

Abstract
Compelling evidence suggests that inflammation, cell survival, and cancer are linked, with a central role played by NF-κB. Recent studies implicate some TLRs in tumor development based on their ability to facilitate tumor growth; however, to our knowledge, involvement of neither TLR7 nor TLR78 has yet been demonstrated. Here we have demonstrated expression of TLR7 and TLR8, the natural receptors for single-stranded RNA, by tumor cells in human lung cancer in situ and in human lung tumor cell lines. Stimulation with TLR7 or TLR8 agonists led to activated NF-κB, upregulated expression of the antiapoptotic protein Bcl-2, increased tumor cell survival, and chemoresistance. Transcriptional analysis performed on human primary lung tumor cells and TLR7- or TLR8-stimulated human lung tumor cell lines revealed a gene expression signature suggestive of chronic stimulation of tumor cells by TLR ligands in situ. Together, these data emphasize that TLR signaling can directly favor tumor development and further suggest that researchers developing anticancer immunotherapy using TLR7 or TLR8 agonists as adjuvants should take into account the expression of these TLRs in lung tumor cells.
Introduction
The concept that inflammatory responses and chronic inflammation contribute to carcinogenesis, tumor progression, and neovascularization is supported by epidemiological studies and experimental findings (1–4). Chronic inflammation can result from viral or bacterial infections or from long-term exposure to noninfectious agents such as asbestos and tobacco (3, 5–8). However, the mechanisms by which it contributes to tumor growth are not fully understood, although a major role for TNF-α has been proposed (9).
TLRs allow for recognition of pathogen- and damaged-associated molecular patterns (PAMPs and DAMPs; refs. 10, 11) and trigger inflammatory responses through activation of NF-κB, a master switch for inflammation (12). NF-κB plays a critical role in the development of tumors in the context of chronic inflammation (13, 14). Mice deficient for inhibitor of NF-κB kinase β (Iκκβ) in intestinal epithelial cells exhibit a striking 80% decline in colitis-associated cancer after chronic exposure to azoxymethane or dextran sulfate sodium (15). Moreover, mice deficient for Iκκα show reduced prostate tumor development (16). In addition, NF-κB induces genes whose products prevent apoptosis, such as Bcl-2 family members, and thus exerts prosurvival activity (17, 18). These observations provide conclusive evidence for a prominent role of NF-κB signaling pathway in inflammation-promoted cancer and tumor cell survival.
Indeed, TLR signaling pathways could promote cancer initiation and progression (19, 20). Sequence variants of TLR1, TLR4, TLR6, and TLR10 are associated with increased risk of prostate and gastric cancer (21, 22). Moreover, the signaling through the adaptor protein MyD88 has a critical role in spontaneous tumor development in mice with heterozygous mutation in the adenomatous polyposis coli gene (23). In addition, deficiency in the single Ig IL-1 receptor–related molecule, a negative regulator of TLR signaling, results in increased intestinal inflammation and colitis-associated tumorigenesis after challenge with dextran sulfate sodium (24). These results emphasize the role of TLR signaling pathways in the promotion of cancer.
Although TLR expression was first observed in immune cells, several reports have described the expression of TLRs in nonmalignant and malignant epithelial cells. TLR1–TLR6 are expressed by colon, lung, prostate, and melanoma mouse tumor cell lines (25), TLR3 is expressed by human breast cancer cells (26), TLR2 and TLR4 are expressed by hepatocarcinoma and gastric carcinoma cells (27), and TLR9 (28) and TLR4 (29) are expressed by human lung cancer cells. Listeria monocytogenes and Helicobacter pylori promote tumor growth of gastric carcinoma through TLR2 and TLR4 signaling, respectively (27). In addition to a direct effect on tumor growth, TLR4 stimulation can also lead to tumor evasion from immune surveillance in colon and lung cancer through the production of immunosuppressive cytokines and resistance to apoptosis induced by TNF-α or TNF-related apoptosis–inducing ligand (TRAIL; refs. 25, 29). Interestingly, stimulation of TLR3 by poly I:C in breast cancer and melanoma cells directly triggers apoptosis of tumor cells (26, 30). Together, these data provide evidence that TLR stimulation in tumor cells can lead to either survival or cell death.
The human lung is in contact with inhaled airborne pathogens, and, via expression of a large panel of TLRs, the airway epithelial cells represent the first barrier against invading microbes (31, 32). Several studies strongly suggest that chronic inflammation (i.e., chronic bronchitis, chronic obstructive diseases, emphysema, asbestos, or tobacco smoke) increases the risk of carcinogenesis (5, 6, 33, 34). Lungs are frequently exposed to RNA viruses, such as respiratory syncytial and influenza viruses, that are recognized by TLR7 and TLR8 (35, 36), which suggests these TLRs are present on lung epithelial cells.
In the present study, we demonstrated a link of TLR7 and TLR8 signaling with inflammation, tumor growth, and chemoresistance. We demonstrated the expression of TLR7 and TLR8 in lung cancer cells and that TLR7 ligation with loxoribine or TLR8 ligation with poly U resulted in activation of NF-κB and upregulation of Bcl-2 expression. This was associated with increased tumor cell survival and resistance to apoptosis induced by chemotherapy in vitro. Our transcriptomic data obtained with fresh tumor cells showed that human lung cancer cells had a gene expression profile similar to that of TLR7- or TLR8-stimulated cell lines, indicative of chronic tumor stimulation. These data emphasize that TLR signaling can directly interfere with the tumor cell either by increasing cell survival or by inducing resistance to cell death, which suggests that the use of TLR ligands as adjuvants could be a double-edged sword in anticancer immunotherapy.
Results
TLR7 and TLR8 are expressed in human lung tumors.
Detection of TLR7 and TLR8 expression by malignant and nonmalignant lung tissues was performed by immunohistochemistry in cells of 13 patients with adenocarcinoma (ADC) or squamous cell carcinoma (SCC). TLR7 was expressed by tumor cells in approximately 70% of the patients tested (Table 1); Figure 1, A and C, shows examples of positive tumor cell TLR7 expression in SCC and ADC, respectively. In contrast, TLR8 was expressed by tumor cells in all lung cancer patients tested, independent of histological type (Table 1 and Figure 1, B and D). Comparison of the precise cellular localization of TLR7 and TLR8 revealed major differences, with TLR7 expression being mainly perinuclear (Figure 1E) and TLR8 being more diffuse and cytoplasmic (Figure 1F). Interestingly, no TLR7 or TLR8 expression was detected in epithelial alveolar cells from the same patients (Figure 1, G and H), whereas strong expression of TLR7 and TLR8 was detected in bronchial epithelial cells (Figure 1, I and J), an observation that we believe to be novel. Similar results were observed in lung emphysema, in which TLR7 and TLR8 were expressed in bronchial epithelial cells, but not in alveolar epithelial cells (Supplemental Figure 1; supplemental material available online with this article; doi:10.1172/JCI36551DS1). Immune cells, including NK, plasmacytoid, and myeloid DCs and B and T lymphocytes, express TLR7 and/or TLR8 (37). Immune cell infiltration has been described in lung tumors. In some patients, the immune cells are organized into lymphoid-like structures called tumor-induced bronchus-associated lymphoid tissues (Ti-BALTs; ref. 38). Accordingly, we observed that most of the immune cells present in the Ti-BALT strongly expressed TLR7 (Figure 1K), whereas fewer of these cells expressed TLR8 (Figure 1L). Moreover, we also observed the presence of TLR7-positive immune cells outside of the Ti-BALT (data not shown). Interestingly, in emphysema sections, some immune cells were present that expressed TLR7 and, weakly, TLR8 (data not shown). These observations demonstrated distinct profiles of TLR7 and TLR8 expression: TLR7 was expressed by immune and bronchial cells and, in 70% of the patients, in tumoral cells, whereas TLR8 was mainly expressed by bronchial and tumor cells.
Table 1 .
TLR7 and TLR8 expression in the lung tumor and microenvironment of 13 patients

Figure 1. TLR7 and TLR8 are expressed in human lung tumors.
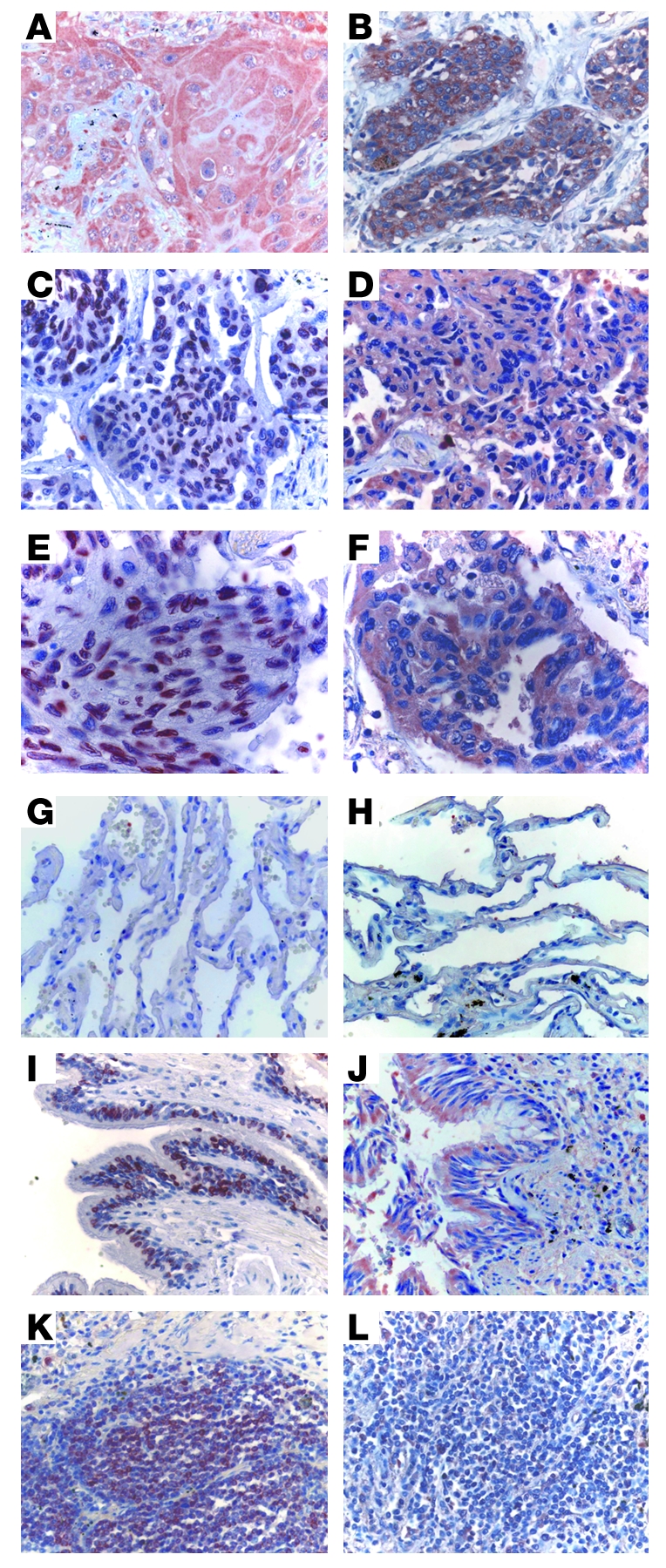
TLR7 (A, C, E, G, I, and K) and TLR8 (B, D, F, H, J, and L) protein expression was analyzed in the tumoral area — SCC (A and B) and ADC (C–F) — as well as in the alveolar epithelium (G and H), bronchial epithelium (I and J), and Ti-BALT (K and L) by immunohistochemical labeling of paraffin-embedded lung tumors, as described in Methods. (E and F) The subcellular localization of TLR7 and TLR8 was detected in tumor cells. Original magnification, ×40 (A–D and G–L); ×65 (E and F).
TLRs are expressed by human lung cell lines.
We characterized the expression of TLRs in several human lung tumor cell lines, including ADC lines A549 and H1355 and SCC line SK-MES, and in human bronchial epithelial cell lines 16HBE and BEAS-2B. All tumor cell lines expressed TLR1–TLR10 (Figure 2A), and the expression of TLR7 and TLR8 was confirmed at the protein level by flow cytometry analysis (Figure 2, B and C) and immunohistochemistry (data not shown) in all cell lines, except in 16HBE cells, which did not express TLR7 (Figure 2C).
Figure 2. Human lung cancer cell lines express TLRs.

(A) Expression of TLR1–TLR10 in A549, H1355, and SK-MES cell lines was analyzed by RT-PCR. Shown is 1 representative of 3 independent experiments. (B and C) Expression of TLR7 and TLR8 was determined in the cells by flow cytometry after intracellular staining in lung tumoral cell lines (B) or in nontumoral bronchial epithelial cell lines (C). Results are representative of 3 independent experiments. Gray histograms represent isotype controls; black histograms represent TLR7 or TLR8 Abs.
TLR7 or TLR8 stimulation of human lung cancer cell lines induces atypical NF-κB activation.
In order to determine whether TLR7 and TLR8 stimulation activates intracellular signaling pathways, we studied NF-κB activation in response to stimulation of A549 cells with IL-1β, the homodimeric TLR7 ligand loxoribine, and the homodimeric TLR8 ligand poly U. As expected, IL-1β stimulation induced canonical activation of NF-κB. We detected 2 waves of inhibitor of NF-κB α (IκBα) phosphorylation: the first occurred at 10 minutes and was associated with IκBα degradation, and the second occurred at 60 minutes and was independent of IκBα degradation (Figure 3, A and B). Interestingly, TLR7 or TLR8 stimulation also led to activation of the NF-κB pathway, and IκBα was phosphorylated 30 minutes after loxoribine or poly U addition. However, no significant degradation of IκBα was detected (Figure 3, C–F). A similar phenomenon was observed in response to the TLR3 ligand poly I:C and the TLR4 ligand LPS. TLR3 or TLR4 stimulation of A549 cells led to IκBα phosphorylation with the same kinetics as those of TLR7 or TLR8 stimulation, and IκBα was not degraded (Supplemental Figure 2). These results indicate that TLR7 and TLR8, which are expressed by human lung cancer cell lines, induce atypical NF-κB activation and that the signaling pathways might be different from those activated by IL-1β, as recently described by Qin et al. (39).
Figure 3. TLR7 and TLR8 lead to IκBα phosphorylation, but not degradation.

A549 cells were untreated or treated with IL-1β (10 ng/ml; A and B), loxoribine (10 μg/ml; C and D), or poly U (10 μg/ml; E and F) for the indicated periods of time. (A, C, and E) Cell lysates were analyzed by immunoblot with anti–phospho-specific IκBα, anti-IκBα, and anti-actin antibodies. (B, D, and F) Quantification of the bands was realized using Image J software. Histograms represent the intensity of the respective band, expressed as relative intensity normalized to actin, for both phospho-IκBα and IκBα. Results are representative of 3 independent experiments.
A further set of experiments was performed to investigate whether NF-κB was translocated to the nucleus. First, p50 and p65 NF-κB subunits were quantified by ELISA on nuclear extracts of A549 cells. A marked amount of p50 and p65 was present in the nuclear extracts after stimulation with IL-1β or loxoribine. However, the amounts of p50 and p65 NF-κB were about 2-fold lower after loxoribine stimulation than after IL-1β stimulation (Figure 4A). Immunofluorescence experiments confirmed the nuclear translocation of p65 NF-κB subunit 120 minutes after stimulation by IL-1β or loxoribine (Figure 4B). Finally, we performed a NF-κB gene reporter assay and found that both IL-1β and loxoribine induced the expression of the gene reporter (Figure 4C), demonstrating NF-κB activity in the nucleus. However, the gene reporter activity in response to loxoribine was 2-fold lower than that in response to IL-1β. Taken together, these data demonstrate that TLR7 ligand induces NF-κB activation even in absence of IκBα degradation, but with a lower efficiency than IL-1β.
Figure 4. TLR7 and TLR8 lead to NF-κB activation.

A549 cells were untreated or treated with IL-1β (10 ng/ml) or loxoribine (10 μg/ml) for the indicated periods of time. (A) Nuclear fractions were analyzed by ELISA, and the relative quantity of p50 and p65 NF-κB subunits in nucleus is expressed in arbitrary units normalized to unstimulated nuclear extracts. (B and C) The intracellular localization of the p65 NF-κB subunit was determined by immunofluorescence staining (B), and NF-κB activation was determined by NF-κB gene reporter assay after transfection with the pNIFTY2 plasmid (C). Data in C represent mean ± SD from 3 independent experiments. *P < 0.05, **P < 0.01.
TLR7 or TLR8 stimulation induces survival of lung cancer cell lines.
It has been previously described that ligation of TLR2, TLR4, TLR5, or TLR9 can induce proliferation of tumor cells, including ovarian, breast, prostate, and gastric tumors (25, 27–29, 40, 41). We therefore examined whether TLR7 or TLR8 stimulation of lung tumor cells increases the proliferation of A549 cells. Stimulation with loxoribine, poly U, or the heterodimeric TLR7/TLR8 ligand Gardiquimod for 10 days induced a 20% reduction of Alamar blue compared with unstimulated cells, suggesting an increase in cell proliferation and/or viability (Figure 5A). This effect could be attributed to either protection against cell death or modification of the cell cycle. Treatment with loxoribine, poly U, or Gardiquimod induced a 1.5- to 2-fold decrease in the percentage of dead cells compared with unstimulated cells. Incubation with cycloheximide or etoposide, used as positive controls, led to a significant 2.5-fold increase in the percentage of dead cells (Figure 5B). No significant differences between the proportion of cells in the G0/G1 and G2/M phases of the cell cycle were detected between unstimulated and stimulated cells, whereas etoposide or cycloheximide treatment resulted in a significant reduction in the percentage of cells in G0/G1 and G2/M (Figure 5C). An increased expression of Bcl-2 was observed in parallel, by RT-PCR, in A459 cells stimulated by loxoribine, poly U, or Gardiquimod (Figure 5D). A significant increase in Bcl-2 protein levels was also detected after addition of loxoribine or Gardiquimod (Figure 5, E and F). Similar results were obtained in SK-MES cells (data not shown). These results suggest that TLR7 or TLR8 stimulation induce a prosurvival rather than a proliferative effect.
Figure 5. TLR7 and TLR8 induce survival of lung tumor cells.

(A) A549 cells were unstimulated or stimulated with loxoribine (10 μg/ml), poly U (10 μg/ml), or Gardiquimod (10 μg/ml) and incubated with Alamar blue for 10 days. Reduction of Alamar blue was then determined by spectrophotometry at 570 and 600 nm. The percentage of reduced Alamar blue was calculated as described by the manufacturer. (B) A549 cells were unstimulated or stimulated with loxoribine, poly U, Gardiquimod, etoposide, or cycloheximide for 24 hours. The percentage of dead cells was determined by Trypan blue exclusion. (C) DNA content was measured by IP staining after RNAse A treatment. Percentages indicate the proportion of subdiploid (G0/G1) and diploid cells (G2/M). Data in A–C represent mean ± SD from 3 independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001 versus medium, Student’s t test. (D) A549 cells were cultured for 6 hours in the presence of loxoribine, poly U, or Gardiquimod, and the expression of Bcl-2 was assayed using TaqMan Low-Density Array technology. The fold increase (arbitrary units) was obtained by 2–ΔΔCT. (E and F) A549 cells were cultured for 36 hours in the presence of loxoribine, poly U, or Gardiquimod. (E) Cell lysates were analyzed by immunoblot with anti–Bcl-2 and anti-actin antibodies. (F) Quantification of the bands was realized using Image J software. Histograms represent the intensity of the respective band, expressed as relative intensity normalized to actin. Results are representative of 3 independent experiments.
TLR7 or TLR8 stimulation induces chemoresistance in lung cancer cell lines.
Polychemotherapy including platinium salt associated with a drug of second generation is often used as treatment for lung cancer (42). Given our observation that TLR7 or TLR8 stimulation increased tumor cell viability, we hypothesized that it interferes with the induction of apoptosis caused by chemotherapy. Therefore, we analyzed the induction of cell death by chemotherapeutic agents currently used for the treatment of non–small-cell lung cancer (NSCLC) in A549 or SK-MES cells previously stimulated with loxoribine or poly U. We first determined the LD50 concentration for each drug (i.e., 2 nM for doxorubine, 50 μM for Navelbine, 15 μM for cycloheximide, 100 and 50 μM for cisplatine for A549 and SK-MES cells, respectively, and 100 μM for carboplatine; Supplemental Figure 3) using Alamar Blue reduction assay. Treatment with cycloheximide, cisplatine, carboplatine, doxorubicine, or Navelbine at the LD50 concentration induced A549 and SK-MES cell death. When the cells were stimulated with loxoribine or poly U prior to treatment with chemotherapeutic agents, cell death was reduced by about 2-fold. Similar results were observed using trypan blue assay (data not shown), which suggests that tumoral cells are less sensitive to chemotherapy-induced cell death after TLR7 or TLR8 stimulation.
To confirm the chemoresistance of TLR-stimulated cell lines, colony assay experiments were performed. Stimulation of A549 and SK-MES cells with loxoribine prior to treatment with mono- or polychemotherapy increased the surviving fraction (Figure 6), confirming that TLR7 stimulation induces chemoresistance in lung cancer cell lines. Similar results were obtained with poly U stimulation of A549 and SKMES cells (data not shown). To determine the magnitude of increased chemoresistance, various doses of cisplatine and carboplatine were used, ranging 1–1,000 and 1–10,000 mM, respectively. We observed a dose-response effect in both A549 and SK-MES cells conferred by loxoribine against cell death, requiring carboplatine and cisplatine doses 10–100 times larger in order to reach the LD50 (Figure 7). As a consequence, these results suggest that stimulation of TLR7 induces chemoresistance through decreased sensibility toward chemotherapeutic agents. Taken together, these data indicate that stimulation of tumor cell lines with either TLR7 or TLR8 ligands could induce chemoresistance and protect tumor cells against apoptosis.
Figure 6. TLR7 and TLR8 induce chemoresistance of lung tumor cells.

A549 (A, C, and E) or SK-MES cells (B, D, and F) were cultured in 6-well plates with or without loxoribine (added at days 0, 3, 6, and 9). Cells were then treated or not with cycloheximide, cisplatine, carboplatine, doxorubicine, or Navelbine at day 12 (A–D), or treated or not with cisplatine or carboplatine in association with Navelbine or doxorubicine (E and F). (A and B) The colony number (shown below) was determined after Crystal Violet coloration. (C–F) Cell viability was analyzed at day 15 by the surviving fraction, calculated as [no. colonies after chemotherapy treatment/(no. cells seeded at day 0 × PE)] × 100, where PE is plating efficiency (calculated as no. colonies/no. cells seeded at day 0). Data represent mean ± SD from 3 independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001 versus unstimulated, Student’s t test.
Figure 7. TLR7 and TLR8 induce dose-dependent chemoresistance of lung tumor cells.

A549 (A and B) or SK-MES cells (C and D) were cultured in 6-well plates with or without loxoribine (added at days 0, 3, 6 and 9), and were treated at day 12 with carboplatine and cisplatine at doses ranging 0–1,000 μM and 0–10,000 μM, respectively. The colony number was determined after Crystal Violet coloration at the day 15, and cell viability was analyzed by the surviving fraction, calculated as described in Figure 6. Data represent mean ± SD from a duplicate experiment. *P < 0.05; **P < 0.01, ***P < 0.001, Student’s t test.
Finally, to determine whether chemoresistance was mediated by the unique signaling downstream of TLR7 or TLR8, similar experiments were performed in TLR3- or TLR4-stimulated A549 and SK-MES cells. LPS stimulation induced chemoresistance to cycloheximide and doxorubicine — and, to a lower extent, to cisplatine and carboplatine — in A549 cells. In contrast, no significant modification of chemosensitivity was observed in SK-MES cells cultured with LPS (Supplemental Figure 4, A and C). Moreover, poly I:C did not induce chemoresistance in A549 or SK-MES cells. On the contrary, it induced a strong and significant reduction of surviving fraction in control A549 cells (Supplemental Figure 4, B and D). These results suggest that chemoresistance induction after TLR stimulation is not specific to TLR7 or TLR8, but can also be induced by TLR4 stimulation, in A549 cells.
Loxoribine-induced chemoresistance is dependent on the TLR7-MyD88 pathway.
In order to determine whether loxoribine-induced chemoresistance is dependent on the TLR7-MyD88 signaling pathway, siRNA knockdown of TLR7 and MyD88 was performed. TLR7 siRNA induced 25- and 5-fold decreases in TLR7 expression at 3 and 5 days after transfection, respectively (Figure 8A). Similarly, MyD88 siRNA induced 70- and 10-fold decreases in MyD88 expression 3 and 5 days after transfection, respectively (Figure 8B). Expression of TLR7 was not modified by MyD88 or scrambled siRNAs (Figure 8A), and expression of MyD88 was not modified by TLR7 or scrambled siRNAs (Figure 8B), demonstrating the specificity of TLR7 or MyD88 knockdown.
Figure 8. Loxoribine-induced chemoresistance is dependent on the TLR7-MyD88 signaling pathway.

A549 cells were transfected or not with TLR7 siRNA, MyD88 siRNA, or scrambled siRNA. (A and B) At 3 and 5 days after transfection, the mRNA expression levels of TLR7 (A) and MyD88 (B) were determined by quantitative RT-PCR. The fold decrease (arbitrary units) was obtained by 2–ΔΔCT. (C) 2 days after transfection, the cells were stimulated with loxoribine or LPS for 48 hours. Cells were then treated or not with cisplatine for additional 36 hours, and cell viability was analyzed by annexin V staining and flow cytometry analysis. The percentages of dead cells were determined as annexin V–positive cells relative to cells treated with cisplatine only. Values shown are calculated as relative percentage of dead cells subtracted from 100%. Data represent mean ± SD from 3 independent experiments.
We then performed chemoresistance experiments in A549 cells after TLR7 or MyD88 knockdown using annexin V assay. Consistent with previous results, treatment with loxoribine and cisplatine increased the cell viability by 50% both in untransfected and in control siRNA–transfected cells compared with cells treated with cisplatine only. Interestingly, the same treatment increased cell viability by only 10% in cells transfected with MyD88 or TLR7 siRNAs (Figure 8C). These data show that loxoribine-induced chemoresistance was reduced 5-fold after siRNA knockdown of TLR7 or MyD88. To determine if these effects were specific to TLR7 signaling, similar experiments were performed after LPS stimulation. LPS-induced chemoresistance was drastically reduced after MyD88 siRNA transfection, but was not altered by TLR7 or scrambled siRNAs transfections. Taken together, these results demonstrate that loxoribine-induced chemoresistance is dependent on the TLR7-MyD88 signaling pathway.
TLR7 or TLR8 stimulation modulates gene transcription.
In immune cells, TLR stimulation leads to upregulation of proinflammatory cytokines. However, there is little information available regarding the molecules that are modulated when tumoral cells are stimulated with TLR ligands. Therefore, we analyzed the modulation of gene expression in A549 and SK-MES cells in response to TLR7 or TLR8 stimulation in comparison to TLR3 or TLR4 stimulation by screening a large panel of immune and angiogenic genes. We confirmed the reproducibility of the experiment by analyzing gene modulation in A549 and SK-MES cells unstimulated or stimulated with loxoribine in triplicate (Supplemental Figure 5). Thereafter, cells were stimulated with loxoribine, poly U, Gardiquimod, poly I:C, or LPS, and the expression of 182 RNAs by TaqMan Low-Density array was analyzed. The ΔΔCT values were obtained by comparison of unstimulated cells with cells stimulated by loxoribine, poly U, Gardiquimod, poly I:C, or LPS. We analyzed the global profile of genes that are modulated by clustering (see Methods). Figure 9 shows the cluster of 73 genes most differentially modulated in TLR3-, TLR4-, TLR7-, and TLR8-stimulated cells in comparison to unstimulated cells. We observed some differences in the modulation of gene expression between the 2 cell lines in response to TLR stimulation. Whatever TLR was being stimulated in A549 cells, some genes, including vasohibin-1 (VASH1), angiopoietin-like 3 (ANGPTL3), CCR4, and Bcl-2 were upregulated, whereas 7 genes, including thrombospondin motif 1 (ADAMTS1), CD86, or ACE, were downregulated. Interestingly, some genes, such as ANGPTL1, IL-15, IL-8, IL-7, CXCL10, and ICAM-1, were upregulated in response to TLR3, but downregulated in response to TLR4, TLR7, and TLR8; others, such as angiopoietin 1 (ANGPT1) and serpin peptidase inhibitor clade F1 (SERPINF1), were downregulated in response to TLR3, but upregulated in response to TLR4, TLR7, and TLR8. In addition, CD80, IL-12A, and IFN-β were upregulated in response to TLR4 stimulation only, whereas 4 genes — FOXC2, ECGF1, ITGB3, and MMP2 — were upregulated in response to TLR3 and TLR4, but not TLR7 or TLR8. In SCC SK-MES cells, we also observed that some genes were upregulated (GNLY, ITGB3, IL-12A, CCR4, and CSF2) and others downregulated (e.g., CD80 and CCL3) in response to TLR3, TLR4, TLR7, and TLR8 stimulation, whereas some genes, including IL-15, IL-7, and CXCL10, were upregulated only in response to TLR3 triggering, and SERPINC1 was upregulated only in response to TLR4 stimulation (Figure 9).
Figure 9. TLR3, TLR4, TLR7, and TLR8 induce regulation of gene transcription.

A549 or SK-MES cells were cultured for 6 hours in the presence of poly I:C, LPS, loxoribine, poly U, or Gardiquimod. Total RNA was extracted and analyzed for the expression of 182 genes using TaqMan Low-Density Array technology. All genes with a CT value greater than 35 were excluded from the analysis. The ΔCT, ΔΔCT, and 2–ΔΔCT values were calculated, and values clustered, as described in Methods; shown is the fold increase (arbitrary units) obtained by 2–ΔΔCT.
These results show that TLR7 and TLR8 stimulation of lung tumor cells modulates the transcription of a number of genes implicated in several functions, including cell survival, angiogenesis, and immune escape. However, despite the fact that the stimulation of these different TLRs leads to a common atypical activation of NF-κB, the gene expression profile is partially different between cells stimulated by TLR7 or TLR8 and those stimulated by TLR3 or TLR4.
Human primary tumor cells and TLR7- or TLR8-stimulated cell lines have the same gene expression profile.
In order to determine whether primary human lung tumor cells that can express high levels of TLR7 or TLR8 display gene expression profiles similar those of TLR7- or TLR8-stimulated cell lines, we analyzed the same 182 RNAs using the TaqMan Low-Density Array in tumor cells obtained from fresh surgical tissues. The ΔΔCT values were obtained by comparison of unstimulated A549 cells with primary ADC tumor cells or unstimulated SK-MES cells with primary SCC tumor cells. This analysis was performed in simplicate for each patient, but included 3 different patients of each histological type of lung tumor. A very strong correlation of gene expression among the ADC patients (r = 0.78) and among the SCC patients (r = 0.7) was observed (Supplemental Figure 6). Interestingly, the profile of ADC was similar to the profile of stimulated A549, and the profile of SCC was similar to that of stimulated SK-MES. We compared the expression levels of the 11 genes that were most differentially expressed by the tumor cell lines. Bcl-2, VASH1, and CCR4 were always upregulated in stimulated cell lines and primary human cells, whereas SERPINC1 was always downregulated (Figure 10). Moreover, some genes that were differentially expressed between A549 and SK-MES cells were also differentially expressed in primary tumor cells in a similar way. This effect was independent of histological type, since the results were compared with unstimulated cell lines for each histological origin as a reference. Consequentially, this clustering of the 11 most differentially expressed genes suggests that primary lung tumor cells might have been stimulated in vivo within the tumor by agonist ligands for TLR7 or TLR8.
Figure 10. Top 11 genes modulated by TLR7 and TLR8 in human primary lung cancer cells.

Total RNA was extracted from primary tumor cells of 3 ADC patients (patients 33, 84, and 97) and 3 SCC patients (patients 23, 25, and 31) or from SK-MES and A549 cells stimulated with loxoribine, poly U, or Gardiquimod, and the expression of 182 genes was assayed. The 11 genes found to be highly modulated in Figure 9 are shown for comparison. The ΔCT, ΔΔCT, and 2–ΔΔCT values were calculated, and values clustered, as described in Methods; shown is the fold increase (arbitrary units) obtained by 2–ΔΔCT.
Discussion
In this study, we demonstrated for the first time to our knowledge that TLR7 and TLR8 were highly expressed by primary human lung tumor cells in NSCLC. Several studies have described the role of TLR signaling pathways in the promotion of epithelial cancers, but we believe TLR7 and TLR8 have not previously been implicated. We showed that TLR7 or TLR8 stimulation of lung tumor cells led to increased cell viability and resistance to apoptosis induced by chemotherapy.
We observed that tumor cells, both in ADC and in SCC, strongly expressed TLR7 and TLR8 and that nonmalignant bronchial cells expressed high levels of TLR7 and moderate levels of TLR8, which indicates that they are able to recognize pathogenic viruses potentially present in the airway. This observation strongly suggests that TLR7 expressed by bronchial cells plays a role in the first line of defense in the respiratory tract. In contrast, normal alveolar epithelial cells did not express TLR7 and TLR8. In lung tumor sections, normal epithelial cells in the adjacent or distant nontumoral area did not express TLR7 or TLR8. On the contrary, bronchial epithelial cells expressed TLR7 and TLR8. In lung cancer, tumor cells arise from malignant transformation of either alveolar or bronchial epithelial cells. Thus, we suspect that tumor cells arising from alveolar epithelial cells can acquire TLR expression during or upon tumorigenesis, whereas tumor cells arising from bronchial epithelial cells continue to express TLR7 and TLR8. A possible explanation for TLR induction in epithelial cells is that these TLRs are acquired during chronic infections. Such induction has been described for TLR7 in the lung tumoral A549 cell line and in the lungs of mice infected with Haemophilus influenzae (43). In agreement with these observations, we detected upregulation of TLR7 expression in A549 cells after stimulation with TLR7 or TLR8 ligands (data not shown). In addition, chronic inflammation or chronic infection induces lesions in the lung epithelium and engages a homeostatic repair program necessary for the maintenance of epithelium integrity (44). It is well accepted that the malignant transformation of epithelial cells can be caused by a defect in the epithelial cell repair program. Thus, the resulting malignant epithelial cells may express TLR7 or TLR8.
Interestingly, we observed that TLR7 had an unexpected intracellular distribution: its localization was mainly perinuclear, whereas TLR8 was cytoplasmic. A similar observation was made for TLR9 and TLR3 (45, 46). Accumulation of TLR3 has been observed in cytoplasmic and perinuclear inclusions in human neurons (46), and TLR9 was observed in a perinuclear reticular compartment that colocalized with endoplasmic reticulum markers (45).
TLR7 or TLR8 stimulation led to the release of inflammatory mediators, mainly through activation of the NF-κB pathway. We observed that TLR7 stimulation induced IκBα phosphorylation, nuclear translocation of the p50 and p65 NF-κB subunits, and specific activation of gene transcription, demonstrating NF-κB activation in response to loxoribine. However, this activation was about 2 times weaker than that induced by IL-1β. Surprisingly, we observed that IκBα was not degraded. This absence of IκBα degradation is unusual, as IκBα is classically degraded after being phosphorylated. Our observation is consistent with the report by Qin et al. (39) describing a similar atypical activation of NF-κB in a model of HEK293 tumor cells that overexpress TLR8. Similar to our observation using a gene reporter assay, the authors demonstrate that this atypical phosphorylation of IκBα without its degradation leads to the activation of NF-κB. Moreover, they show that IκBα is phosphorylated by MEKK3. This is different from IkBα activation in immune cells, which depends on TGF-activated kinase 1 (TAK1). Furthermore, MEKK3 is implicated in resistance to apoptosis through activation of NF-κB by increasing the expression of Bcl-2 (47). As a consequence, IκBα phosphorylation is probably caused by recruitment of the MEKK3 kinase instead of the TAK1 kinase.
We observed strong regulation of several genes, including Bcl-2, which increased at least 20- to 100-fold in lung tumor cell lines stimulated with agonists of TLR7 or TLR8. Interestingly, we also observed in these conditions a strong chemoresistance in both A549 and SK-MES cells, which suggests that the upregulation of Bcl-2 could contribute to cell survival. However, our observations also suggest that others mechanisms are implicated in chemoresistance. Indeed, TLR8 stimulation did not upregulate Bcl-2 protein, whereas it induced cell survival and chemoresistance, and TLR3 stimulation induced upregulation of Bcl-2 mRNA in A549 cells, which was not associated with chemoresistance. We also observed, in response to TLR7 or TLR8 stimulation, the upregulation of CCR4, VASH1 (a negative feedback regulator of angiogenesis; ref. 48), and Cyp7A1 (a member of the cytochrome P450 family implicated in drug metabolism and detoxifying system; refs. 49, 50) and the downregulation of prolactin, CD80, FN1, and serpin-C1 in the 2 cell lines. Upregulation of CCR4 and downregulation of FN1 could increase the migratory and metastatic capacities of lung tumor cells. CCR4 has previously been demonstrated to be essential for metastasis of lung tumor cells in the bone marrow (51), and fibronectin is frequently downregulated in cancer, which is thought to be a factor underlying metastatic behavior (52). In some experimental conditions, we also observed upregulation of the proinflammatory cytokines IL-6, IL-8, CSF-2, IL-1α, and IL-12 or molecules like NOS-2 or SerpinF1, which are expressed under the control of NF-κB. Interestingly, IL-6, IL-8, CSF-2 (GM-CSF), and NOS-2 have been implicated in survival or proliferation of tumor cells (53–58). Upregulation of KDR (VEGFR2) and downregulation of SerpinC1 were observed after TLR7 or TLR8 stimulation. Interestingly, upregulation of VEGFR2 in patients has been associated with a decrease of disease free survival (59), and a decrease of SerpinC1 has been associated with poor prognosis in lung cancer patients (60). Together, these observations led us to conclude that TLR7 or TLR8 stimulation could increase tumor viability and metastasis.
The analysis of gene expression in response to other TLR ligands, such as TLR3 and TLR4 ligands, revealed a profile that was partially different between cells stimulated by TLR7 or TLR8 and those stimulated by TLR3 or TLR4. Indeed, despite the fact that the stimulation of these different TLRs leads to a common atypical activation of NFκB, the gene expression profile was different. These observations suggest that other signaling pathways could be implicated in the modulation of immune and angiogenesis gene expression, such as MAPK or IRF pathways that are also activated in response to TLR stimulation.
In several cell types, including fibroblasts and epithelial cells, TLR stimulation leads to cell cycle entry and proliferation (61, 62). We observed that lung cancer cells displayed prolonged survival and reduced cell death when stimulated with TLR7 or TLR8 agonists. These effects were not caused by modification of the cell cycle, demonstrating tumor cell survival rather than proliferation, and were consistent with the observations of Lindemans et al. (63), who demonstrated that TLR7 or TLR8 stimulation by respiratory syncitial virus inhibits apoptosis of neutrophils via a NF-κB–dependent mechanism. Thus, we conclude that the expression of TLR7 or TLR8 by human lung cancer cells represents an advantage to tumor growth when TLRs are stimulated.
Therefore, characterization of those TLR7 and TLR8 ligands present in the tumor microenvironment in vivo is a crucial point. Because single-stranded RNAs are natural agonists for these receptors, either viruses (PAMPs) or endogenous nucleic acids released by altered tissues in the tumor (DAMPs) could stimulate the tumor cells. Comparison of the gene expression of highly purified primary human lung cancer cells and in vitro–stimulated cell lines as well as clustering analysis revealed a similar gene profile, which suggests that tumor cells are stimulated by TLR7 or TLR8 ligands in vivo. The common profile between stimulated cell lines and human lung cancer cells revealed a common gene expression signature for TLR-stimulated lung cancer cells.
In lung cancer, chemotherapy is frequently associated with surgery (42). We observed that TLR7 or TLR8 stimulation increased tumor cell survival by modulating a set of genes implicated in cell survival and conferred resistance to apoptosis induced by chemotherapeutic agents that are currently used to treat patients. Furthermore, siRNA experiments demonstrated that TLR7-induced chemoresistance depended on TLR7 and MyD88 expression. The chemoresistance induced by TLR stimulation could be explained by the increase of cell viability through IL-6, IL-8, CSF-2, or Bcl-2, but also by activation of detoxifying system through Cyp7A1, which may induce the degradation of xenobiotics (49, 50). Interestingly, LPS stimulation induced chemoresistance, but in a lineage- and drug-dependent manner. This is consistent with Bcl-2 and Cyp7A1 induction in LPS-stimulated A549 cells, but not in SK-MES cells. Interestingly, chemoresistance was observed in neither A549 nor SK-MES cells in response to poly I:C stimulation, since it induces cell death, as has been described in breast and melanoma tumor cells (26, 30). The differences among different TLRs in stimulation effects on chemoresistance could be explained by the fact that TLRs use different molecular adaptors. The TLR3 signaling pathway is TRIF dependent, and the TLR4 pathway is TRIF and MyD88 dependent, whereas TLR7 and TLR8 signaling pathways are MyD88 dependent. This hypothesis was confirmed by our siRNA experiments. The LPS-induced chemoresistance was abolished by siRNA MyD88, which suggests that the chemoresistance may involve mainly the MyD88 signaling pathway, whereas apoptosis induction may involve the TRIF signaling pathway. Another possibility is that poly I:C binds receptors other than TLR3, such as RIG-I/MDA-5, and could consequentially induce different signaling pathways leading to different biological effects.
Chemoresistance was also observed for ovarian cancer after TLR4 stimulation, which induced paclitaxel chemoresistance (41). These studies clearly demonstrate that TLR stimulation decreases sensitivity to chemotherapy. Thus, TLR7 or TLR8 expression by lung tumor cells in patients should be taken in account in order to adapt the chemotherapeutic protocols. Another treatment currently being investigated is the use of TLR7 or TLR8 agonists as adjuvants for priming antitumoral immune responses. For example, the TLR7 ligand imiquimod is used for the treatment of skin cancer and metastatic melanoma. Efficacy in the treatment of skin cancer is relative, mainly because of increased immune functions, particularly enhancement of NK cell antitumor activity, DC maturation, and T cell immunity to tumor antigens (64–66). In contrast, treatment with TLR7 agonists does not influence the clinical outcome in metastatic melanoma (67). In lung tumors, we observed significant infiltration of immune cells, including NK cells (S. Platonova, unpublished observation), CD3+ T lymphocytes, and antigen-presenting cells (38). Thus, TLR7 or TLR8 agonists in vivo could act on cells of the immune system, tumor cells, or both, with opposing effects. As a consequence, it would be interesting to determine the expression profile of TLR7 or TLR8 on tumor cells to evaluate the potential of such agonists in the treatment of lung cancer. Specifically, TLR7-negative tumor–bearing patients could benefit from TLR7 agonist treatment, which could stimulate antitumoral immune responses and result in a favorable clinical outcome. Together, these data emphasize the particular role of TLR7 and TLR8 in cancer and underscore the importance of determining their expression and functionality in cancer with the goal of improving therapeutic protocols.
Methods
Patient tumors.
Human primary lung tumor tissues were obtained from the Institut Mutualiste Montsouris after medical surgery of patients with TNM-classified stage I–III tumors. All patients underwent complete surgical resection of their tumors, including multilevel lymph node sampling or lymphadenectomy, but none received preoperative chemotherapy or radiotherapy. Patients with mixed histologic features, metastasis, or pleural invasion were ineligible. All patients were smokers. Lung samples, including tumoral and nontumoral tissues, were obtained the day of surgery, from informed patients, with the agreement of the French ethic committee (agreement 2007-A00845-48) in accordance with article L.1121-1 of French law.
Reagents.
LPS from E. coli (055:B5) were obtained from Sigma-Aldrich. Poly I:C, loxoribine, poly U, and Gardiquimod were purchased from Invivogen. Recombinant TNF-α and IL-1β were from R&D Systems. Anti-TLR7 (rabbit polyclonal) and anti-TLR8 (clone 44C143) were from Alexis. Anti-actin and anti-IκBα (rabbit polyclonal) were from Santa Cruz Technologies. Anti–phospho-IκBα and anti–Bcl-2 were purchased from Cell Signaling Technology, and F(ab′)2 goat anti-mouse IgM or IgG and goat anti-rabbit were from Jackson Immunoresearch. Cisplatine, carboplatine, doxorubicine, Navelbine, and cycloheximide were gifts of C. Bardin (Hôpital Hôtel Dieu).
Cell lines.
The A549 ADC cell lines and SK-MES SCC were obtained from the American Type Culture Collection (ATCC). The H1355 ADC cell line was obtained from S. Rogers (Brigham and Women’s Hospital, Boston, Massachusetts). The 16HBE and BEAS-2B bronchial epithelial cell lines were obtained from M. Si-Tahar (Pasteur Institute, Paris, France). All cell lines were cultured in DMEM/F12 medium (Invitrogen) containing 10% heat-inactivated FCS, 1% Ultroser G, and 1% penicillin-streptomycin (Invitrogen) at 37°C in a humidified atmosphere with 5% CO2.
RNA isolation and RT-PCR.
Total RNA was isolated from lung tumor cell lines or from freshly purified human lung cancer cells using the Qiagen RNeasy kit, and the quality of the extracted RNA was determined by BioAnalyser 2100 array (Agilent). Retrotranscription was performed using 1 μg RNA (Applied Biosystem), and PCR to amplify TLR1–TLR10 was performed using primers designed by Invivogen.
Flow cytometry.
A549, H1355, SK-MES, 16HBE, and BEAS-2B cells were fixed with 1% paraformaldehyde in PBS and permeabilized in PBS buffer containing 0.1% saponin (Sigma-Aldrich) and 2% FCS. Cells were stained with polyclonal anti-TLR7 or monoclonal PE-conjugated anti-TLR8 (clone 44C143) antibodies (Alexis). Staining was assessed with a FACScalibur cytometer, and flow cytometry data were analyzed using Cellquest Pro software (BD Biosciences).
Immunohistochemistry.
Paraffin-embedded tumor biopsies with representative areas of tumor and adjacent lung parenchyma were retrieved retrospectively from 13 NSCLC patients. The expression and subcellular localization of TLR7 and TLR8 by tumor cells, bronchial cells, and immune cells was performed as follows. Serial 5-μm tissue sections were deparaffinized, rehydrated, and pretreated in 10 mM citrate buffer, pH 6, for antigen retrieval. Sections were incubated with H2O2 for 15 minutes, then blocked in 5% human serum for 30 minutes at room temperature. Samples were incubated with either polyclonal anti-TLR7 or monoclonal anti-TLR8 antibody (Alexis) for 1 hour at room temperature. Control isotype antibodies served as negative controls. Specific staining was detected by incubation with Envision rabbit or mouse HRP (Dako Cytomation) for 30 minutes followed by a 5-minute incubation with 3-amino-9-ethylcarbazole substrate (Vector Laboratories). Tissue sections were then counterstained with Harris hematoxylin (Sigma-Aldrich), and slides were mounted with Glycergel Mounting Medium (Dako Cytomation) and visualized by light microscopy (Leica Axiovert II).
Western blot analysis.
A549 cells (1 × 106 cells) were starved for 4 hours in medium containing 2% FCS and then stimulated with loxoribine, poly U, or TNF-α (10 ng/ml) for the indicated time periods. Cells were lysed in 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 2 mM EDTA, 0.5% Triton X100, 2 mM Na3VO4, 10 mM NaF, and 10 mM sodium pyrophosphate supplemented with a complete protease inhibitor cocktail (Boehringer). Protein contents in the cell lysates were quantified using the quick start Bradford protein assay kit (Biorad) to ensure that all samples contained similar amounts of protein. Proteins (30 μg) were resolved in 10% Tris-glycine polyacrylamide gels (Invitrogen) and transferred to PVDF immobilon-P membranes (Millipore). Blots were probed with monoclonal antibody against phospho-IκBα, polyclonal antibody against Bcl-2, or polyclonal antibody against total IκBα or actin.
NF-κB activation analysis by ELISA.
A549 cells (1 × 106 cells) were cultured for 4 hours in medium containing 2% FCS and then stimulated with loxoribine or IL1-β for the indicated time periods. Nuclear protein extraction was performed using the Nuclear Extract Kit (Active Motif) according to the manufacturer’s recommendations. Briefly, cells were incubated for 15 minutes on ice in hypotonic buffer. After addition of detergents, cell lysates were centrifuged, and supernatants were collected. Protein contents in the cell lysates were quantified using the quick start Bradford protein assay kit (Biorad) to ensure that all samples contained similar amounts of protein. Next, nuclear contents were assayed to determine the relative quantity of p50 and p65 NF-κB subunits contained in the nucleus using TransAm NF-κB ELISA Kit (Active Motif) according to the manufacturer’s recommendations. Briefly, NF-κB consensus sequence containing oligonucleotides was coated in plates before adding the nuclear extract containing activated transcription factor. After the binding of NF-κB to its consensus sequence, the relative quantity of NF-κB subunits was determined by colorimetric reaction.
NF-κB gene reporter assays.
NF-κB gene reporter assays were carried out with the pNIFTY2-SEAP plasmid (Invivogen) following the recommendations of the manufacturer. Briefly, A549 cells (5 × 104 cells) were cultured in 12-well plates and transfected using lyovec (Invivogen) and 1 μg/ml pNIFTY2-SEAP. At 48 hours after transfection, cells were stimulated or not with loxoribine or IL-1β for 24 hours. Supernatants were collected and incubated with pNpp. NF-κB activity was analyzed by measuring the hydrolysis of pNpp in the supernatants with a spectrophotometer at 405 nm.
Immunofluorescence analysis of p65 NF-κB subunit nuclear translocation.
A549 cells (5 × 104 cells) were cultured in labteck chambers (Nalge Nunc) for 4 hours in medium containing 2% FCS, and then stimulated with loxoribine or IL-1β for the indicated time periods. Cells were then washed twice in PBS and fixated with 4% paraformaldehyde in PBS for 10 minutes. After 3 washes with PBS containing 1 mg/ml BSA, cells were permeabilized in PBS buffer containing 0.1% SDS (Sigma-Aldrich) for 10 minutes and then blocked in 10% FCS in PBS for 20 minutes at room temperature. Samples were incubated with anti-p65 (clone SC-109; Santa Cruz) for 1 hour at room temperature. Specific staining was detected by incubation with Alexa Fluor 594–coupled goat anti-rabbit antibody (Invitrogen) for 30 minutes and DAPI. Slides were mounted with Fluoromount mounting medium (Dako Cytomation) and visualized by fluorescence microscopy (Leica Axiovert II).
Tumor cell proliferation assay.
Human lung cancer cell lines were seeded in 96-well plates (800 cells/well) and left unstimulated or stimulated with loxoribine, poly U, or Gardiquimod. The proliferation assay was performed with Alamar blue (AbD Serotec) according to the manufacturer’s instructions. Briefly, cells were seeded and stimulated with TLR ligands in 100 μl medium, and 10 μl Alamar Blue was added. The optical density in each well was determined at 540 nm and 570 nm after 10 days of culture, and the percentage of reduced Alamar blue in stimulated cells was determined relative to that in unstimulated cells (considered 100%).
Chemoresistance assay.
A549 cells or SK-MES cells were seeded in 6-well plates (200 cells/ml) and stimulated or not with loxoribine at days 0, 3, 6, and 9. At day 12, the cells were treated or not with chemotherapeutic agents at LD50, and the number of colonies in each culture condition was determined at day 15 after Crystal violet coloration. Cells were washed twice with PBS and then stained with glutaraldehyde (6.0%, vol/vol) and crystal violet (0.5%, wt/vol) in H2O for 30 minutes. Finally, cells were washed in distillated water, and the colony number was determined by software analysis (VisionCapt software).
siRNA transfection and chemoresistance assay.
siRNA assays were carried out with MyD88- and TLR7-specific siRNA or with scrambled siRNA coupled with AF488 (Qiagen). Briefly, A549 cells (1.5 × 105 cells) were cultured in 6-well plates and transfected with HiPerfect (Qiagen) and 2 μM siRNA. After 48 hours, the transfection efficiency was determined by the fluorescence intensity, and the expression level of MyD88 and TLR7 was determined by RT-PCR. A549 cells were stimulated or not with loxoribine or LPS for 48 hours and then incubated with cisplatine at LD50 for 36 hours. The percentage of apoptotic cells was determined after annexin V staining and was assessed with a FACScalibur cytometer. Flow cytometry data were analyzed using Cellquest Pro software (BD Biosciences). The percentage of dead cells in untransfected and unstimulated cells was considered 100%, and the percentages of the transfected and stimulated cells were determined relative to this value.
Cell cycle analysis.
A549 cells were cultured in the presence of loxoribine, poly U, Gardiquimod, etoposide, or cycloheximide. After 48 hours, cells were harvested, washed in cold PBS, and fixed with 2 ml of 70% cold ethanol in PBS for 2 hours at 4°C. After centrifugation, the supernatant was removed, and the pellet was rinsed with 0.5% PBS Tween and incubated with 50 μg RNAse A (Sigma-Aldrich) and 50 μg propidium iodide (BD Bioscience) in PBS. Propidium iodide staining was determined with a FACScalibur cytometer.
Purification of primary lung tumor cells.
Tumoral tissues, obtained after surgery, were mechanically dilacerated, and single-cell suspensions were obtained after digestion with collagenase A (1 mg/ml) and DNAse (100 IU/ml) (Sigma-Aldrich) for 1 hour at 37°C under magnetic agitation in serum-free RPMI1640, as previously described (68). Cells were incubated with anti–epithelial membrane antigen (anti-EMA), anti-EpCam (clone BrEp4), and anti-cytokeratin (clone AE1-AE3) monoclonal antibodies (BD Biosciences) for 20 minutes at 4°C. Labeled cells were then incubated with microbeads coupled to goat anti-mouse antibody. Tumor cells expressing EMA, BrEP4, and AE1-AE3 were then sorted by magnetic isolation according to the instructions of the manufacturer (Miltenyi Biotech). The purity, as determined by flow cytometry, was greater than 90%.
Transcriptomic analysis.
Total RNA was extracted from A549 or SK-MES cells stimulated with loxoribine, poly U, Gardiquimod, poly I:C, or LPS for 6 hours or from purified human lung cancer cells using the RNeasy isolation kit RNA (Qiagen). The integrity and quantity of the RNA was evaluated with a bioanalyzer-2100 (Agilent Technologies). All samples were assessed for gene expression analysis using the TaqMan Low-Density Array angiogenesis and immune panel (Applied Biosystems). RT-PCR experiments were carried out according to the manufacturer’s instructions (Applied Biosystems). Quantitative real-time TaqMan PCR was performed using Low-Density Arrays and the 7900 robotic real-time PCR system (Applied Biosystems). The 18S primers and probes were used as internal controls. Data were analyzed using SDS Software (version 2.2; Applied Biosystems). The ΔCT values were calculated for each gene by subtraction of the CT value of the 18S housekeeping gene from the CT of the gene of interest. The ΔΔCT values were obtained by subtraction of the ΔCT values for unstimulated cells from the CT obtained for stimulated cells. The fold increase (shown in arbitrary units) was obtained by 2–ΔΔCT. Average-linkage hierarchical clustering was applied, and the results were displayed using the GENESIS program (http://www.genome.tugraz.at) in order to perform Pearson uncentered hierarchical clustering.
Statistics.
Data are represented as mean ± SD of 3 separate experiments performed in triplicate. Statistical analysis was performed using 2-tailed Student’s t test. P values less than 0.05 were considered to be significant.
Supplementary Material
Acknowledgments
This work was supported by the Institut National du Cancer and the Association pour la Recherche contre le Cancer (INCA/ARC grant R07120DP to C. Sautès-Fridman), the Institut National de la Santé et de la Recherche Médicale, the University Pierre et Marie Curie, and the University Paris Descartes. J. Cherfils-Vicini was a beneficiary of study grant from the Cancéropôle Ile de France and ARC. We thank Robert Weil (Institut Pasteur) for helpful discussions on NF-κB activation and Maud Teyssandier and Sebastien André (Team 16 of Centre de Recherche des Cordeliers) for the gift of pNIFTY2 plasmid.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2010;120(4):1285–1297. doi:10.1172/JCI36551.
References
- 1.Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357(9255):539–545. doi: 10.1016/S0140-6736(00)04046-0. [DOI] [PubMed] [Google Scholar]
- 2.Clevers H. At the crossroads of inflammation and cancer. Cell. 2004;118(6):671–674. doi: 10.1016/j.cell.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 3.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420(6917):860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Keller SA, et al. NF-kappaB is essential for the progression of KSHV- and EBV-infected lymphomas in vivo. Blood. 2006;107(8):3295–3302. doi: 10.1182/blood-2005-07-2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Littman AJ, Jackson LA, Vaughan TL. Chlamydia pneumoniae and lung cancer: epidemiologic evidence. Cancer Epidemiol Biomarkers Prev. 2005;14(4):773–778. doi: 10.1158/1055-9965.EPI-04-0599. [DOI] [PubMed] [Google Scholar]
- 6.Littman AJ, et al. Prior lung disease and risk of lung cancer in a large prospective study. Cancer Causes Control. 2004;15(8):819–827. doi: 10.1023/B:CACO.0000043432.71626.45. [DOI] [PubMed] [Google Scholar]
- 7.Littman AJ, et al. Chlamydia pneumoniae infection and risk of lung cancer. Cancer Epidemiol Biomarkers Prev. 2004;13(10):1624–1630. [PubMed] [Google Scholar]
- 8.Philip M, Rowley DA, Schreiber H. Inflammation as a tumor promoter in cancer induction. Semin Cancer Biol. 2004;14(6):433–439. doi: 10.1016/j.semcancer.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 9.Balkwill F. TNF-alpha in promotion and progression of cancer. Cancer Metastasis Rev. 2006;25(3):409–416. doi: 10.1007/s10555-006-9005-3. [DOI] [PubMed] [Google Scholar]
- 10.Killeen SD, Wang JH, Andrews EJ, Redmond HP. Exploitation of the Toll-like receptor system in cancer: a doubled-edged sword? Br J Cancer. 2006;95(3):247–252. doi: 10.1038/sj.bjc.6603275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lotze MT, et al. The grateful dead: damage-associated molecular pattern molecules and reduction/oxidation regulate immunity. Immunol Rev. 2007;220:60–81. doi: 10.1111/j.1600-065X.2007.00579.x. [DOI] [PubMed] [Google Scholar]
- 12.Trinchieri G, Sher A. Cooperation of Toll-like receptor signals in innate immune defence. Nat Rev Immunol. 2007;7(3):179–190. doi: 10.1038/nri2038. [DOI] [PubMed] [Google Scholar]
- 13.Karin M, Greten FR. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005;5(10):749–759. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- 14.Pikarsky E, et al. NF-κB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004;431(7007):461–466. doi: 10.1038/nature02924. [DOI] [PubMed] [Google Scholar]
- 15.Greten FR, et al. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 2004;118(3):285–296. doi: 10.1016/j.cell.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 16.Luo JL, et al. Nuclear cytokine-activated IKKalpha controls prostate cancer metastasis by repressing Maspin. Nature. 2007;446(7136):690–694. doi: 10.1038/nature05656. [DOI] [PubMed] [Google Scholar]
- 17.Karin M, Lin A. NF-kappaB at the crossroads of life and death. Nat Immunol. 2002;3(3):221–227. doi: 10.1038/ni0302-221. [DOI] [PubMed] [Google Scholar]
- 18.Luo JL, Kamata H, Karin M. IKK/NF-kappaB signaling: balancing life and death--a new approach to cancer therapy. J Clin Invest. 2005;115(10):2625–2632. doi: 10.1172/JCI26322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen YC, Giovannucci E, Kraft P, Lazarus R, Hunter DJ. Association between Toll-like receptor gene cluster (TLR6, TLR1, and TLR10) and prostate cancer. Cancer Epidemiol Biomarkers Prev. 2007;16(10):1982–1989. doi: 10.1158/1055-9965.EPI-07-0325. [DOI] [PubMed] [Google Scholar]
- 20.Tsan MF. Toll-like receptors, inflammation and cancer. Semin Cancer Biol. 2006;16(1):32–37. doi: 10.1016/j.semcancer.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 21.Chen YC, Giovannucci E, Lazarus R, Kraft P, Ketkar S, Hunter DJ. Sequence variants of Toll-like receptor 4 and susceptibility to prostate cancer. Cancer Res. 2005;65(24):11771–11778. doi: 10.1158/0008-5472.CAN-05-2078. [DOI] [PubMed] [Google Scholar]
- 22.Sun J, et al. Interactions of sequence variants in interleukin-1 receptor-associated kinase4 and the toll-like receptor 6-1-10 gene cluster increase prostate cancer risk. Cancer Epidemiol Biomarkers Prev. 2006;15(3):480–485. doi: 10.1158/1055-9965.EPI-05-0645. [DOI] [PubMed] [Google Scholar]
- 23.Rakoff-Nahoum S, Medzhitov R. Regulation of spontaneous intestinal tumorigenesis through the adaptor protein MyD88. Science. 2007;317(5834):124–127. doi: 10.1126/science.1140488. [DOI] [PubMed] [Google Scholar]
- 24.Xiao H, et al. The Toll-interleukin-1 receptor member SIGIRR regulates colonic epithelial homeostasis, inflammation, and tumorigenesis. Immunity. 2007;26(4):461–475. doi: 10.1016/j.immuni.2007.02.012. [DOI] [PubMed] [Google Scholar]
- 25.Huang B, et al. Toll-like receptors on tumor cells facilitate evasion of immune surveillance. Cancer Res. 2005;65(12):5009–5014. doi: 10.1158/0008-5472.CAN-05-0784. [DOI] [PubMed] [Google Scholar]
- 26.Salaun B, Coste I, Rissoan MC, Lebecque SJ, Renno T. TLR3 can directly trigger apoptosis in human cancer cells. J Immunol. 2006;176(8):4894–4901. doi: 10.4049/jimmunol.176.8.4894. [DOI] [PubMed] [Google Scholar]
- 27.Huang B, et al. Listeria monocytogenes promotes tumor growth via tumor cell toll-like receptor 2 signaling. Cancer Res. 2007;67(9):4346–4352. doi: 10.1158/0008-5472.CAN-06-4067. [DOI] [PubMed] [Google Scholar]
- 28.Droemann D, et al. Human lung cancer cells express functionally active Toll-like receptor 9. Respir Res. 2005;6:1. doi: 10.1186/1465-9921-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.He W, Liu Q, Wang L, Chen W, Li N, Cao X. TLR4 signaling promotes immune escape of human lung cancer cells by inducing immunosuppressive cytokines and apoptosis resistance. Mol Immunol. 2007;44(11):2850–2859. doi: 10.1016/j.molimm.2007.01.022. [DOI] [PubMed] [Google Scholar]
- 30.Salaun B, Lebecque S, Matikainen S, Rimoldi D, Romero P. Toll-like receptor 3 expressed by melanoma cells as a target for therapy? Clin Cancer Res. 2007;13(15 Pt 1):4565–4574. doi: 10.1158/1078-0432.CCR-07-0274. [DOI] [PubMed] [Google Scholar]
- 31.Guillot L, et al. Response of human pulmonary epithelial cells to lipopolysaccharide involves Toll-like receptor 4 (TLR4)-dependent signaling pathways: evidence for an intracellular compartmentalization of TLR4. J Biol Chem. 2004;279(4):2712–2718. doi: 10.1074/jbc.M305790200. [DOI] [PubMed] [Google Scholar]
- 32.Mayer AK, et al. Differential recognition of TLR-dependent microbial ligands in human bronchial epithelial cells. J Immunol. 2007;178(5):3134–3142. doi: 10.4049/jimmunol.178.5.3134. [DOI] [PubMed] [Google Scholar]
- 33.Ardies CM. Inflammation as cause for scar cancers of the lung. Integr Cancer Ther. 2003;2(3):238–246. doi: 10.1177/1534735403256332. [DOI] [PubMed] [Google Scholar]
- 34.Pons J, et al. Expression of Toll-like receptor 2 is up-regulated in monocytes from patients with chronic obstructive pulmonary disease. Respir Res. 2006;7:64. doi: 10.1186/1465-9921-7-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science. 2004;303(5663):1529–1531. doi: 10.1126/science.1093616. [DOI] [PubMed] [Google Scholar]
- 36.Heil F, et al. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science. 2004;303(5663):1526–1529. doi: 10.1126/science.1093620. [DOI] [PubMed] [Google Scholar]
- 37.Smits EL, Ponsaerts P, Berneman ZN, Van Tendeloo VF. The use of TLR7 and TLR8 ligands for the enhancement of cancer immunotherapy. Oncologist. 2008;13(8):859–875. doi: 10.1634/theoncologist.2008-0097. [DOI] [PubMed] [Google Scholar]
- 38.Dieu-Nosjean MC, et al. Long-term survival for patients with non-small-cell lung cancer with intratumoral lymphoid structures. J Clin Oncol. 2008;26(27):4410–4417. doi: 10.1200/JCO.2007.15.0284. [DOI] [PubMed] [Google Scholar]
- 39.Qin J, et al. TLR8-mediated NF-kappaB and JNK activation are TAK1-independent and MEKK3-dependent. J Biol Chem. 2006;281(30):21013–21021. doi: 10.1074/jbc.M512908200. [DOI] [PubMed] [Google Scholar]
- 40.Jego G, Bataille R, Geffroy-Luseau A, Descamps G, Pellat-Deceunynck C. Pathogen-associated molecular patterns are growth and survival factors for human myeloma cells through Toll-like receptors. Leukemia. 2006;20(6):1130–1137. doi: 10.1038/sj.leu.2404226. [DOI] [PubMed] [Google Scholar]
- 41.Kelly MG, et al. TLR-4 signaling promotes tumor growth and paclitaxel chemoresistance in ovarian cancer. Cancer Res. 2006;66(7):3859–3868. doi: 10.1158/0008-5472.CAN-05-3948. [DOI] [PubMed] [Google Scholar]
- 42.Spira A, Ettinger DS. Multidisciplinary management of lung cancer. N Engl J Med. 2004;350(4):379–392. doi: 10.1056/NEJMra035536. [DOI] [PubMed] [Google Scholar]
- 43.Sakai A, et al. The bacterium, nontypeable Haemophilus influenzae, enhances host antiviral response by inducing Toll-like receptor 7 expression: evidence for negative regulation of host anti-viral response by CYLD. FEBS J. 2007;274(14):3655–3668. doi: 10.1111/j.1742-4658.2007.05899.x. [DOI] [PubMed] [Google Scholar]
- 44.Swann JB, Smyth MJ. Immune surveillance of tumors. J Clin Invest. 2007;117(5):1137–1146. doi: 10.1172/JCI31405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leifer CA, et al. TLR9 is localized in the endoplasmic reticulum prior to stimulation. J Immunol. 2004;173(2):1179–1183. doi: 10.4049/jimmunol.173.2.1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Préhaud C, Mégret F, Lafage M, Lafon M. Virus infection switches TLR-3-positive human neurons to become strong producers of beta interferon. J Virol. 2005;79(20):12893–12904. doi: 10.1128/JVI.79.20.12893-12904.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Samanta AK, Huang HJ, Bast RC, Liao WS. Overexpression of MEKK3 confers resistance to apoptosis through activation of NFkappaB. J Biol Chem. 2004;279(9):7576–7583. doi: 10.1074/jbc.M311659200. [DOI] [PubMed] [Google Scholar]
- 48.Watanabe K, et al. Vasohibin as an endothelium-derived negative feedback regulator of angiogenesis. J Clin Invest. 2004;114(7):898–907. doi: 10.1172/JCI21152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rodriguez-Antona C, Ingelman-Sundberg M. Cytochrome P450 pharmacogenetics and cancer. Oncogene. 2006;25(11):1679–1691. doi: 10.1038/sj.onc.1209377. [DOI] [PubMed] [Google Scholar]
- 50.Oyama T, et al. Expression of cytochrome P450 in non-small cell lung cancer. Front Biosci. 2008;13:5787–5793. doi: 10.2741/3116. [DOI] [PubMed] [Google Scholar]
- 51.Nakamura ES, et al. RANKL-induced CCL22/macrophage-derived chemokine produced from osteoclasts potentially promotes the bone metastasis of lung cancer expressing its receptor CCR4. Clin Exp Metastasis. 2006;23(1):9–18. doi: 10.1007/s10585-006-9006-1. [DOI] [PubMed] [Google Scholar]
- 52.Görögh T, et al. Transcriptional repression of the human fibronectin gene in laryngeal squamous cell carcinoma cells. J Cancer Res Clin Oncol. 2001;127(3):166–172. doi: 10.1007/s004320000198. [DOI] [PubMed] [Google Scholar]
- 53.Gao SP, et al. Mutations in the EGFR kinase domain mediate STAT3 activation via IL-6 production in human lung adenocarcinomas. J Clin Invest. 2007;117(12):3846–3856. doi: 10.1172/JCI31871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Naugler WE, Karin M. The wolf in sheep’s clothing: the role of interleukin-6 in immunity, inflammation and cancer. Trends Mol Med. 2008;14(3):109–119. doi: 10.1016/j.molmed.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 55.Qian Y, et al. Regulation of TLR4-induced IL-6 response in bladder cancer cells by opposing actions of MAPK and PI3K signaling. J Cancer Res Clin Oncol. 2009;135(3):379–386. doi: 10.1007/s00432-008-0478-z. [DOI] [PubMed] [Google Scholar]
- 56.Zhu YM, Webster SJ, Flower D, Woll PJ. Interleukin-8/CXCL8 is a growth factor for human lung cancer cells. Br J Cancer. 2004;91(11):1970–1976. doi: 10.1038/sj.bjc.6602227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Uemura Y, et al. Effects of GM-CSF and M-CSF on tumor progression of lung cancer: roles of MEK1/ERK and AKT/PKB pathways. Int J Mol Med. 2006;18(2):365–373. [PubMed] [Google Scholar]
- 58.Davie SA, et al. Effects of FVB/NJ and C57Bl/6J strain backgrounds on mammary tumor phenotype in inducible nitric oxide synthase deficient mice. Transgenic Res. 2007;16(2):193–201. doi: 10.1007/s11248-006-9056-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Carrillo de Santa Pau E, et al. Prognostic significance of the expression of vascular endothelial growth factors A, B, C, and D and their receptors R1, R2, and R3 in patients with nonsmall cell lung cancer. Cancer. 2009;115(8):1701–1712. doi: 10.1002/cncr.24193. [DOI] [PubMed] [Google Scholar]
- 60.Unsal E, Atalay F, Atikcan S, Yilmaz A. Prognostic significance of hemostatic parameters in patients with lung cancer. Respir Med. 2004;98(2):93–98. doi: 10.1016/j.rmed.2003.07.001. [DOI] [PubMed] [Google Scholar]
- 61.Hasan UA, et al. Cell proliferation and survival induced by Toll-like receptors is antagonized by type I IFNs. Proc Natl Acad Sci U S A. 2007;104(19):8047–8052. doi: 10.1073/pnas.0700664104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hasan UA, Trinchieri G, Vlach J. Toll-like receptor signaling stimulates cell cycle entry and progression in fibroblasts. J Biol Chem. 2005;280(21):20620–20627. doi: 10.1074/jbc.M500877200. [DOI] [PubMed] [Google Scholar]
- 63.Lindemans CA, et al. Respiratory syncytial virus inhibits granulocyte apoptosis through a phosphatidylinositol 3-kinase and NF-kappaB-dependent mechanism. J Immunol. 2006;176(9):5529–5537. doi: 10.4049/jimmunol.176.9.5529. [DOI] [PubMed] [Google Scholar]
- 64.Kanzler H, Barrat FJ, Hessel EM, Coffman RL. Therapeutic targeting of innate immunity with Toll-like receptor agonists and antagonists. Nat Med. 2007;13(5):552–559. doi: 10.1038/nm1589. [DOI] [PubMed] [Google Scholar]
- 65.Schön M, Schön M. TLR7 and TLR8 as targets in cancer therapy. Oncogene. 2008;27(2):190–199. doi: 10.1038/sj.onc.1210913. [DOI] [PubMed] [Google Scholar]
- 66.Stary G, Bangert C, Tauber M, Strohal R, Kopp T, Stingl G. Tumoricidal activity of TLR7/8-activated inflammatory dendritic cells. J Exp Med. 2007;204(6):1441–1451. doi: 10.1084/jem.20070021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dummer R, et al. An exploratory study of systemic administration of the Toll-like receptor-7 agonist 852A in patients with refractory metastatic melanoma. Clin Cancer Res. 2008;14(3):856–864. doi: 10.1158/1078-0432.CCR-07-1938. [DOI] [PubMed] [Google Scholar]
- 68.Perrot I, et al. Dendritic cells infiltrating human non-small cell lung cancer are blocked at immature stage. J Immunol. 2007;178(5):2763–2769. doi: 10.4049/jimmunol.178.5.2763. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.