735. [DOI] [PubMed] [Google Scholar]
- 24.Peltomaki P. DNA mismatch repair and cancer. Mutat Res. 2001;488:77–85. doi: 10.1016/s1383-5742(00)00058-2. [DOI] [PubMed] [Google Scholar]
- 25.Boivin GP, Washington K, Yang K, Ward JM, Pretlow TP, Russell R, Besselsen DG, Godfrey VL, Doetschman T, Dove WF, Pitot HC, Halberg RB, Itzkowitz SH, Groden J, Coffey RJ. Pathology of mouse models of intestinal cancer: consensus report and recommendations. Gastroenterology. 2003;124:762–777. doi: 10.1053/gast.2003.50094. [DOI] [PubMed] [Google Scholar]
- 26.Moser AR, Shoemaker AR, Connelly CS, Clipson L, Gould KA, Luongo C, Dove WF, Siggers PH, Gardner RL. Homozygosity for the Min allele of Apc results in disruption of mouse development prior to gastrulation. Dev Dyn. 1995;203:422–433. doi: 10.1002/aja.1002030405. [DOI] [PubMed] [Google Scholar]
- 27.He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW. Identification of c-MYC as a target of the APC pathway. Science. 1998;281:1509–1512. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- 28.Tetsu O, McCormick F. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 1999;398:422–426. doi: 10.1038/18884. [DOI] [PubMed] [Google Scholar]
- 29.Moser AR, Pitot HC, Dove WF. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science. 1990;247:322–324. doi: 10.1126/science.2296722. [DOI] [PubMed] [Google Scholar]
- 30.Moser AR, Dove WF, Roth KA, Gordon JI. The Min (multiple intestinal neoplasia) mutation: its effect on gut epithelial cell differentiation and interaction with a modifier system. J Cell Biol. 1992;116:1517–1526. doi: 10.1083/jcb.116.6.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fodde R, Edelmann W, Yang K, van Leeuwen C, Carlson C, Renault B, Breukel C, Alt E, Lipkin M, Khan PM. A targeted chain-termination mutation in the mouse Apc gene results in multiple intestinal tumors. Proc Natl Acad Sci U S A. 1994;91:8969–8973. doi: 10.1073/pnas.91.19.8969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smits R, Kartheuser A, Jagmohan-Changur S, Leblanc V, Breukel C, de Vries A, van Kranen H, van Krieken JH, Williamson S, Edelmann W, Kucherlapati R, Khan PM, Fodde R. Loss of Apc and the entire chromosome 18 but absence of mutations at the Ras and Tp53 genes in intestinal tumors from Apc1638N, a mouse model for Apc-driven carcinogenesis. Carcinogenesis. 1997;18:321–327. doi: 10.1093/carcin/18.2.321. [DOI] [PubMed] [Google Scholar]
- 33.Oshima M, Oshima H, Kitagawa K, Kobayashi M, Itakura C, Taketo M. Loss of Apc heterozygosity and abnormal tissue building in nascent intestinal polyps in mice carrying a truncated Apc gene. Proc Natl Acad Sci U S A. 1995;92:4482–4486. doi: 10.1073/pnas.92.10.4482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Quesada CF, Kimata H, Mori M, Nishimura M, Tsuneyoshi T, Baba S. Piroxicam and acarbose as chemopreventive agents for spontaneous intestinal adenomas in APC gene 1309 knockout mice. Jpn J Cancer Res. 1998;89:392–396. doi: 10.1111/j.1349-7006.1998.tb00576.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pretlow TP, Edelmann W, Kucherlapati R, Pretlow TG, Augenlicht LH. Spontaneous aberrant crypt foci in Apc1638N mice with a mutant Apc allele. Am J Pathol. 2003;163:1757–1763. doi: 10.1016/S0002-9440(10)63535-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Edelmann W, Yang K, Kuraguchi M, Heyer J, Lia M, Kneitz B, Fan K, Brown AM, Lipkin M, Kucherlapati R. Tumorigenesis in Mlh1 and Mlh1/Apc1638N mutant mice. Cancer Res. 1999;59:1301–1307. [PubMed] [Google Scholar]
- 37.Greiner JW, Zeytin H, Anver MR, Schlom J. Vaccine-based therapy directed against carcinoembryonic antigen demonstrates antitumor activity on spontaneous intestinal tumors in the absence of autoimmunity. Cancer Res. 2002;62:6944–6951. [PubMed] [Google Scholar]
- 38.Zeytin HE, Patel AC, Rogers CJ, Canter D, Hursting SD, Schlom J, Greiner JW. Combination of a poxvirus-based vaccine with a cyclooxygenase-2 inhibitor (celecoxib) elicits antitumor immunity and long-term survival in CEA.Tg/MIN mice. Cancer Res. 2004;64:3668–3678. doi: 10.1158/0008-5472.CAN-03-3878. [DOI] [PubMed] [Google Scholar]
- 39.Romagnolo B, Berrebi D, Saadi-Keddoucci S, Porteu A, Pichard A, Peuchmaur M, Vandewalle A, Kahn A, Perret C. Intestinal dysplasia and adenoma in transgenic mice after overexpression of an activated B-catenin. Cancer Res. 1999;59:3875–3879. [PubMed] [Google Scholar]
- 40.Wong MH, Rubinfeld B, Gordon JI. Effects of forced expression of an NH2-terminal truncated beta-Catenin on mouse intestinal epithelial homeostasis. J Cell Biol. 1998;141:765–777. doi: 10.1083/jcb.141.3.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Harada N, Tamai Y, Ishikawa T, Sauer B, Takaku K, Oshima M, Taketo MM. Intestinal polyposis in mice with a dominant stable mutation of the beta-catenin gene. EMBO J. 1999;18:5931–5942. doi: 10.1093/emboj/18.21.5931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reitmair AH, Redston M, Cai JC, Chuang TC, Bjerknes M, Cheng H, Hay K, Gallinger S, Bapat B, Mak TW. Spontaneous intestinal carcinomas and skin neoplasms in Msh2-deficient mice. Cancer Res. 1996;56:3842–3849. [PubMed] [Google Scholar]
- 43.Edelmann W, Umar A, Yang K, Heyer J, Kucherlapati M, Lia M, Kneitz B, Avdievich E, Fan K, Wong E, Crouse G, Kunkel T, Lipkin M, Kolodner RD, Kucherlapati R. The DNA mismatch repair genes Msh3 and Msh6 cooperate in intestinal tumor suppression. Cancer Res. 2000;60:803–807. [PubMed] [Google Scholar]
- 44.Shoemaker AR, Haigis KM, Baker SM, Dudley S, Liskay RM, Dove WF. Mlh1 deficiency enhances several phenotypes of Apc(Min)/+ mice. Oncogene. 2000;19:2774–2779. doi: 10.1038/sj.onc.1203574. [DOI] [PubMed] [Google Scholar]
- 45.Kuraguchi M, Yang K, Wong E, Avdievich E, Fan K, Kolodner RD, Lipkin M, Brown AM, Kucherlapati R, Edelmann W. The distinct spectra of tumor-associated Apc mutations in mismatch repair-deficient Apc1638N mice define the roles of MSH3 and MSH6 in DNA repair and intestinal tumorigenesis. Cancer Res. 2001;61:7934–7942. [PubMed] [Google Scholar]
- 46.Markowitz S, Wang J, Myeroff L, Parsons R, Sun L, Lutterbaugh J, Fan RS, Zborowska E, Kinzler KW, Vogelstein B. Inactivation of the type II TGF-beta receptor in colon cancer cells with microsatellite instability. Science. 1995;268:1336–1338. doi: 10.1126/science.7761852. [DOI] [PubMed] [Google Scholar]
- 47.Parsons R, Myeroff LL, Liu B, Willson JK, Markowitz SD, Kinzler KW, Vogelstein B. Microsatellite instability and mutations of the transforming growth factor beta type II receptor gene in colorectal cancer. Cancer Res. 1995;55:5548–5550. [PubMed] [Google Scholar]
- 48.Akiyama Y, Nakasaki H, Nihei Z, Iwama T, Nomizu T, Utsunomiya J, Yuasa Y. Frequent microsatellite instabilities and analyses of the related genes in familial gastric cancers. Jpn J Cancer Res. 1996;87:595–601. doi: 10.1111/j.1349-7006.1996.tb00265.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, Yin M, Allen R, Sidman C, Proetzel G, Calvin D. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature. 1992;359:693–699. doi: 10.1038/359693a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kulkarni AB, Huh CG, Becker D, Geiser A, Lyght M, Flanders KC, Roberts AB, Sporn MB, Ward JM, Karlsson S. Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc Natl Acad Sci U S A. 1993;90:770–774. doi: 10.1073/pnas.90.2.770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Engle SJ, Hoying JB, Boivin GP, Ormsby I, Gartside PS, Doetschman T. Transforming growth factor beta1 suppresses nonmetastatic colon cancer at an early stage of tumorigenesis. Cancer Res. 1999;59:3379–3386. [PubMed] [Google Scholar]
- 52.Engle SJ, Ormsby I, Pawlowski S, Boivin GP, Croft J, Balish E, Doetschman T. Elimination of colon cancer in germ-free transforming growth factor beta 1-deficient mice. Cancer Res. 2002;62:6362–6366. [PubMed] [Google Scholar]
- 53.Eppert K, Scherer SW, Ozcelik H, Pirone R, Hoodless P, Kim H, Tsui LC, Bapat B, Gallinger S, Andrulis IL, Thomsen GH, Wrana JL, Attisano L. MADR2 maps to 18q21 and encodes a TGFbeta-regulated MAD-related protein that is functionally mutated in colorectal carcinoma. Cell. 1996;86:543–552. doi: 10.1016/s0092-8674(00)80128-2. [DOI] [PubMed] [Google Scholar]
- 54.Thiagalingam S, Lengauer C, Leach FS, Schutte M, Hahn SA, Overhauser J, Willson JK, Markowitz S, Hamilton SR, Kern SE, Kinzler KW, Vogelstein B. Evaluation of candidate tumour suppressor genes on chromosome 18 in colorectal cancers. Nat Genet. 1996;13:343–346. doi: 10.1038/ng0796-343. [DOI] [PubMed] [Google Scholar]
- 55.Takagi Y, Kohmura H, Futamura M, Kida H, Tanemura H, Shimokawa K, Saji S. Somatic alterations of the DPC4 gene in human colorectal cancers in vivo. Gastroenterology. 1996;111:1369–1372. doi: 10.1053/gast.1996.v111.pm8898652. [DOI] [PubMed] [Google Scholar]
- 56.Waldrip WR, Bikoff EK, Hoodless PA, Wrana JL, Robertson EJ. Smad2 signaling in extraembryonic tissues determines anterior-posterior polarity of the early mouse embryo. Cell. 1998;92:797–808. doi: 10.1016/s0092-8674(00)81407-5. [DOI] [PubMed] [Google Scholar]
- 57.Zhu Y, Richardson JA, Parada LF, Graff JM. Smad3 mutant mice develop metastatic colorectal cancer. Cell. 1998;94:703–714. doi: 10.1016/s0092-8674(00)81730-4. [DOI] [PubMed] [Google Scholar]
- 58.Taketo MM, Takaku K. Gastro-intestinal tumorigenesis in Smad4 mutant mice. Cytokine Growth Factor Rev. 2000;11:147–157. doi: 10.1016/s1359-6101(99)00038-6. [DOI] [PubMed] [Google Scholar]
- 59.Cubilla AL, Fitzgerald PJ. Morphological patterns of primary nonendocrine human pancreas carcinoma. Cancer Res. 1975;35:2234–2248. [PubMed] [Google Scholar]
- 60.Berger DH, Chang H, Wood M, Huang L, Heath CW, Lehman T, Ruggeri BA. Mutational activation of K-ras in non-neoplastic exocrine pancreatic lesions in relation to cigarette smoking status. Cancer. 1999;85:326–332. doi: 10.1002/(sici)1097-0142(19990115)85:2<326::aid-cncr9>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 61.Brat DJ, Lillemoe KD, Yeo CJ, Warfield PB, Hruban RH. Progression of pancreatic intraductal neoplasias to infiltrating adenocarcinoma of the pancreas. Am J Surg Pathol. 1998;22:163–169. doi: 10.1097/00000478-199802000-00003. [DOI] [PubMed] [Google Scholar]
- 62.Hruban RH, Yeo CJ, Kern SE. Pancreatic Cancer. In: Vogelstein B, Kinzler KW, editors. The Genetic Basis of Human Cancer. McGraw-Hill; New York: 2002. pp. 659–673. [Google Scholar]
- 63.Bardeesy N, Sharpless NE, DePinho RA, Merlino G. The genetics of pancreatic adenocarcinoma: a roadmap for a mouse model. Semin Cancer Biol. 2001;11:201–218. doi: 10.1006/scbi.2000.0371. [DOI] [PubMed] [Google Scholar]
- 64.Ornitz DM, Hammer RE, Messing A, Palmiter RD, Brinster RL. Pancreatic neoplasia induced by SV40 T-antigen expression in acinar cells of transgenic mice. Science. 1987;238:188–193. doi: 10.1126/science.2821617. [DOI] [PubMed] [Google Scholar]
- 65.Tevethia MJ, Bonneau RH, Griffith JW, Mylin L. A simian virus 40 large T-antigen segment containing amino acids 1 to 127 and expressed under the control of the rat elastase-1 promoter produces pancreatic acinar carcinomas in transgenic mice. J Virol. 1997;71:8157–8166. doi: 10.1128/jvi.71.11.8157-8166.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kaufman HL, Di Vito J, Jr, Horig H. Immunotherapy for pancreatic cancer: current concepts. Hematol Oncol Clin North Am. 2002;16:159–197. viii. doi: 10.1016/s0889-8588(01)00002-8. [DOI] [PubMed] [Google Scholar]
- 67.Mukherjee P, Ginardi AR, Madsen CS, Sterner CJ, Adriance MC, Tevethia MJ, Gendler SJ. Mice with spontaneous pancreatic cancer naturally develo MUC-1-specific CTLs that eradicate tumors when adoptively transferred. J Immunol. 2000;165:3451–3460. doi: 10.4049/jimmunol.165.6.3451. [DOI] [PubMed] [Google Scholar]
- 68.McConnell EJ, Pathangey LB, Madsen CS, Gendler SJ, Mukherjee P. Dendritic cell-tumor cell fusion and staphylococcal enterotoxin B treatment in a pancreatic tumor model. J Surg Res. 2002;107:196–202. doi: 10.1006/jsre.2001.6497. [DOI] [PubMed] [Google Scholar]
- 69.Sandgren EP, Quaife CJ, Paulovich AG, Palmiter RD, Brinster RL. Pancreatic tumor pathogenesis reflects the causative genetic lesion. Proc Natl Acad Sci U S A. 1991;88:93–97. doi: 10.1073/pnas.88.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sandgren EP, Luetteke NC, Palmiter RD, Brinster RL, Lee DC. Overexpression of TGF alpha in transgenic mice: induction of epithelial hyperplasia, pancreatic metaplasia, and carcinoma of the breast. Cell. 1990;61:1121–1135. doi: 10.1016/0092-8674(90)90075-p. [DOI] [PubMed] [Google Scholar]
- 71.Wagner M, Luhrs H, Kloppel G, Adler G, Schmid RM. Malignant transformation of duct-like cells originating from acini in transforming growth factor transgenic mice. Gastroenterology. 1998;115:1254–1262. doi: 10.1016/s0016-5085(98)70098-8. [DOI] [PubMed] [Google Scholar]
- 72.Wagner M, Greten FR, Weber CK, Koschnick S, Mattfeldt T, Deppert W, Kern H, Adler G, Schmid RM. A murine tumor progression model for pancreatic cancer recapitulating the genetic alterations of the human disease. Genes Dev. 2001;15:286–293. doi: 10.1101/gad.184701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Aguirre AJ, Bardeesy N, Sinha M, Lopez L, Tuveson DA, Horner J, Redston MS, DePinho RA. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev. 2003;17:3112–3126. doi: 10.1101/gad.1158703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bardeesy N, DePinho RA. Pancreatic cancer biology and genetics. Nat Rev Cancer. 2002;2:897–909. doi: 10.1038/nrc949. [DOI] [PubMed] [Google Scholar]
- 75.Hodge JW, McLaughlin JP, Kantor JA, Schlom J. Diversified prime and boost protocols using recombinant vaccinia virus and recombinant non-replicating avian pox virus to enhance T cell immunity and antitumor responses. Vaccine. 1997;15:759–768. doi: 10.1016/s0264-410x(96)00238-1. [DOI] [PubMed] [Google Scholar]
- 76.McLaughlin JP, Abrams S, Kantor J, Dobrzanski MJ, Greenbaum J, Schlom J, Greiner JW. Immunization with a syngeneic tumor infected with recombinant vaccinia virus expressing granulocyte-macrophage colony-stimulating factor (GM-CSF) induces tumor regression and long-lasting systemic immunity. J Immunother. 1997;20:449–459. doi: 10.1097/00002371-199711000-00004. [DOI] [PubMed] [Google Scholar]
- 77.Hodge JW, Sabzevari H, Yafal AG, Gritz L, Lorenz MG, Schlom J. A triad of costimulatory molecules synergize to amplify T cell activation. Cancer Res. 1999;59:5800–5807. [PubMed] [Google Scholar]
- 78.Morrissey PJ, Bressler L, Park LS, Alpert A, Gillis S. Granulocyte-macrophage colony-stimulating factor augments the primary antibody response by enhancing the function of antigen-presenting cells. J Immunol. 1987;139:1113–1119. [PubMed] [Google Scholar]
- 79.Disis ML, Bernhard H, Shiota FM, Hand SL, Gralow JR, Huseby ES, Gillis S, Cheever MA. Granulocyte-macrophage colony-stimulating factor: an effective adjuvant for protein and peptide-based vaccines. Blood. 1996;88:202–210. [PubMed] [Google Scholar]
- 80.Kass E, Parker J, Schlom J, Greiner JW. Comparative studies of the effects of recombinant GM-CSF and GM-CSF administered via a poxvirus to enhance the concentration of antigen-presenting cells in regional lymph nodes. Cytokine. 2000;12:960–971. doi: 10.1006/cyto.2000.0684. [DOI] [PubMed] [Google Scholar]
- 81.Bronte V, Chappell DB, Apolloni E, Cabrelle A, Wang M, Hwu P, Restifo NP. Unopposed production of granulocyte-macrophage colony-stimulating factor by tumors inhibits CD8+ T cell responses by dysregulating antigen-presenting cell maturation. J Immunol. 1999;162:5728–5737. [PMC free article] [PubMed] [Google Scholar]
- 82.Kass E, Panicali DL, Mazzara G, Schlom J, Greiner JW. Granulocyte/macrophage-colony stimulating factor produced by recombinant avian poxviruses enriches the regional lymph nodes with antigen-presenting cells and acts as an immunoadjuvant. Cancer Res. 2001;61:206–214. [PubMed] [Google Scholar]
- 83.Paulsen JE, Steffensen IL, Loberg EM, Husoy T, Namork E, Alexander J. Qualitative and quantitative relationship between dysplastic aberrant crypt foci and tumorigenesis in the Min/+ mouse colon. Cancer Res. 2001;61:5010–5015. [PubMed] [Google Scholar]
- 84.Smith WL, Garavito RM, DeWitt DL. Prostaglandin endoperoxide H synthases (cyclooxygenases)-1 and -2. J Biol Chem. 1996;271:33157–33160. doi: 10.1074/jbc.271.52.33157. [DOI] [PubMed] [Google Scholar]
- 85.Masferrer JL, Seibert K, Zweifel B, Needleman P. Endogenous glucocorticoids regulate an inducible cyclooxygenase enzyme. Proc Natl Acad Sci U S A. 1992;89:3917–3921. doi: 10.1073/pnas.89.9.3917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kujubu DA, Fletcher BS, Varnum BC, Lim RW, Herschman HR. TIS10, a phorbol ester tumor promoter-inducible mRNA from Swiss 3T3 cells, encodes a novel prostaglandin synthase/cyclooxygenase homologue. J Biol Chem. 1991;266:12866–12872. [PubMed] [Google Scholar]
- 87.Souza RF, Shewmake K, Beer DG, Cryer B, Spechler SJ. Selective inhibition of cyclooxygenase-2 suppresses growth and induces apoptosis in human esophageal adenocarcinoma cells. Cancer Res. 2000;60:5767–5772. [PubMed] [Google Scholar]
- 88.Vane JR. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat New Biol. 1971;231:232–235. doi: 10.1038/newbio231232a0. [DOI] [PubMed] [Google Scholar]
- 89.Smalley WE, DuBois RN. Colorectal cancer and nonsteroidal anti-inflammatory drugs. Adv Pharmacol. 1997;39:1–20. doi: 10.1016/s1054-3589(08)60067-8. [DOI] [PubMed] [Google Scholar]
- 90.Sandler RS, Halabi S, Baron JA, Budinger S, Paskett E, Keresztes R, Petrelli N, Pipas JM, Karp DD, Loprinzi CL, Steinbach G, Schilsky R. A randomized trial of aspirin to prevent colorectal adenomas in patients with previous colorectal cancer. N Engl J Med. 2003;348:883–890. doi: 10.1056/NEJMoa021633. [DOI] [PubMed] [Google Scholar]
- 91.Baron JA, Cole BF, Sandler RS, Haile RW, Ahnen D, Bresalier R, McKeown-Eyssen G, Summers RW, Rothstein R, Burke CA, Snover DC, Church TR, Allen JI, Beach M, Beck GJ, Bond JH, Byers T, Greenberg ER, Mandel JS, Marcon N, Mott LA, Pearson L, Saibil F, van Stolk RU. A randomized trial of aspirin to prevent colorectal adenomas. N Engl J Med. 2003;348:891–899. doi: 10.1056/NEJMoa021735. [DOI] [PubMed] [Google Scholar]
- 92.Cruz-Correa M, Hylind LM, Romans KE, Booker SV, Giardiello FM. Long-term treatment with sulindac in familial adenomatous polyposis: a prospective cohort study. Gastroenterology. 2002;122:641–645. doi: 10.1053/gast.2002.31890. [DOI] [PubMed] [Google Scholar]
- 93.Wolfe MM, Lichtenstein DR, Singh G. Gastrointestinal toxicity of nonsteroidal anti-inflammatory drugs. N Engl J Med. 1999;340:1888–1899. doi: 10.1056/NEJM199906173402407. [DOI] [PubMed] [Google Scholar]
- 94.Koki AT, Leahy KM, Masferrer JL. Potential utility of COX-2 inhibitors in chemoprevention and chemotherapy. Expert Opin Investig Drugs. 1999;8:1623–1638. doi: 10.1517/13543784.8.10.1623. [DOI] [PubMed] [Google Scholar]
- 95.Fosslien E. Biochemistry of cyclooxygenase (COX)-2 inhibitors and molecular pathology of COX-2 in neoplasia. Crit Rev Clin Lab Sci. 2000;37:431–502. doi: 10.1080/10408360091174286. [DOI] [PubMed] [Google Scholar]
- 96.Masferrer JL, Leahy KM, Koki AT, Zweifel BS, Settle SL, Woerner BM, Edwards DA, Flickinger AG, Moore RJ, Seibert K. Antiangiogenic and antitumor activities of cyclooxygenase-2 inhibitors. Cancer Res. 2000;60:1306–1311. [PubMed] [Google Scholar]
- 97.Oshima M, Dinchuk JE, Kargman SL, Oshima H, Hancock B, Kwong E, Trzaskos JM, Evans JF, Taketo MM. Suppression of intestinal polyposis in Apc delta716 knockout mice by inhibition of cyclooxygenase 2 (COX-2) Cell. 1996;87:803–809. doi: 10.1016/s0092-8674(00)81988-1. [DOI] [PubMed] [Google Scholar]
- 98.Reddy BS, Hirose Y, Lubet R, Steele V, Kelloff G, Paulson S, Seibert K, Rao CV. Chemoprevention of colon cancer by specific cyclooxygenase-2 inhibitor, celecoxib, administered during different stages of carcinogenesis. Cancer Res. 2000;60:293–297. [PubMed] [Google Scholar]
- 99.Oshima M, Murai N, Kargman S, Arguello M, Luk P, Kwong E, Taketo MM, Evans JF. Chemoprevention of intestinal polyposis in the Apcdelta716 mouse by rofecoxib, a specific cyclooxygenase-2 inhibitor. Cancer Res. 2001;61:1733–1740. [PubMed] [Google Scholar]
- 100.Jacoby RF, Seibert K, Cole CE, Kelloff G, Lubet RA. The cyclooxygenase-2 inhibitor celecoxib is a potent preventive and therapeutic agent in the min mouse model of adenomatous polyposis. Cancer Res. 2000;60:5040–5044. [PubMed] [Google Scholar]
- 101.Steinbach G, Lynch PM, Phillips RK, Wallace MH, Hawk E, Gordon GB, Wakabayashi N, Saunders B, Shen Y, Fujimura T, Su LK, Levin B. The effect of celecoxib, a cyclooxygenase-2 inhibitor, in familial adenomatous polyposis. N Engl J Med. 2000;342:1946–1952. doi: 10.1056/NEJM200006293422603. [DOI] [PubMed] [Google Scholar]
- 102.Iniguez MA, Punzon C, Fresno M. Induction of cyclooxygenase-2 on activated T lymphocytes: regulation of T cell activation by cyclooxygenase-2 inhibitors. J Immunol. 1999;163:111–119. [PubMed] [Google Scholar]
- 103.Rivero M, Santiago B, Galindo M, Brehmer MT, Pablos JL. Cyclooxygenase-2 inhibition lacks immunomodulatory effects on T cells. Clin Exp Rheumatol. 2002;20:379–385. [PubMed] [Google Scholar]
- 104.Goodwin SD, Glenny RW. Nonsteroidal anti-inflammatory drug-associated pulmonary infiltrates with eosinophilia. Review of the literature and Food and Drug Administration Adverse Drug Reaction reports. Arch Intern Med. 1992;152:1521–1524. doi: 10.1001/archinte.152.7.1521. [DOI] [PubMed] [Google Scholar]
- 105.Takahashi Y, Roman C, Chemtob S, Tse MM, Lin E, Heymann MA, Clyman RI. Cyclooxygenase-2 inhibitors constrict the fetal lamb ductus arteriosus both in vitro and in vivo. Am J Physiol Regul Integr Comp Physiol. 2000;278:R1496–R1505. doi: 10.1152/ajpregu.2000.278.6.R1496. [DOI] [PubMed] [Google Scholar]
- 106.Iniguez MA, Martinez-Martinez S, Punzon C, Redondo JM, Fresno M. An essential role of the nuclear factor of activated T cells in the regulation of the expression of the cyclooxygenase-2 gene in human T lymphocytes. J Biol Chem. 2000;275:23627–23635. doi: 10.1074/jbc.M001381200. [DOI] [PubMed] [Google Scholar]
- 107.Sonoshita M, Takaku K, Sasaki N, Sugimoto Y, Ushikubi F, Narumiya S, Oshima M, Taketo MM. Acceleration of intestinal polyposis through prostaglandin receptor EP2 in Apc(delta 716) knockout mice. Nat Med. 2001;7:1048–1051. doi: 10.1038/nm0901-1048. [DOI] [PubMed] [Google Scholar]
- 108.Takeda H, Sonoshita M, Oshima H, Sugihara K, Chulada PC, Langenbach R, Oshima M, Taketo MM. Cooperation of cyclooxygenase 1 and cyclooxygenase 2 in intestinal polyposis. Cancer Res. 2003;63:4872–4877. [PubMed] [Google Scholar]
- 109.Gilboa E. The risk of autoimmunity associated with tumor immunotherapy. Nat Immunol. 2001;2:789–792. doi: 10.1038/ni0901-789. [DOI] [PubMed] [Google Scholar]
- 110.DeLong P, Tanaka T, Kruklitis R, Henry AC, Kapoor V, Kaiser LR, Sterman DH, Albelda SM. Use of cyclooxygenase-2 inhibition to enhance the efficacy of immunotherapy. Cancer Res. 2003;63:7845–7852. [PubMed] [Google Scholar]
- 111.Marshall JL, Hawkins MJ, Tsang KY, Richmond E, Pedicano JE, Zhu MZ, Schlom J. Phase I study in cancer patients of a replication-defective avipox recombinant vaccine that expresses human carcinoembryonic antigen. J Clin Oncol. 1999;17:332–337. doi: 10.1200/JCO.1999.17.1.332. [DOI] [PubMed] [Google Scholar]
- 112.von Mehren M, Arlen P, Tsang KY, Rogatko A, Meropol N, Cooper HS, Davey M, McLaughlin S, Schlom J, Weiner LM. Pilot study of a dual gene recombinant avipox vaccine containing both carcinoembryonic antigen (CEA) and B7.1 transgenes in patients with recurrent CEA-expressing adenocarcinomas. Clin Cancer Res. 2000;6:2219–2228. [PubMed] [Google Scholar]
- 113.Marshall JL, Hoyer RJ, Toomey MA, Faraguna K, Chang P, Richmond E, Pedicano JE, Gehan E, Peck RA, Arlen P, Tsang KY, Schlom J. Phase I study in advanced cancer patients of a diversified prime-and-boost vaccination protocol using recombinant vaccinia virus and recombinant nonreplicating avipox virus to elicit anti-carcinoembryonic antigen immune responses. J Clin Oncol. 2000;18:3964–3973. doi: 10.1200/JCO.2000.18.23.3964. [DOI] [PubMed] [Google Scholar]
- 114.Guadagni F, Roselli M, Cosimelli M, Spila A, Cavaliere F, Arcuri R, D'Alessandro R, Fracasso PL, Casale V, Vecchione A, Casciani CU, Greiner JW, Schlom J. Quantitative analysis of CEA expression in colorectal adenocarcinoma and serum: lack of correlation. Int J Cancer. 1997;72:949–954. doi: 10.1002/(sici)1097-0215(19970917)72:6<949::aid-ijc5>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 115.Sheehan KM, Sheahan K, O'Donoghue DP, MacSweeney F, Conroy RM, Fitzgerald DJ, Murray FE. The relationship between cyclooxygenase-2 expression and colorectal cancer. JAMA. 1999;282:1254–1257. doi: 10.1001/jama.282.13.1254. [DOI] [PubMed] [Google Scholar]
- 116.Saukkonen K, Nieminen O, van Rees B, Vilkki S, Harkonen M, Juhola M, Mecklin JP, Sipponen P, Ristimaki A. Expression of cyclooxygenase-2 in dysplasia of the stomach and in intestinal-type gastric adenocarcinoma. Clin Cancer Res. 2001;7:1923–1931. [PubMed] [Google Scholar]
- 117.Tucker ON, Dannenberg AJ, Yang EK, Zhang F, Teng L, Daly JM, Soslow RA, Masferrer JL, Woerner BM, Koki AT, Fahey TJ., III Cyclooxygenase-2 expression is upregulated in human pancreatic cancer. Cancer Res. 1999;59:987–990. [PubMed] [Google Scholar]
- 118.Achiwa H, Yatabe Y, Hida T, Kuroishi T, Kozaki K, Nakamura S, Ogawa M, Sugiura T, Mitsudomi T, Takahashi T. Prognostic significance of elevated cyclooxygenase 2 expression in primary, resected lung adenocarcinomas. Clin Cancer Res. 1999;5:1001–1005. [PubMed] [Google Scholar]
- 119.Kulkarni S, Rader JS, Zhang F, Liapis H, Koki AT, Masferrer JL, Subbaramaiah K, Dannenberg AJ. Cyclooxygenase-2 is overexpressed in human cervical cancer. Clin Cancer Res. 2001;7:429–434. [PubMed] [Google Scholar]
- 120.Chan G, Boyle JO, Yang EK, Zhang F, Sacks PG, Shah JP, Edelstein D, Soslow RA, Koki AT, Woerner BM, Masferrer JL, Dannenberg AJ. Cyclooxygenase-2 expression is upregulated in squamous cell carcinoma of the head and neck. Cancer Res. 1999;59:991–994. [PubMed] [Google Scholar]
- 121.Mizobata S, Tompkins K, Simpson JF, Shyr Y, Primus FJ. Induction of cytotoxic T cells and their antitumor activity in mice transgenic for carcinoembryonic antigen. Cancer Immunol Immunother. 2000;49:285–295. doi: 10.1007/s002620000116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Xu X, Clarke P, Szalai G, Shively JE, Williams LE, Shyr Y, Shi E, Primus FJ. Targeting and therapy of carcinoembryonic antigen-expressing tumors in transgenic mice with an antibody-interleukin 2 fusion protein. Cancer Res. 2000;60:4475–4484. [PubMed] [Google Scholar]
- 123.Xiang R, Primus FJ, Ruehlmann JM, Niethammer AG, Silletti S, Lode HN, Dolman CS, Gillies SD, Reisfeld RA. A dual-function DNA vaccine encoding carcinoembryonic antigen and CD40 ligand trimer induces T cell-mediated protective immunity against colon cancer in carcinoembryonic antigen-transgenic mice. J Immunol. 2001;167:4560–4565. doi: 10.4049/jimmunol.167.8.4560. [DOI] [PubMed] [Google Scholar]
- 124.Xiang R, Silletti S, Lode HN, Dolman CS, Ruehlmann JM, Niethammer AG, Pertl U, Gillies SD, Primus FJ, Reisfeld RA. Protective immunity against human carcinoembryonic antigen (CEA) induced by an oral DNA vaccine in CEA-transgenic mice. Clin Cancer Res. 2001;7:856s–864s. [PubMed] [Google Scholar]
- 125.Niethammer AG, Xiang R, Becker JC, Wodrich H, Pertl U, Karsten G, Eliceiri BP, Reisfeld RA. A DNA vaccine against VEGF receptor 2 prevents effective angiogenesis and inhibits tumor growth. Nat Med. 2002;8:1369–1375. doi: 10.1038/nm1202-794. [DOI] [PubMed] [Google Scholar]
- 126.Chakrabarti R, Chang Y, Song K, Prud'homme GJ. Plasmids encoding membrane-bound IL-4 or IL-12 strongly costimulate DNA vaccination against carcinoembryonic antigen (CEA) Vaccine. 2004;22:1199–1205. doi: 10.1016/j.vaccine.2003.09.023. [DOI] [PubMed] [Google Scholar]
- 127.Grosenbach DW, Barrientos JC, Schlom J, Hodge JW. Synergy of vaccine strategies to amplify antigen-specific immune responses and antitumor effects. Cancer Res. 2001;61:4497–4505. [PubMed] [Google Scholar]
- 128.Hodge JW, Grosenbach DW, Aarts WM, Poole DJ, Schlom J. Vaccine therapy of established tumors in the absence of autoimmunity. Clin Cancer Res. 2003;9:1837–1849. [PubMed] [Google Scholar]
- 129.Zhou H, Luo Y, Mizutani M, Mizutani N, Becker JC, Primus FJ, Xiang R, Reisfeld RA. A novel transgenic mouse model for immunological evaluation of carcinoembryonic antigen-based DNA minigene vaccines. J Clin Invest. 2004;113:1792–1798. doi: 10.1172/JCI21107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Reali E, Canter D, Zeytin H, Schlom J, Greiner JW. Comparative studies of avipox-GM-CSF versus recombinant GM-CSF protein as immune adjuvants with different vaccine platforms. Vaccine. 2005;23:2909–2921. doi: 10.1016/j.vaccine.2004.11.060. [DOI] [PubMed] [Google Scholar]