

Abstract
Prenylation inhibitors have gained increasing attention as potential therapeutics for cancer. Initial work focused on inhibitors of farnesylation, but more recently geranylgeranyl transferase inhibitors (GGTIs) have begun to be evaluated for their potential antitumor activity in vitro and in vivo. In this study, we have developed a nonpeptidomimetic GGTI, termed GGTI-2Z [(5-nitrofuran-2-yl)methyl-(2Z,6E,10E)-3,7,11,15-tetramethylhexadeca-2,6,10,14-tetraenyl 4-chlorobutyl(methyl)phosphoramidate], which in combination with lovastatin inhibits geranylgeranyl transferase I (GGTase I) and GGTase II/RabGGTase, without affecting farnesylation. The combination treatment results in a G0/G1 arrest and synergistic inhibition of proliferation of cultured STS-26T malignant peripheral nerve sheath tumor cells. We also show that the antiproliferative activity of drugs in combination occurs in the context of autophagy. The combination treatment also induces autophagy in the MCF10.DCIS model of human breast ductal carcinoma in situ and in 1c1c7 murine hepatoma cells, where it also reduces proliferation. At the same time, there is no detectable toxicity in normal immortalized Schwann cells. These studies establish GGTI-2Z as a novel geranylgeranyl pyrophosphate derivative that may work through a new mechanism involving the induction of autophagy and, in combination with lovastatin, may serve as a valuable paradigm for developing more effective strategies in this class of antitumor therapeutics.
Lipid modifications of proteins are known to facilitate their membrane association, cellular localization, and protein–protein interactions, which are related to their functions including cell survival, proliferation, differentiation, and invasion. Most GTPases, like Ras, contain a C-terminal CaaX motif that undergoes prenylation catalyzed by prenyl transferase enzymes, including farnesyl transferase (FTase) or geranylgeranyl transferase I (GGTase I), followed by serial modification via several other enzymes (Konstantinopoulos et al., 2007). The substrate specificities of FTase and GGTase I are not mutually exclusive and, therefore, cross-prenylation can occur. For example, inhibition of N-Ras and K-Ras farnesylation using a FTase inhibitor (FTI) leads to alternative geranylgeranylation by GGTase I (Lerner et al., 1997). On the other hand, Rab GTPases have a CXC or a CC motif at their C termini, which is geranylgeranylated by RabGGTase or GGTase II (Leung et al., 2006).
Several different strategies have been explored to develop inhibitors of GGTase (GGTIs). Peptidomimetic inhibitors and small-molecule inhibitors of GGTase I developed in different laboratories have shown promise as antitumor agents in vitro and in vivo (Vogt et al., 1996; Peterson et al., 2006). For instance, geranylgeranylation inhibition was found to regress breast tumor xenografts in vivo via nuclear accumulation of hypophosphorylated p27Kip1 (Kazi et al., 2009). Although GGTIs were expected to target more proteins in a cell than FTIs, the toxicity of GGTase I inhibition and genetic ablation was surprisingly less significant than initially expected (Sun et al., 2003).
Statins, the cholesterol-lowering drugs, inhibit the rate-limiting enzyme 3-hydroxy-3-methylglutaryl coenzyme A reductase of the mevalonate pathway of lipid synthesis, which further leads to inhibition of downstream isoprenoids farnesyl pyrophosphate and geranylgeranyl pyrophosphate (GGPP). Several statins by themselves have been shown to inhibit geranylgeranylation of different target proteins, including Rac1 and Rap1, in diverse cellular systems at concentrations of 10 and 20 μM, respectively (Hamadmad and Hohl, 2007; Kou et al., 2009). One approach to achieving effective GGTase I inhibition includes simultaneously targeting the prenylation pathway at two distinct steps, one to deplete the cellular GGPP pools via statin treatment and the other using a GGPP competitive GGTI that will be more effective when competition from the endogenous GGPP is alleviated (Konstantinopoulos et al., 2007). The underlying rationale is that the two compounds demonstrate modest action by themselves, but when combined they exhibit much more pronounced effects because a lower concentration of an isoprenoid-competitive GGTI can better target GGTase I in an environment with reduced endogenous GGPP (Wojtkowiak et al., 2009). We have previously demonstrated the utility of this approach for the inhibition of cellular farnesylation (Wojtkowiak et al., 2008).
In the current study, we tested a novel compound, GGTI-2Z, which is a GGPP derivative designed to block geranylgeranylation in an isoprenoid-competitive manner, to determine whether it effectively blocks protein geranylgeranylation in a malignant peripheral nerve sheath tumor (MPNST) cell line. We tested this compound either alone or in combination with lovastatin. We demonstrate that GGTI-2Z, in combination with lovastatin, causes G0/G1 cell-cycle arrest, inhibits proliferation, and induces autophagy in cultured STS-26T MPNST cells.
Materials and Methods
Synthetic Chemistry
Compound 2.
In an argon-flushed flask, compound 1 (Gibbs et al., 1999) (3200 mg, 9.58 mmol) was dissolved in 65 ml of dimethylformamide (DMF) (Fig. 1). The solution was cooled to −60°C, and potassium bis(trimethylsilyl)amide [(Me3Si)2NK; 0.5 M in toluene, 11.5 mmol, 23 ml] was added dropwise. The solution was warmed to 0°C for 5 min and then recooled to −60°C for 30 min. A slurry of 2-[N,N-bis(trifluoromethylsulfonyl)amide]-5-chloropyridine (4500 mg, 11.5 mmol) in 8 ml of DMF was added to the potassium enolate solution. The reaction was allowed to warm from −60°C to room temperature over 3 h. It was then taken up in 100 ml of ether, washed with 10% aqueous citric acid (2 × 75 ml) and water (4 × 100 ml), dried over MgSO4, and concentrated. Purification by flash chromatography (95:5 hexanes/ethyl acetate) gave 3130 mg (70%) of triflate compound 2 as a pale yellow oil. 1H NMR (300 MHz, CDCl3): δ 1.25 (t, J = 7.05 Hz, 3H), 1.56 (m, 9H), 1.60 (s, 3H), 1.97 (m, 8H), 2.25 (m, 2H), 2.89 (t, J = 7.5 Hz, 2H), 4.15 (q, J = 7.125 Hz, 2H), 5.06 (m, 3H), 5.87 (s, 1H).
Fig. 1.

Synthesis of prodrug GGTI-2Z (compound 7) and structures of 2Z-GGMP (compound 8) and 2Z-GGPP (compound 9). TEA, triethylamine; DCM, dichloromethane.
Compound 3.
In a flame-dried, argon-flushed flask were placed triflate compound 2 (750 mg, 1.6 mmol), Pd(PPh3)2Cl2 (11.2 mg, 0.016 mmol), and 15 ml of tetrahydrofuran (THF). The reaction mixture was heated to reflux at 70°C, and (CH3)3In (Perez et al., 2001) (15 ml, 1.5 mmol in THF) was added dropwise. After 4 h, 2 ml of MeOH was added, and the reaction mixture was concentrated. The reaction mixture was next taken up in 30 ml of ether, washed with 10% HCl (10 ml), aqueous NaHCO3 (15 ml), and brine, dried over MgSO4, and concentrated. Purification by flash chromatography (hexanes/ethyl acetate 95:5) gave 313 mg (59%) of compound 3 as a colorless oil. Note that this procedure using trimethylindium (Perez et al., 2001) affords superior results to the Stille coupling procedure with tetramethyltin used in previous work (Zahn et al., 2001) on 2Z-GGPP (compound 9). 1H NMR (300 MHz, CDCl3): δ 1.55 (t, J = 6.9 Hz, 3H), 1.87 (m, 9H), 1.94 (s, 3H), 2.15 (s, 3H), 2.27- 2.4 (m, 10H), 2.9 (t, J = 7.8 Hz, 2H), 4.41 (q, J = 6.9 Hz, 2H), 5.44 (m, 3H), 5.92 (s, 1H).
Compound 4.
To the solution of ester compound 3 (313 mg, 0.94 mmol) in 7 ml of toluene was added diisobutyl aluminum hydride (1.0 M solution in toluene, 2.82 ml, 2.82 mmol) under argon at −78°C. The reaction was stirred at −78°C for 1 h. The reaction was quenched by adding 30 ml of ethyl acetate and allowed to warm to room temperature. Thirty milliliters of water was added, and the aqueous solution was extracted with ethyl acetate (2 × 20 ml). The combined organic layers were washed with brine (2 × 20 ml) and dried over MgSO4. Concentration followed by flash chromatography (hexanes/ethyl acetate 4:1) afforded alcohol compound 4 (210 mg, 76%) as a colorless oil. 1H NMR (300 MHz, CDCl3): δ 1.64 (m, 15H), 1.95 (m, 12H), 4.03 (d, J = 7.2 Hz 2H), 5.03 (s, 3H), 5.34 (t, J = 7.25, 1H).
Compound 5.
A solution of 4-chloro-N-methylbutanamine (2.4 g, 15.6 mmol) in CH2Cl2 (21 ml) was cooled to 0°C. POCl3 (1.4 ml, 10.4 mmol) was added followed by a solution of triethylamine (4.3 ml, 31.2 mmol) in CH2Cl2 (7.75 ml, 4 M). It was left to warm gradually to room temperature over 3 h. The reaction mixture was quenched with saturated ammonium chloride (30 ml) and diluted with CH2Cl2 (50 ml). The aqueous layer was extracted with CH2Cl2 (2 × 50 ml), and the combined organic layers were dried over MgSO4. Concentration followed by flash chromatography (hexane/ethyl acetate 4:1) gave compound 5 (1.57 g, 42%) as an oil. 1H NMR (300 MHz, CDCl3): δ 1.67 (m, 4H), 2.74 (m, 3H), 3.18 (m, 2H), 3.48 (m, 2H). 31P NMR (CDCl3, 121 MHz): −7.8 ppm.
Compound 6.
Lithium bis(trimethylsilyl)amide [(Me3Si)2NLi; 3.16 ml, 1 M solution in THF, 3.16 mmol] was added dropwise to a solution of 2-hydroxymethyl-5-nitrofuran (452 mg, 3.16 mmol) at −78°C. After 10 min at −78°C, a solution of compound 5 (750 mg, 3.16 mmol) was added. The reaction was warmed to −65°C within 1 h, then quenched with 20 ml of saturated ammonium chloride solution. It was extracted with CH2Cl2 (2 × 20 ml) and dried over Na2SO4. It was purified by flash chromatography (hexane/ethyl acetate 9:1) to give compound 6 (692 mg, 63%). 1H NMR (300 MHz, CDCl3): δ 1.78(m, 4H), 2.7 (m, 3H), 3.10 (m, 2H), 3.50 (m, 2H), 5.12 (m, 2H), 6.75 (s, 1H), 7.29 (s, 1H). 31P NMR (CDCl3, 121 MHz): −8.2 ppm.
GGTI-2Z: Compound 7.
To a solution of compound 6 (129 mg, 0.375 mmol), 2Z-geranylgeraniol (compound 4) (120 mg, 0.413 mmol) and TEA (68 μl, 0.487 mmol) in CH2Cl2 (2 ml) was added TiCl4 (1 M solution in CH2Cl2, 37.5 μl, 0.0375 mmol) at room temperature. The reaction mixture was stirred for 1 h, filtered, concentrated, and after flash chromatography (hexanes/ethyl acetate 1:1) afforded compound 7 (63 mg, 28%) as colorless oil. 1H NMR (500 MHz, CDCl3): δ 1.58 (s, 9H), 1.66 (m, 5H, N-CH2-CH2 under CH3), 1.75 (m, 5H, CH2-CH2-Cl under CH3), 1.95 (m, 4H), 2.05 (m, 8H), 2.6 (m, 3H), 3.03 (m, 2H), 3.55 (t, J = 6Hz, 2H), 4.46 (m, 2H), 4.96 (d, J = 8.5Hz, 2H), 5.08 (m, 3H), 5.34 (t, J = 7.5, 1H), 6.6 (d, J = 4Hz, 1H), 7.2 (d, J = 3.5Hz, 1H). 13C NMR (CDCl3, 125 Hz): 16.27, 17.94, 23.83, 25.38, 25.95, 26.86, 26.99, 29.70, 32.39, 33.45, 39.95, 44.91, 48.48, 59.44, 63.28 (d, P-C, J = 5.1 Hz), 112.30, 112.97, 120.04 (d, P-C, J = 6.9Hz), 123.48, 124.32, 124.59, 131.55, 135.32, 136.25, 143.04, 153.55. 31P NMR (CDCl3, 121 MHz): −14 ppm. MS: ESI 621/623 +Na (Fig. 1).
In Vitro GGTase I Inhibition Assay
2Z-GGMP [compound 8; synthesized from 2Z-geranylgeraniol compound 4 in a similar manner to that described for 3-allylfarnesyl monophosphate (Clark et al., 2007)] and 2Z-GGPP [compound 9 (Zahn et al., 2001)] were evaluated against mammalian GGTase I (Hartman et al., 2005) in a modified version of the previously published FTase biochemical continuous fluorescent assay (Reigard et al., 2005). For all assays, the excitation wavelength used was 340 nm, and emission was monitored at 500 nm. The inhibitor assay mixture consisted of assay buffer [52 mM Tris-HCl (pH 7.0), 5.8 mM dithiothreitol, 12 mM MgCl2, and 12 M ZnCl2], 0.25% detergent (0.4% n-dodecyl-d-maltoside in 52 mM Tris-HCl, pH 7.0), 0.7 M dansyl-GCVLL, 1.35 M GGPP, varying analog concentrations (0.1–5.0 M), and 0.02 M GGTase I. The first five assay components were added to a 750-μl quartz cuvette for a total volume of 500.5 μl in the order listed; the reaction was initiated by the addition of enzyme, and change in fluorescence intensity was monitored over 300 s. Inhibitor data were expressed as a reaction velocity vs. substrate concentration plot, and the IC50 was determined by using GraphPad Prism software (GraphPad Software Inc., San Diego, CA). Using this procedure, we demonstrated that 2Z-GGMP (compound 8) inhibits GGTase I with an IC50 value of 21 nM, and 2Z-GGPP (compound 9) exhibited the same IC50 value (100 nM) as that previously reported (Zahn et al., 2001). Using the analogous FTase assay (Clark et al., 2007), we demonstrated that the IC50 of compound 8 for this enzyme is ≫ 10 μM.
Reagents
GGTI-2Z aliquots were prepared in dimethyl sulfoxide (DMSO) and stored at −80°C. HA14-1 (Ryan Scientific Inc., Mount Pleasant, SC) and lovastatin (Sigma-Aldrich, St. Louis, MO) aliquots were prepared and stored similarly. A plasmid (pRK7.GFP.H-Ras.CaaX) encoding green fluorescent protein (GFP) fused to the C-terminal 10 amino acids of rat H-Ras sequence, which encompasses its CaaX sequence, was constructed by subcloning into the pRK7.GFP plasmid (Yang and Mattingly, 2006), a forward primer with the sequence 5′-GATCCGGCTGCATGAGCTGCAAATGTGTGCTGTCCTG-3′ and a reverse primer with the sequence 5′-AATTCAGGACAGCACACATTTGCAGCTCATGCAGCCG-3′ using the sticky-end ligation method.
Cell Culture
STS-26T MPNST cells and normal, spontaneously immortalized rat Schwann cells (iSC) were obtained and maintained as described previously (Wojtkowiak et al., 2008). The murine hepatoma 1c1c7 cell line was obtained from Dr. J. Whitlock, Jr. (Stanford University, Palo Alto, CA) and cultured in α-minimal essential medium containing 5% fetal bovine serum with 100 units/ml of penicillin and 100 μg/ml of streptomycin. Derivatives of 1c1c7 cells that stably expressed GFP-LC3 were generated by transfection of an expression plasmid obtained from N. Mizushima (Tokyo Medical and Dental University, Tokyo, Japan). These cells have been stably transfected to express a GFP fusion construct of LC3. The MCF10.DCIS cell line was obtained from the Cell Lines Resource (Karmanos Cancer Institute, Detroit, MI) and maintained as a monolayer in Dulbecco's modified Eagle's medium/F12 containing 5% horse serum, 20 ng/ml epidermal growth factor, 0.5 μg/ml hydrocortisone, 10 μg/ml insulin, 50 U/ml penicillin, and 50 μg/ml streptomycin at 37°C and 5% CO2.
Western Blot Analysis
Lysates were prepared from monolayers of cells in 2× Laemmli sample buffer by boiling for 5 min and cleared by centrifugation (Mattingly et al., 2001). Samples were then separated on SDS-polyacrylamide gels and electrophoretically transferred to nitrocellulose. Membranes were then probed with 1:200 dilution of anti-RhoA, 1:500 dilution of anti-Rab5, 1:600 dilution of anti-unprenylated Rap1A antibodies, and caspase-3 antibody at 1:1000 dilution (Santa Cruz Biotechnology, Santa Cruz, CA). LC3-I and LC3-II were detected by using 1:2000 dilution of anti-LC3 (gift from Dr. David Kessel, Wayne State University, Detroit, MI).
Live-Cell Imaging Assays
Human embryonic kidney (HEK) 293 cells were plated into 35-mm culture plates 24 h before transfection with pRK7.GFP.H-Ras.CaaX by using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA) as described previously (Norum et al., 2005). Four hours after transfection, fresh media were added along with vehicle or drug at the appropriate concentrations as stated in the figure legends. At the end of treatment, nuclei were stained with Hoechst 33342, followed by confocal live-cell imaging on a LSM-510 microscope (Carl Zeiss Inc., Thornwood, NY) at 40× magnification. A similar protocol was used to study the nuclear morphology of STS-26T cells with and without drug treatment.
Immunofluorescence Assays
STS-26T cells were plated onto glass coverslips and treated as indicated in the figure legends. The cells were fixed and processed for confocal immunofluorescence analysis by using anti-LAMP-2 mouse monoclonal (BD Biosciences, San Jose, CA) and anti-LC3 rabbit polyclonal antibodies at 1:50 dilution followed by appropriate fluorescently coupled secondary antibodies. The number of LC3-positive puncta were quantified by using Volocity software 5.2.1 (PerkinElmer Life and Analytical Sciences, Waltham, MA).
Cell Proliferation Assay
STS-26T cells and iSC were plated at ∼20,000 cells per 35-mm dish 24 h before drug treatment. At the appropriate time points, attached cells were trypsinized and combined with media containing detached cells. The cells were collected by centrifugation for 5 min at 50g and counted with a hemacytometer.
MTT Assay
Cells were plated at a density of 2500 cells per well containing 200 μl of growth media with inhibitors or vehicle in 96-well plates and cultured for 72 h. Twenty microliters of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) (MTT) (Invitrogen) stock solution (5 mg/ml in phosphate-buffered saline) was then added, and the plates were incubated for 4 h. The medium was removed, and the formazan precipitate formed was dissolved in 150 μl of DMSO. Absorbance values were measured with a plate reader (SpectraFluor Plus; Tecan, Salzburg, Australia) at 485-nm wavelength. After normalizing the absorbance values for media and vehicle controls, the data were analyzed with GraphPad Prism version 4.0c by nonlinear regression (curve fit) and plotting sigmoidal dose-response to obtain GI50 (concentration of drug for 50% inhibition of growth) values, which were further plotted on an isobologram for synergy analysis.
Flow Cytometric Analysis
STS-26T cells were treated and collected for DNA analysis as described previously (Mattingly et al., 2006). DNA content was analyzed with a FACScalibur instrument (BD Biosciences). A minimum of 104 cells per sample was analyzed to determine the percentage of apoptotic cells and cells in G1, S and G2/M phases (Modfit; Variety Software, Topsham, ME).
DEVDase Activity Assay
Lysates of STS-26T cells were prepared and used for DEVDase assays as described previously (Wojtkowiak et al., 2008). Changes in fluorescence over time were converted to picomoles of product by comparison with a standard curve made with 7-amino-4-methylcoumarin. DEVDase-specific activities are reported as nanomoles of product made per minute per milligram of protein. The bicinchoninic acid assay, using bovine serum albumin as a standard, was used to estimate protein concentrations.
Results
Inhibition of Geranylgeranylation of GTPases by GGTI-2Z and Lovastatin Combination.
Recently, we demonstrated that the monophosphate derivatives of certain farnesyl pyrophosphate analogs are potent FTIs, and that prodrugs derived from these analogs block protein farnesylation (Clark et al., 2007; Wojtkowiak et al., 2008). We have also synthesized and evaluated novel GGPP analogs and found several analogs that are in vitro inhibitors of GGTase I (Gibbs et al., 1999; Zahn et al., 2001; Maynor et al., 2008). In particular, we demonstrated that the 2Z-GGPP analog, compound 9, from which we synthesized the prodrug GGTI-2Z, is an excellent inhibitor of geranylgeranylation of dansyl-GCVLL peptide by GGTase I (Zahn et al., 2001). We have now demonstrated that the corresponding monophosphate compound 8 is the most potent GGPP-based GGTI yet reported (IC50 = 21 nM). In the cellular evaluation of GGTI-2Z, we sought to confirm whether the compound also inhibits geranylgeranylation in vivo. STS-26T cells were treated with 3 μM GGTI-2Z either alone or in combination with 500 nM lovastatin. DMSO treatment was used as a vehicle control. We first tested whether this novel GGTI could inhibit geranylgeranylation of Rap1A via GGTase I. We achieved this by probing with an antibody that only recognizes the unprenylated form of Rap1A. GGTI-2Z alone was unable to inhibit Rap1A geranylgeranylation even after 48 h of treatment. A distinct band representing unprenylated Rap1A appeared within 24 h in whole-cell lysates treated with lovastatin alone, and this unprenylated Rap1A was strikingly increased upon treatment with a combination of GGTI-2Z and lovastatin (Fig. 2A). The amount of unprenylated Rap1A in comparison with the total Rap1 levels (Fig. 2A) increased over time with the combination treatment. This result suggests that GGTI-2Z, when combined with lovastatin, inhibits Rap1A geranylgeranylation.
Fig. 2.

Inhibition of prenylation in STS-26T cells by GGTI-2Z/lovastatin combination treatment. STS-26T cultures were treated as indicated for 24 or 48 h. Whole-cell lysates were probed for prenylation status of Ras superfamily GTPases via Western analysis. A, detection of Rap1A via an antibody directed toward the unprenylated form of Rap1A and detection of total Rap1. B, detection of RhoA, which has been reported to be overexpressed after block of GGTase. C, detection of Rab5. Unprenylated GTPases migrate more slowly on SDS-polyacrylamide gel electrophoresis gels. β-Tubulin was used as a loading control in all Western blots. Results shown are representative of at least three independent experiments.
Another geranylgeranylated protein that has been inhibited in the past by laboratories using other GGTI compounds is RhoA. Delarue et al. (2007) have shown that GGTI treatment of pancreatic cancer cells results in an increase in RhoA expression levels. In our study with the GGTI-2Z and lovastatin combination, we saw a similar marked increase in the expression level of RhoA within 24 h compared with vehicle control, and this increase was maintained even at 48 h of treatment (Fig. 2B).
We also tested for inhibition of RabGGTase or GGTase II by looking for reduced prenylation of Rab5. Strikingly, we observed a clear up-shift caused by the appearance of unprenylated Rab5 upon combination treatment (Fig. 2C) as opposed to vehicle or single compound treatments.
Combination of GGTI-2Z and Lovastatin Does Not Inhibit FTase.
Prenylation of Ras proteins helps target them to the plasma membrane where their site of action lies. These membrane proteins can be fluorescently tagged to visualize their cellular localization patterns in the presence or absence of prenylation inhibitors (Maurer-Stroh et al., 2007). We transfected HEK293 cells with a construct that encodes GFP fused to the CaaX motif of H-Ras (an exclusively farnesylated protein) and then treated the cells with our compounds alone or in combination. The nuclei were then stained followed by live-cell imaging via confocal microscopy for localization of GFP. As seen in Fig. 3, we observed that in the case of vehicle-treated cells GFP.H-Ras.CaaX localizes to the plasma membrane along with some intracellular expression that may represent the Golgi (Choy et al., 1999). Treatment with a low-dose combination of lovastatin and a FTI that we have previously shown to inhibit farnesylation (Wojtkowiak et al., 2008) inhibits the membrane localization and induces a diffuse cytosolic distribution of GFP.H-Ras.CaaX. In contrast, a combination of as high as 6 μM GGTI-2Z plus 500 nM lovastatin failed to prevent membrane localization of the GFP.H-Ras.CaaX protein. These data indicate that GGTase inhibition by GGTI-2Z and lovastatin does not inhibit prenylation of the exclusively farnesylated GFP.H-Ras.CaaX protein (Fig. 3).
Fig. 3.

Lack of effect of GGTI-2Z/lovastatin treatment on membrane localization of a farnesylated GFP construct. HEK293 cells were transiently transfected with pRK7.GFP.H-Ras.CaaX plasmid, followed by treatment with prenylation inhibitors as shown for 16 h. FTI-1/lovastatin treatment inhibited H-Ras.CaaX localization at the plasma membrane, whereas GGTI-2Z/lovastatin treatment did not affect the localization even at 6 μM GGTI-2Z concentration. Results are representative of three independent experiments. DAPI, 4′,6-diamidino-2-phenylindole.
GGTI-2Z in Combination with Lovastatin Inhibits Proliferation of STS-26T Cells without Significant Loss of Cell Viability.
Next, we sought to test the effect of inhibition of geranylgeranylation by the two compounds on growth and proliferation of STS-26T cells. We treated the cells with the compounds alone or in combination and found that 1 μM concentration of GGTI-2Z or lovastatin alone had little effect on proliferation (Fig. 4A). However, 45 h of exposure of the cells to a combination of 3 μM GGTI-2Z and 1 μM lovastatin caused a significant inhibition of proliferation of the cells, and this inhibition was similar to the extent of 10 μM GGTI-2Z treatment alone (Fig. 4B). In addition, as shown in Fig. 4, there was a dose-dependent and a time-dependent inhibition of proliferation of these cells (Fig. 4, A and B). In addition to proliferation, we examined the percentage viability of the cells. At all the time points tested, there was little effect on cell viability with single or combination treatments (Fig. 4C). We further tested whether there was synergy between the two compounds when used in combination via an MTT assay. After 72 h of treatment, the data analysis showed that these compounds were indeed synergistic in their growth inhibitory effect (Fig. 4D). The synergistic inhibition was indicated by the GI50 values for the combination treatment lying below the theoretical line connecting the GI50 values for GGTI-2Z and lovastatin alone.
Fig. 4.

A and B, inhibition of proliferation of STS-26T cells after GGTI-2Z/lovastatin treatment. Samples were collected every 24 h after treatment for analysis of cell number. Data represent means ± S.D. of three independent experiments. C, number of nonviable cells was analyzed with respect to total number of cells to calculate percentage of viability at the given time points. D, cells were treated with the compounds at several different concentrations as shown, alone, or in combination, and the data were tested for synergy using isobologram analysis. Data represent means ± S.D. of two independent experiments.
GGTI-2Z in Combination with Lovastatin Arrests STS-26T MPNST Cells in G0/G1 Phase of the Cell Cycle.
We observed significant inhibition of proliferation of STS-26T cells by cotreatment with GGTI-2Z and lovastatin. We therefore next determined which point of the cell cycle these compounds targeted to inhibit proliferation. We performed flow cytometry analysis of STS-26T cells treated with GGTI-2Z and lovastatin singly or in combination (Fig. 5). Our results showed that treatment with 3 μM GGTI-2Z or 1 μM lovastatin alone did not affect cell-cycle progression. Interestingly, the same concentrations of the drugs, when used in combination, induced a G0/G1 cell-cycle arrest and a subsequent reduction in the percentage of cells in G2/M and S phases.
Fig. 5.

GGTI-2Z/lovastatin treatment arrests STS-26T cells in G0/G1. STS-26T cells were treated as shown. Cultures were harvested 48 h after treatment for DNA content by staining with propidium iodide. Histograms represent 104 events, and the cell-cycle profile was determined by using Modfit. Results are representative of two independent experiments.
GGTI-2Z Alone or in Combination with Lovastatin Does Not Induce Apoptosis in STS-26T Cells.
Although analyses of cell viability by Trypan blue exclusion assay suggested no cytotoxicity by combined GGTI-2Z and lovastatin treatment, we wanted to re-examine whether the treatment induced any apoptosis in the cells. For this purpose, we used N-acetyl-Asp-Glu-Val-Asp-amino-4-methylcoumarin to assay the activity of caspases-3 and -7 (Wojtkowiak et al., 2008). HA14-1, a known inducer of apoptosis through inhibition of Bcl-2, was used as a positive control for this assay. When treated with HA14-1 for 2 h, STS-26T cells showed significant induction of apoptosis as demonstrated by a strong increase in DEVDase activation compared with untreated control cells. There was even stronger induction of caspase-like activity after 4 h of treatment with HA14-1. In contrast, treatment with the prenylation inhibitors alone or in combination did not yield any detectable DEVDase activation (Fig. 6A). As an additional test for caspase activation in these cells, we probed lysates of cells treated with the drugs alone or in combination for expression of cleaved caspase-3. As shown in Fig. 6B, procaspase-3, which is the uncleaved form of caspase-3, was evident in all of the cell lysates at 35 kDa. However, in contrast to HA14-1, the prenylation inhibitors did not induce any cleaved caspase-3, indicating lack of detectable apoptosis in these cells by this measure. Finally, we also performed nuclear morphology assays using Hoechst 33342 dye to monitor chromatin condensation as an indicator of apoptosis. Within 45 min of HA14-1 treatment, we observed nuclear condensation in the form of bright blue spots as seen in Fig. 6C. Conversely, with GGTI and/or lovastatin treatment, we did not we see any nuclear morphological changes (Fig. 6C) or any appearance of DNA laddering (data not shown), which further confirms the lack of apoptosis in the cells.
Fig. 6.

Lack of apoptosis in STS-26T cells after GGTI-2Z/lovastatin treatment. HA14-1-treated cells were used as a positive control. A, STS-26T cells were treated as indicated. Data represent means of triplicate samples and are representative of two independent experiments. B, STS-26T cells were treated as indicated. Attached and detached cells were pooled, and whole-cell lysates were separated and probed for cleaved caspase-3. C, STS-26T cells were treated for 48 h with the indicated concentrations of lovastatin and/or GGTI-2Z or for 30 min with HA14-1. Nuclei were stained with Hoechst 33342, and live-cell imaging was performed on a LSM-510 microscope at 40× magnification.
GGTI-2Z and Lovastatin Combination Treatment Induces Autophagy in STS-26T Cells.
We further investigated whether autophagy was involved in determining the response of the cells after prenylation inhibition. We achieved this by assaying LC3, the classical marker of autophagy (Klionsky et al., 2008). During the formation of autophagosomes, LC3-I is processed to LC3-II via phosphatidylethanolamine attachment. The presence of LC3-II is, therefore, associated with occurrence of autophagy. We observed a subtle increase with single treatments and a marked increase with combination treatment in the appearance of LC3-II in STS-26T cells within 24 h compared with vehicle treatment. This increase was sustained at 48 h (Fig. 7A). This result suggests that the drugs may be inducing or up-regulating the autophagic process in these cells.
Fig. 7.

Induction of autophagy in STS-26T cells by GGTI-2Z/lovastatin treatment. STS-26T cells were treated with the indicated concentrations of GGTI-2Z and lovastatin alone or in combination. Results are representative of three independent experiments. A, treatments were for 24 or 48 h as shown. Whole-cell lysates were then probed for LC3 and β-tubulin. B, cells were subject to 2-h pretreatment with protease inhibitors, 10 μM pepstatin A, and 10 μM E64D as indicated. Whole-cell lysates were then probed for LC3 and β-tubulin. C, cells were subject to 48 h of drug treatment, with addition of 50 nM bafilomycin A1 for the last 2 h of the incubation as indicated. Whole-cell lysates were then probed for LC3 and β-tubulin. D, STS-26T cells were treated as indicated for 48 h followed by methanol fixation. Cells were then stained for LC3 and LAMP-2. Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI), and cells were visualized under a LSM-510 microscope at 40× magnification. E, quantitative analysis of LC3-positive puncta treated with either DMSO or the drug combination for 48 h.
Increase in LC3-II levels may be associated with either of two possible mechanisms: an increase in formation of autophagosomes or a decrease in processing/degradation of LC3-II caused by an absence of autophagosome/lysosome fusion or depression of lysosomal protease activities. To distinguish between the effects on synthesis and degradation, we pretreated the cells with the protease inhibitors E64D and pepstatin A. These compounds inhibit lysosomal proteases, which would prevent degradation of LC3-II in the autophagolysosome. The results showed that the protease inhibitors did induce a further increase in LC3-II levels (Fig. 7B), which is consistent with there being autophagosome and lysosome fusion and subsequent proteolytic processing in the autophagolysosome. We also used bafilomycin A1, which blocks the maturation of autophagosomes by its inhibition of the vacuolar ATPase. In this case, we treated the cells with the vehicle, single compounds, or combination treatment for 48 h, with bafilomycin A1 also present for the final 2 h of the incubation. The results show that the single treatments do not have much effect on autophagic flux compared with vehicle control, whereas the combination treatment again increases LC3-II accumulation either in the presence or absence of bafilomycin A1 (Fig. 7C). Furthermore, immunocytochemical staining of LC3 along with the lysosomal marker, LAMP-2, was also performed after treatments with the compounds as indicated (Fig. 7D). Vehicle or single compound treatments did not result in colocalization of the two proteins. In contrast, a very distinct punctate colocalization of LC3 and LAMP-2 was observed in cells after the combination treatment. Quantitative analysis of LC3-positive puncta, using two independent LC3 antibodies, revealed an approximately 5-fold increase in the number of punctate structures upon combination treatment compared with DMSO treatment (Fig. 7E). These results indicate that the autophagic process is both induced and proceeds to completion in STS-26T cells cotreated with GGTI-2Z and lovastatin.
GGTI-2Z and Lovastatin Also Effectively Inhibit Proliferation of 1c1c7 Cells and Induce Autophagy in DCIS Cells.
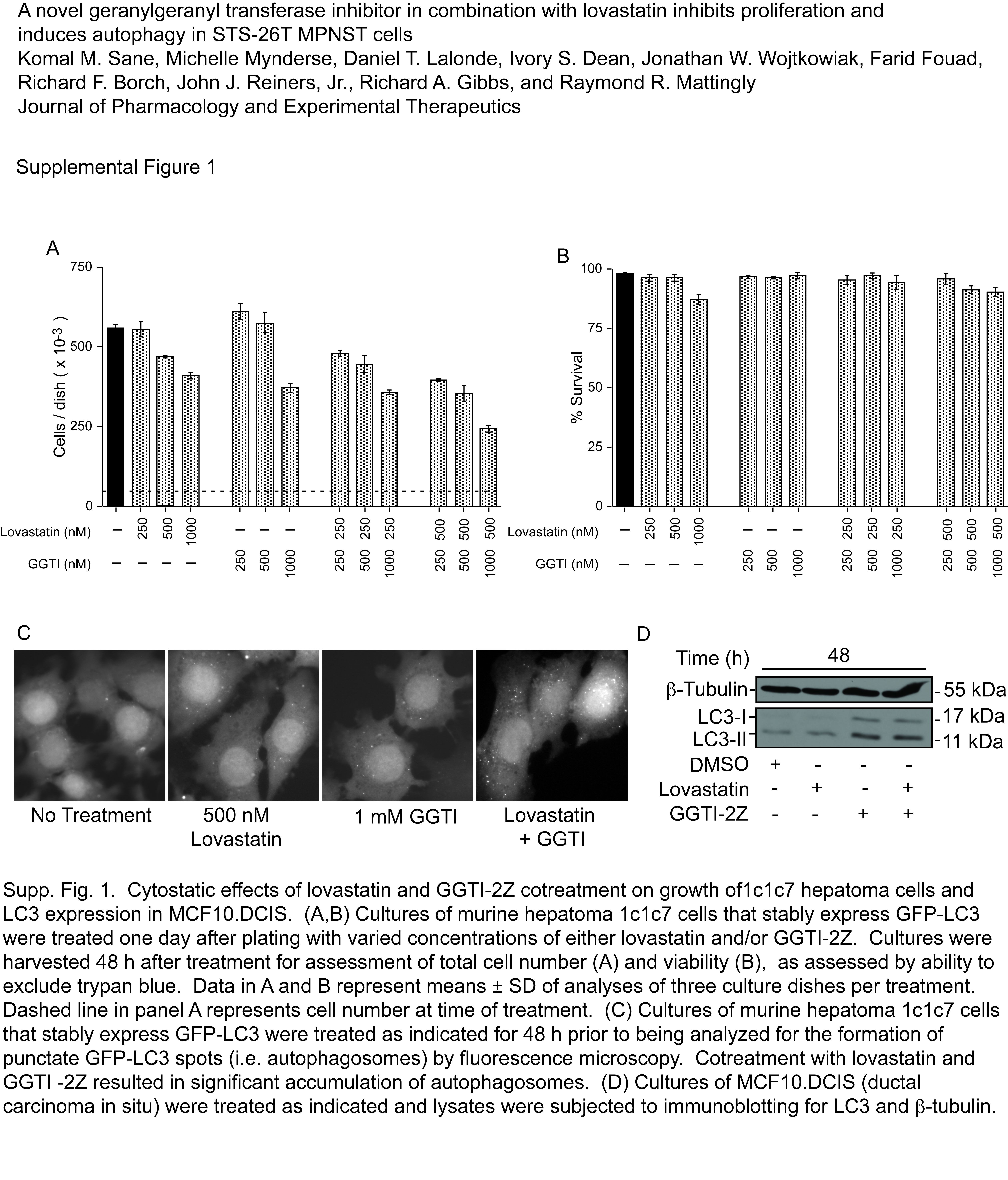
To test the effects of the drugs in cell lines that model other cancers, we performed cell proliferation and viability assays in a murine hepatoma cell line, 1c1c7. We found that GGTI-2Z and lovastatin combination also inhibits proliferation of these cells without significantly affecting their viability (Supplemental Fig. 1, A and B). This cell line stably expresses a GFP fusion construct of LC3. Upon combination treatment, there was increased appearance of LC3-positive vesicles, indicating induction of autophagy (Supplemental Fig. 1C). We also treated cultures of MCF10.DCIS cells with the drugs and assayed for LC3-II accumulation by Western blotting. The results showed increased LC3 processing, confirming autophagic induction in these cells as well (Supplemental Fig. 1D).
Normal Immortalized Rat Schwann Cells Are Resistant to GGTI-2Z and Lovastatin Cotreatment.
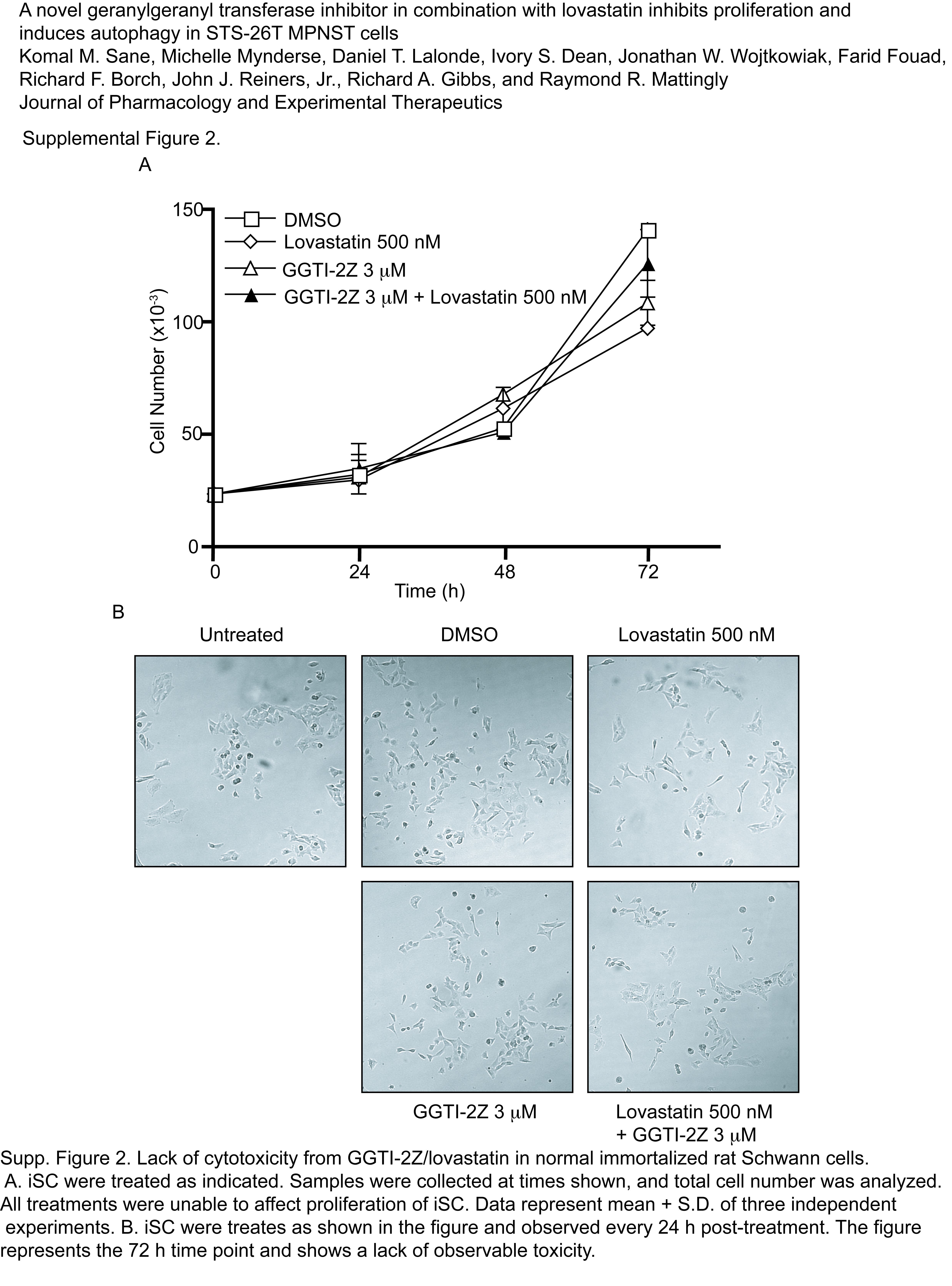
We then tested whether GGTI-2Z and lovastatin in combination affected proliferation of iSC. As shown in Supplemental Fig. 2A, the compounds either alone or in combination had little to no effect on the proliferation of iSC. In addition, we found no detectable morphological changes in these cells upon treatment with the compounds (Supplemental Fig. 2B).
Discussion
Prenylation inhibitors of different classes have been tested preclinically and clinically for therapy of many types of cancer. Statins were among the first of such compounds to be tested for their potential antitumor activity in preclinical models. Nevertheless, in general, statins have shown limited promise when combined with conventional chemotherapy (reviewed in Konstantinopoulos et al., 2007). Great effort has been focused on the development and testing of selective inhibitors of FTase. As previously mentioned, however, FTI-treated cells can bypass this inhibition for certain substrates (notably, K-Ras and N-Ras) through alternative geranylgeranylation. In addition, FTIs are capable of targeting prenylation of multiple proteins. There has been little success of these compounds when used alone in clinical trials.
In the past our group has tested prenylation inhibitors, including FTIs and statins, as potential therapies for neurofibromatosis type 1 (NF1) and other hyperproliferative disorders (Mattingly et al., 2002). We recently showed that a novel FTI compound, FTI-1, in combination with lovastatin induces apoptosis in two different NF1 MPNST cell lines (Wojtkowiak et al., 2008). Moreover, we observed little to no detectable toxicity of the treatment in normal iSC, indicating the potential use of this combination treatment for NF1 MPNSTs. The FTase substrates that are affected by FTI treatment to produce inhibition of cell proliferation and survival are still unclear. However, an interesting study involving a chemical genetics approach revealed RabGGTase or GGTase II as a target of FTIs (Lackner et al., 2005). This finding supported the idea that FTIs have many different targets that may be responsible for their activity and side effects and also identified a potential role for Rab proteins and RabGGTase in p53-independent apoptosis induced by FTIs.
In this study, we have developed a novel GGTI whose action is potentiated by cotreatment with lovastatin, resulting in inhibition of proliferation and cell-cycle arrest associated with induction of autophagy in STS-26T MPNST cell line. The strategy that we used to develop the GGPP-based inhibitor compound GGTI-2Z is analogous to that previously described for the development of FTIs (Maynor et al., 2008). Of all the GGPP analogs evaluated, compound 8 served as the best inhibitor with an impressive IC50 value of approximately 21 nM for GGTase I enzyme in vitro. Encouragingly, this analog also exhibited no significant binding to mammalian FTase, further confirming its promise as a tool for evaluating cellular GGTase I inhibition. In addition, as seen by immunocytochemistry, GGTI-2Z does not affect the membrane localization of a GFP construct that is exclusively farnesylated, suggesting that it acts to solely inhibit geranylgeranylation, whereas farnesylation remains unaffected. Furthermore, the fact that it did not have any effect on either morphology or proliferation of normal iSC implies a lower risk of toxicity to normal cells.
Our rationale for the combinatorial approach was to achieve more efficient GGTase I inhibition via GGTI-2Z by simultaneously depleting endogenous GGPP pools. Rap1A is a Ras family GTPase that is known to be solely geranylgeranylated presumably by GGTase I (Casey et al., 1991). We observed inhibition of Rap1A prenylation, thus confirming our in vitro result that GGTI-2Z effectively inhibits GGTase I, although it does so only when combined with lovastatin. Conversely, lovastatin alone is sufficient to modestly inhibit Rap1A geranylgeranylation. These data suggest that Rap1A may not be a critical target for the inhibition of STS-26T cell proliferation, because even higher levels of lovastatin (up to 1 μM) do not affect cell-cycle distribution.
The combination of GGTI-2Z/lovastatin synergistically inhibits proliferation of STS-26T cells. This antiproliferative activity is consistent with the induction of cell-cycle arrest in the G1 phase. Other GGTIs have also been shown to block cell-cycle progression of several tumor cell lines and subsequently induce apoptosis (Vogt et al., 1997). However, with GGTI-2Z/lovastatin there was little effect on cell viability and we did not observe the classical apoptotic morphology or apoptotic markers in STS-26T cells.
Another potential determinant of cell survival is the phenomenon of autophagy. In recent years, autophagy has been discovered to be an important mechanism adopted by many different cell lines for determining their fate, and it is still a topic of debate whether autophagy is a mechanism of cell survival or cell death (Apel et al., 2009). Interestingly, there is increasing evidence suggesting that several cancer cells show up-regulation of the process leading to cell survival and cancer progression (Rubinsztein et al., 2007). Recently, three FTIs were found to induce autophagy in two different human cancer cell lines (Pan et al., 2008). In addition, some statins can induce autophagy in a cell type-specific manner owing to their ability to inhibit protein prenylation rather than cholesterol synthesis (Araki and Motojima, 2008). For instance, cerivastatin or simvastatin are capable of inducing autophagy in rhabdomyosarcoma cells (Araki and Motojima, 2008), whereas lovastatin or simvastatin fail to do so in hepatocytes (Samari and Seglen, 1998). We examined our cultures treated with the GGTI/lovastatin combination to determine whether autophagy occurred. Based on the analysis of LC3-I conversion to LC3-II via Western blot, and the colocalization of LC3 with the lysosomal protein LAMP-2 via immunocytochemistry, we confirmed that autophagy was induced and driven to completion in STS-26T cultures cotreated with GGTI-2Z and lovastatin.
One of our observations shows that the compounds inhibit prenylation of Rab5, a GGTase II or RabGGTase substrate. These data indicate that the compounds not only inhibit GGTase I, but also serve as substrates for GGTase II. This was not surprising because the two enzymes share strikingly similar active sites (Lackner et al., 2005), and, therefore, a compound designed to bind the GGPP binding pocket of either of the two can be expected to bind similarly to the other. The preference of binding in that case will be determined by relative affinity of the compound for the enzymes. We have not yet tested the in vitro ability of GGTI-2Z to bind to GGTase II to determine the KM value. An alternative explanation for the dual inhibition of GGTase I and RabGGTase-mediated prenylation is that GGTI-2Z inhibits GGPP synthase, blocking the production of the GGPP substrate needed for both processes (Wiemer et al., 2007). In addition, there is a noticeable decrease in the expression level of Rab5 upon GGTI-2Z/lovastatin treatment. Potentially, this RabGGTase inhibition and/or enhanced Rab5 turnover may contribute to the effects of the compounds on MPNSTs.
Rab proteins have been shown to play an important role in carcinogenesis (Cheng et al., 2004). Traditionally, Rab5 is known to have a well-established role in endocytosis and vesicular transport of proteins (Bucci et al., 1992). More recently, however, an interesting study in cell culture and fly models of Huntington's disease suggested a role for Rab5 in the early stages of the process of macroautophagy that is independent of its endocytic function (Ravikumar et al., 2008). This study showed that Rab5 inhibition via expression of dominant-negative Rab5 results in a decrease in LC3-positive autophagic vacuoles and also enhances polyglutamine toxicity. In our study we saw that inhibition of Rab5 prenylation via GGTase inhibition is correlated with an increase in LC3-II levels. Prenylation inhibitors may be capable of only partially blocking Rab5 activity, and thus, the partially prenylated and active Rab5 may still be sufficient for autophagic progression. Additional evidence that may support such a connection is that fluvastatin and pravastatin-induced RabGGTase inhibition causes vacuolation in rat skeletal myofibers (Sakamoto et al., 2007). Alternatively, the GGTI may target one or more other proteins that may potentially contribute to its action. Further studies would be required to better elucidate the role of Rab5 and other proteins in GGTI/lovastatin-induced autophagy in MPNSTs.
In conclusion, we have developed a novel compound, GGTI-2Z, that blocks prenylation mediated by both GGTase I and RabGGTase and exerts cytostatic activities in STS-26T MPNST cells in a caspase-3-independent manner. The action of GGTI-2Z is potentiated by a low-dose statin combination treatment and strongly correlates with the induction of autophagy. This combination treatment does not block proliferation of or induce toxicity in normal, immortalized Schwann cells, but it does have activity against two other transformed cell lines, 1c1c7 murine hepatoma cells and MCF10.DCIS cells that model human breast ductal carcinoma in situ. Further studies testing therapeutic efficacy of GGTI-2Z may serve to develop better understanding of geranylgeranylation inhibitors and evaluate their potential in the context of cancer therapy and some Rab-associated protein-trafficking disorders.
Supplementary Material
Acknowledgments
We thank T. Glover for MPNST cell lines; E. M. Shooter for immortalized Schwann cells; P. Mathieu for technical assistance and advice; K. Zukowski for assistance with flow cytometry; and members of the Imaging and Cytometry Core Facilities at the Department of Pharmacology, Wayne State University for assistance with confocal imaging.
This work was supported by the Department of the Army [Grants DAMD17-03-1-0182, W81XWH-05-1-0193]; the National Institutes of Health [Grant R01-CA131990]; and the National Institutes of Health National Institutes of Environmental Health Sciences [Grant P30-ES06639] (Environmental Health Sciences Center Pilot Project). This project was aided by the Imaging and Cytometry Core Facilities at Wayne State University, which were supported by the National Institute of Environmental Health Sciences [Grant P30-ES06639] and the National Cancer Institute [Grant P30-CA22453]. J.W.W. was supported by the National Institutes of Health [Grant T32-ES012163]. Work at Purdue was supported in part by the National Institutes of Health [Grants R01-CA78819, P30-CA23168].
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
doi:10.1124/jpet.109.160192.
The online version of this article (available at http://jpet.aspetjournals.org) contains supplemental material.
- GGTI
- geranylgeranyl transferase inhibitor
- GGTI-2Z (compound 7)
- (5-nitrofuran-2-yl)methyl-(2Z,6E,10E)-3,7,11,15-tetramethylhexadeca-2,6,10,14-tetraenyl 4-chlorobutyl(methyl)phosphoramidate
- GGPP
- geranylgeranyl pyrophosphate
- GGMP
- geranylgeranyl monophosphate
- FTase
- farnesyl transferase
- RabGGTase
- Rab geranylgeranyl transferase
- FTI
- farnesyl transferase inhibitor
- GGTase
- geranylgeranyl transferase
- MPNST
- malignant peripheral nerve sheath tumor
- iSC
- immortalized Schwann cells
- NF1
- neurofibromatosis type I
- GI50
- concentration of drug for 50% inhibition of growth
- compound 1
- (6E,10E)-ethyl 7,11,16-trimethyl-3-oxohexadeca-6E,10E,14-trienoate
- compound 2
- (2E,6E,10E)-ethyl 7,11,15-trimethyl-3-(trifluoromethylsulfonyloxy)hexadeca-2,6,10,14-tetraenoate
- compound 3
- (2Z,6E,10E)-ethyl 3,7,11,15-tetramethylhexadeca-2,6,10,14-tetraenoate
- compound 4
- (2Z,6E,10E)-3,7,11,15-tetramethylhexadeca-2,6,10,14-tetraen-1-ol (2Z-geranylgeraniol)
- compound 5
- 4-chlorobutyl(methyl)phosphoramidic dichloride
- compound 6
- (5-nitrofuran-2-yl)methyl 4-chlorobutyl(methyl)phosphoramidochloridate
- 2Z-GGMP (compound 8)
- 2Z geranylgeranyl monophosphate
- 2Z-GGPP (compound 9)
- 2Z-geranylgeranyl pyrophosphate
- DMSO
- dimethyl sulfoxide
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- DMF
- dimethylformamide
- THF
- tetrahydrofuran
- GFP
- green fluorescent protein
- HEK
- human embryonic kidney
- E64D
- l-3-trans-carboxyrane2
- LC3
- microtubule-associated protein-1 light chain 3
- LAMP-2
- lysosomal-associated membrane protein 2
- TEA
- triethylamine.
References
- Apel A, Zentgraf H, Buchler MW, Herr I. (2009) Autophagy-a double-edged sword in oncology. Int J Cancer 125:991–995 [DOI] [PubMed] [Google Scholar]
- Araki M, Motojima K. (2008) Hydrophobic statins induce autophagy in cultured human rhabdomyosarcoma cells. Biochem Biophys Res Commun 367:462–467 [DOI] [PubMed] [Google Scholar]
- Bucci C, Parton RG, Mather IH, Stunnenberg H, Simons K, Hoflack B, Zerial M. (1992) The small GTPase rab5 functions as a regulatory factor in the early endocytic pathway. Cell 70:715–728 [DOI] [PubMed] [Google Scholar]
- Casey PJ, Thissen JA, Moomaw JF. (1991) Enzymatic modification of proteins with a geranylgeranyl isoprenoid. Proc Natl Acad Sci U S A 88:8631–8635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng KW, Lahad JP, Kuo WL, Lapuk A, Yamada K, Auersperg N, Liu J, Smith-McCune K, Lu KH, Fishman D, et al. (2004) The RAB25 small GTPase determines aggressiveness of ovarian and breast cancers. Nat Med 10:1251–1256 [DOI] [PubMed] [Google Scholar]
- Choy E, Chiu VK, Silletti J, Feoktistov M, Morimoto T, Michaelson D, Ivanov IE, Philips MR. (1999) Endomembrane trafficking of ras: the CAAX motif targets proteins to the ER and Golgi. Cell 98:69–80 [DOI] [PubMed] [Google Scholar]
- Clark MK, Scott SA, Wojtkowiak J, Chirco R, Mathieu P, Reiners JJ, Jr, Mattingly RR, Borch RF, Gibbs RA. (2007) Synthesis, biochemical, and cellular evaluation of farnesyl monophosphate prodrugs as farnesyltransferase inhibitors. J Med Chem 50:3274–3282 [DOI] [PubMed] [Google Scholar]
- Delarue FL, Adnane J, Joshi B, Blaskovich MA, Wang DA, Hawker J, Bizouarn F, Ohkanda J, Zhu K, Hamilton AD, et al. (2007) Farnesyltransferase and geranylgeranyltransferase I inhibitors up-regulate RhoB expression by HDAC1 dissociation, HAT association, and histone acetylation of the RhoB promoter. Oncogene 26:633–640 [DOI] [PubMed] [Google Scholar]
- Gibbs BS, Zahn TJ, Mu Y, Sebolt-Leopold JS, Gibbs RA. (1999) Novel farnesol and geranylgeraniol analogues: a potential new class of anticancer agents directed against protein prenylation. J Med Chem 42:3800–3808 [DOI] [PubMed] [Google Scholar]
- Hamadmad SN, Hohl RJ. (2007) Lovastatin suppresses erythropoietin receptor surface expression through dual inhibition of glycosylation and geranylgeranylation. Biochem Pharmacol 74:590–600 [DOI] [PubMed] [Google Scholar]
- Hartman HL, Hicks KA, Fierke CA. (2005) Peptide specificity of protein prenyltransferases is determined mainly by reactivity rather than binding affinity. Biochemistry 44:15314–15324 [DOI] [PubMed] [Google Scholar]
- Kazi A, Carie A, Blaskovich MA, Bucher C, Thai V, Moulder S, Peng H, Carrico D, Pusateri E, Pledger WJ, et al. (2009) Blockade of protein geranylgeranylation inhibits Cdk2-dependent p27Kip1 phosphorylation on Thr187 and accumulates p27Kip1 in the nucleus: implications for breast cancer therapy. Mol Cell Biol 29:2254–2263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A, et al. (2008) Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 4:151–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstantinopoulos PA, Karamouzis MV, Papavassiliou AG. (2007) Posttranslational modifications and regulation of the RAS superfamily of GTPases as anticancer targets. Nat Rev Drug Discov 6:541–555 [DOI] [PubMed] [Google Scholar]
- Kou R, Sartoretto J, Michel T. (2009) Regulation of Rac1 by simvastatin in endothelial cells: differential roles of AMP-activated protein kinase and calmodulin-dependent kinase kinase-β. J Biol Chem 284:14734–14743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lackner MR, Kindt RM, Carroll PM, Brown K, Cancilla MR, Chen C, de Silva H, Franke Y, Guan B, Heuer T, et al. (2005) Chemical genetics identifies Rab geranylgeranyl transferase as an apoptotic target of farnesyl transferase inhibitors. Cancer Cell 7:325–336 [DOI] [PubMed] [Google Scholar]
- Lerner EC, Zhang TT, Knowles DB, Qian Y, Hamilton AD, Sebti SM. (1997) Inhibition of the prenylation of K-Ras, but not H- or N-Ras, is highly resistant to CAAX peptidomimetics and requires both a farnesyltransferase and a geranylgeranyltransferase I inhibitor in human tumor cell lines. Oncogene 15:1283–1288 [DOI] [PubMed] [Google Scholar]
- Leung KF, Baron R, Seabra MC. (2006) Thematic review series: lipid posttranslational modifications. Geranylgeranylation of Rab GTPases. J Lipid Res 47:467–475 [DOI] [PubMed] [Google Scholar]
- Mattingly RR, Gibbs RA, Menard RE, Reiners JJ., Jr (2002) Potent suppression of proliferation of a10 vascular smooth muscle cells by combined treatment with lovastatin and 3-allylfarnesol, an inhibitor of protein farnesyltransferase. J Pharmacol Exp Ther 303:74–81 [DOI] [PubMed] [Google Scholar]
- Mattingly RR, Kraniak JM, Dilworth JT, Mathieu P, Bealmear B, Nowak JE, Benjamins JA, Tainsky MA, Reiners JJ., Jr (2006) The mitogen-activated protein kinase/extracellular signal-regulated kinase kinase inhibitor PD184352 (CI-1040) selectively induces apoptosis in malignant schwannoma cell lines. J Pharmacol Exp Ther 316:456–465 [DOI] [PubMed] [Google Scholar]
- Mattingly RR, Milstein ML, Mirkin BL. (2001) Down-regulation of growth factor-stimulated MAP kinase signaling in cytotoxic drug-resistant human neuroblastoma cells. Cell Signal 13:499–505 [DOI] [PubMed] [Google Scholar]
- Maurer-Stroh S, Koranda M, Benetka W, Schneider G, Sirota FL, Eisenhaber F. (2007) Toward complete sets of farnesylated and geranylgeranylated proteins. PLoS Comput Biol 3:e66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynor M, Scott SA, Rickert EL, Gibbs RA. (2008) Synthesis and evaluation of 3- and 7-substituted geranylgeranyl pyrophosphate analogs. Bioorg Med Chem Lett 18:1889–1892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norum JH, Methi T, Mattingly RR, Levy FO. (2005) Endogenous expression and protein kinase A-dependent phosphorylation of the guanine nucleotide exchange factor Ras-GRF1 in human embryonic kidney 293 cells. FEBS J 272:2304–2316 [DOI] [PubMed] [Google Scholar]
- Pan J, Chen B, Su CH, Zhao R, Xu ZX, Sun L, Lee MH, Yeung SC. (2008) Autophagy induced by farnesyltransferase inhibitors in cancer cells. Cancer Biol Ther 7:1679–1684 [DOI] [PubMed] [Google Scholar]
- Perez I, Sestelo JP, Sarandeses LA. (2001) Atom-efficient metal-catalyzed cross-coupling reaction of indium organometallics with organic electrophiles. J Am Chem Soc 123:4155–4160 [DOI] [PubMed] [Google Scholar]
- Peterson YK, Kelly P, Weinbaum CA, Casey PJ. (2006) A novel protein geranylgeranyltransferase-I inhibitor with high potency, selectivity, and cellular activity. J Biol Chem 281:12445–12450 [DOI] [PubMed] [Google Scholar]
- Ravikumar B, Imarisio S, Sarkar S, O'Kane CJ, Rubinsztein DC. (2008) Rab5 modulates aggregation and toxicity of mutant huntingtin through macroautophagy in cell and fly models of Huntington disease. J Cell Sci 121:1649–1660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reigard SA, Zahn TJ, Haworth KB, Hicks KA, Fierke CA, Gibbs RA. (2005) Interplay of isoprenoid and peptide substrate specificity in protein farnesyltransferase. Biochemistry 44:11214–11223 [DOI] [PubMed] [Google Scholar]
- Rubinsztein DC, Gestwicki JE, Murphy LO, Klionsky DJ. (2007) Potential therapeutic applications of autophagy. Nat Rev Drug Discov 6:304–312 [DOI] [PubMed] [Google Scholar]
- Sakamoto K, Honda T, Yokoya S, Waguri S, Kimura J. (2007) Rab-small GTPases are involved in fluvastatin and pravastatin-induced vacuolation in rat skeletal myofibers. FASEB J 21:4087–4094 [DOI] [PubMed] [Google Scholar]
- Samari HR, Seglen PO. (1998) Inhibition of hepatocytic autophagy by adenosine, aminoimidazole-4-carboxamide riboside, and N6-mercaptopurine riboside. Evidence for involvement of amp-activated protein kinase. J Biol Chem 273:23758–23763 [DOI] [PubMed] [Google Scholar]
- Sun J, Ohkanda J, Coppola D, Yin H, Kothare M, Busciglio B, Hamilton AD, Sebti SM. (2003) Geranylgeranyltransferase I inhibitor GGTI-2154 induces breast carcinoma apoptosis and tumor regression in H-Ras transgenic mice. Cancer Res 63:8922–8929 [PubMed] [Google Scholar]
- Vogt A, Qian Y, McGuire TF, Hamilton AD, Sebti SM. (1996) Protein geranylgeranylation, not farnesylation, is required for the G1- to S-phase transition in mouse fibroblasts. Oncogene 13:1991–1999 [PubMed] [Google Scholar]
- Vogt A, Sun J, Qian Y, Hamilton AD, Sebti SM. (1997) The geranylgeranyltransferase-I inhibitor GGTI-298 arrests human tumor cells in G0/G1 and induces p21(WAF1/CIP1/SDI1) in a p53-independent manner. J Biol Chem 272:27224–27229 [DOI] [PubMed] [Google Scholar]
- Wiemer AJ, Tong H, Swanson KM, Hohl RJ. (2007) Digeranyl bisphosphonate inhibits geranylgeranyl pyrophosphate synthase. Biochem Biophys Res Commun 353:921–925 [DOI] [PubMed] [Google Scholar]
- Wojtkowiak JW, Fouad F, LaLonde DT, Kleinman MD, Gibbs RA, Reiners JJ, Jr, Borch RF, Mattingly RR. (2008) Induction of apoptosis in neurofibromatosis type 1 malignant peripheral nerve sheath tumor cell lines by a combination of novel farnesyl transferase inhibitors and lovastatin. J Pharmacol Exp Ther 326:1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojtkowiak JW, Gibbs RA, Mattingly RR. (2009) Working together: farnesyl transferase inhibitors and statins block protein prenylation. Mol Cell Pharmacol 1:1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Mattingly RR. (2006) The Ras-GRF1 exchange factor coordinates activation of H-Ras and Rac1 to control neuronal morphology. Mol Biol Cell 17:2177–2189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahn TJ, Whitney J, Weinbaum C, Gibbs RA. (2001) Synthesis and evaluation of GGPP geometric isomers: divergent substrate specificities of FTase and GGTase I. Bioorg Med Chem Lett 11:1605–1608 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}