

Abstract
Resistance to thyroid hormone (RTH), a dominantly inherited disorder usually associated with mutations in thyroid hormone receptor β (THRB), is characterized by elevated levels of circulating thyroid hormones (including thyroxine), failure of feedback suppression of thyrotropin, and variable tissue refractoriness to thyroid hormone action. Raised energy expenditure and hyperphagia are recognized features of hyperthyroidism, but the effects of comparable hyperthyroxinemia in RTH patients are unknown. Here, we show that resting energy expenditure (REE) was substantially increased in adults and children with THRB mutations. Energy intake in RTH subjects was increased by 40%, with marked hyperphagia particularly evident in children. Rates of muscle TCA cycle flux were increased by 75% in adults with RTH, whereas rates of ATP synthesis were unchanged, as determined by 13C/31P magnetic resonance spectroscopy. Mitochondrial coupling index between ATP synthesis and mitochondrial rates of oxidation (as estimated by the ratio of ATP synthesis to TCA cycle flux) was significantly decreased in RTH patients. These data demonstrate that basal mitochondrial substrate oxidation is increased and energy production in the form of ATP synthesis is decreased in the muscle of RTH patients and that resting oxidative phosphorylation is uncoupled in this disorder. Furthermore, these observations suggest that mitochondrial uncoupling in skeletal muscle is a major contributor to increased REE in patients with RTH, due to tissue selective retention of thyroid hormone receptor α sensitivity to elevated thyroid hormone levels.
Introduction
Thyroid hormone action is mediated by the products of 2 human genes (thyroid hormone receptor α [THRA] and thyroid hormone receptor β [THRB]), which are alternately spliced to generate 3 highly homologous nuclear receptor isoforms (TRα1, TRβ1, and TRβ2) with differing tissue distributions (1). Resistance to thyroid hormone (RTH) is an uncommon disorder, characterized by elevated circulating thyroid hormones with nonsuppressed thyrotropin (TSH) levels, reflecting resistance within the hypothalamic-pituitary-thyroid axis but variable refractoriness to hormone action in peripheral tissues (2). Usually dominantly inherited, RTH is associated with diverse, heterozygous THRB gene mutations impairing hormone binding and/or transcriptional activity of receptors (3–5). In addition, mutant receptors inhibit the action of their wild-type counterparts in a dominant-negative manner when they are coexpressed (6, 7). The clinical phenotype of RTH is variable: most subjects are either asymptomatic or have nonspecific symptoms and are deemed to be in a compensated euthyroid state termed generalized RTH (GRTH); in contrast, a subset of affected individuals can exhibit some clinical features of hyperthyroidism, suggesting greater central or pituitary RTH (PRTH) than in peripheral tissues. Although imprecise, this clinical distinction may remain useful in the management of the disorder (8). The basis of and mechanisms underlying variable tissue resistance in RTH are not fully understood.
It is well recognized that the thyroid hormone is a major determinant of energy expenditure, including basal metabolic rate (BMR) (9); indeed, even small changes in thyroid hormone levels in patients receiving hormone replacement are associated with effects on resting energy expenditure (REE) (10). Markedly increased appetite is considered to be a clinical feature of hyperthyroidism, and hyperphagia and enhanced energy intake have indeed been formally documented in thyrotoxicosis (11).
We have identified a large cohort of subjects with RTH, defined genetically with THRB gene mutations, with lifelong elevation of circulating thyroid hormones to levels comparable to conventional thyrotoxicosis. To determine whether such hyperthyroxinemia is associated with changes in energy balance, we have evaluated whole-body energy expenditure by indirect calorimetry, food intake using an ad libitum test meal (12), and quantified hunger and satiety using visual analogue scores during energy balance feeding in adults and children with RTH. As skeletal muscle is the major determinant of energy expenditure in humans (13), we have used 13C/31P magnetic resonance spectroscopy (MRS) to assess in vivo resting mitochondrial oxidative phosphorylation activity noninvasively in a subset of this RTH cohort. The rate of flux through the TCA cycle, a direct measure of mitochondrial oxidative function, was determined using 13C MRS during an infusion of [2-13C] acetate. The unidirectional rates of muscle ATP synthase flux, an assessment of overall energy production, were estimated during the same session using 31P saturation-transfer MRS (14). We believe these data provide a unique insight into the effects that RTH may have on resting mitochondrial function in vivo.
Results
Characteristics of the RTH cohort.
We studied an unselected group of adult subjects with RTH (n = 54) from 35 unrelated families, harboring 25 different THRB gene mutations. The demographic, anthropometric, and biochemical characteristics of this cohort, together with these parameters in control and thyrotoxic subjects, are shown in Table 1. Control and RTH subjects were matched for age, gender, and BMI. The nonsignificant increase in BMI in RTH subjects is, at least in part, a reflection of the fact that affected individuals can be shorter, as has been noted previously (15). However, measurement of body composition by dual energy x-ray densitometry (DEXA) indicated significantly greater fat to lean body mass (LBM) ratio in subjects with RTH, compared with the control group (P < 0.05; Table 1). As expected, RTH subjects exhibited the characteristic abnormal pattern of elevated free thyroxine (fT4) and free triiodothyronine (fT3), with near normal TSH levels; in contrast, comparable elevation of fT4 and fT3 in thyrotoxicosis was associated with suppressed TSH. Consistent with the known positive, cardiac chronotropic effect of thyroid hormone excess, mean sleeping heart rate was significantly elevated in the thyrotoxic patients and, to a lesser extent, in the RTH subjects, compared with the euthyroid controls. Conversely, levels of serum sex hormone binding globulin (SHBG) were significantly lower in RTH than thyrotoxic (P < 0.01) or control (P < 0.01) subjects, confirming that the liver is refractory to thyroid hormone action in this disorder as has been described previously (16).
Table 1 .
Demographic, anthropometric, and biochemical characteristics of control, adult patients with RTH, and thyrotoxic patients studied by ventilated hood calorimetry

Energy expenditure in RTH subjects.
REE, measured by indirect calorimetry and expressed per kilogram LBM to allow for differences in body composition between groups, was significantly elevated (~18%; P = 3 × 10–11) in the RTH subjects, compared with the euthyroid controls, but not as high as in comparably hyperthyroxinemic, thyrotoxic patients (P = 0.0003) (Figure 1A). In all 3 groups the relationship between REE and LBM, was similar with RTH subjects being intermediate between control and thyrotoxic groups (Figure 1B). The regression slopes did not differ significantly, though intercepts did (P = 0.82 and 0.001, respectively). REE did correlate with serum fT4 and fT3 levels in RTH, albeit not as strongly as in thyrotoxic subjects (Figure 1, C and D). However, when we compared mean sleeping heart rate and REE in RTH and thyrotoxicosis (Figure 1E), both groups showed a significant positive correlation between these parameters. A previous study has shown that clinical phenotype does correlate with the THRB gene defect in RTH, but only in subjects harbouring TRβ mutations, whose transcriptional properties are closely linked to the magnitude of impaired T3 binding (referred to herein as type 1 mutations); other TRβ mutations, whose T3 binding properties are not related to transcriptional activity (referred to herein as type 2 mutations) do not correlate with phenotype (17). We undertook similar analyses in subjects harboring type 1 (R320H, R320C, R338W, R438H, R438C, P453T, and P453S) or type 2 (R243W, R383C, and R383H) mutations from our RTH cohort, confirming the correlation of circulating fT4 levels with the severity of the receptor defect noted previously (Figure 1F) (17). Interestingly, REE showed a similar correlation with genotype, being more elevated in cases with markedly deleterious (type 1) TRβ mutations (Figure 1G). It has also been suggested that a subgroup of TRβ mutations (R338W, R383H, and R383C) are associated with PRTH (18, 19). However, mean REE in individuals harboring such mutations (R338W, R383H, and R383C; n = 9; 0.172 MJ/d/kg) was not significantly different compared with individuals harboring receptor mutations not associated with this phenotype (R438C, R438H, P453T, P453S, and P453C; n = 13; 0.178 MJ/d/kg) (P = 0.43).
Figure 1. Energy expenditure in patients with RTH.

(A) REE expressed per unit LBM in control (gray circles, n = 45), adult RTH (black circles, n = 52), and thyrotoxic (white circles, n = 11) subjects. (B) Relationship between REE and LBM in control, RTH, or thyrotoxic subjects. (C and D) Relationship between REE/LBM and fT4 (C) or fT3 levels (D) in control (n = 45), RTH (n = 46), and thyrotoxic (n = 10) subjects, excluding subjects on exogenous levothyroxine or triiodothyronine (RTH, n = 6; thyrotoxic, n = 1). (E) Relationship between mean sleeping heart rate and REE/LBM in RTH (black circles, n = 42) and thyrotoxic subjects (white circles, n = 11). Data was not available for 10 RTH subjects. (F) Correlation of the mean T3 binding affinity of the type 1 mutant TRβs with mean fT4 level in RTH subjects, excluding subjects on exogenous levothyroxine. Type 1 mutants (black circles, n = 7) follow the regression line describing this correlation; type 2 mutants (white circles, n = 3) have mutations significantly deviating from this relationship. Each data point represents mean ± SD for all subjects with that particular THRB mutation. Ka indicates the binding affinity constant of thyroid receptor (WT or mutant) for T3. (G) Correlation of mean REE/LBM with T3 binding affinity of type 1 (black circles, n = 6) and type 2 mutant TRβs (white circles, n = 3) in RTH subjects. Each data point represents mean ± SD for all subjects with that particular THRB mutation. (H) BMR expressed per unit LBM in control (gray circles, n = 22) and RTH subjects (black circles, n = 13), matched for age, gender, weight and BMI. Diagonal lines indicate the regression lines (B–G). Horizontal lines indicate the mean (A and H). r, Pearson product moment correlation coefficient.
A subgroup of RTH subjects (n = 13) also underwent chamber calorimetry, and BMR measurements (expressed per kg LBM) confirmed significantly raised energy expenditure (P = 0.0005) compared with age-, gender-, weight-, and BMI-matched control subjects (n = 22; Figure 1H). In children with RTH (n = 13), measurements of REE by indirect calorimetry were compared with values expected from several different predictive equations and found to be significantly elevated (18% to 25% above predicted) (Table 2).
Table 2 .
Thyroid status and REE in children with RTH

Appetite and food intake in RTH subjects.
Food intake was evaluated by using an ad libitum test meal, which has been previously validated in both normal and appetite-disordered individuals (12, 20). Energy intake in adults with RTH was significantly greater than in control subjects (P < 0.01; Figure 2A). During chamber calorimetry studies, food intake of RTH and control subjects was regulated and matched to their predicted total daily energy expenditure based on their REE measured previously by ventilated hood calorimetry. When fed to energy balance, subjects with RTH reported increased hunger and decreased satiety, assessed using self-completed visual analogue scales, compared with control subjects (Figure 2, B and C, respectively).
Figure 2. Energy intake in adult patients with RTH.

(A) Ad libitum energy intake (expressed as energy intake per unit LBM) assessed by a test meal in RTH adults (black circles, n = 36) compared with control subjects (gray circles, n = 35). Horizontal lines indicate the mean. (B) Visual analogue scores (VASs) of hunger in RTH (black squares, black line; n = 11) and control subjects (gray squares, gray line; n = 10) for energy neutral feeding during chamber calorimetry. (C) Visual analogue scores of satiety in RTH (black squares, black line; n = 11) and control subjects (gray squares, gray line; n = 10) for energy neutral feeding during chamber calorimetry. Arrows denote timing of meals; asterisks denote significant P values (P ≤ 0.05). Mean ± SEM.
Food intake, measured using the ad libitum test meal, was markedly increased in children with RTH versus controls (P < 0.001; Figure 3), to an extent that is comparable with individuals with monogenic obesity syndromes associated with hyperphagia (21). Furthermore, the degree by which energy intake was increased in RTH children (75% greater than control subjects) was disproportionate relative to the observed increase (~18%–25% greater than predicted; Table 2) in their REE. The macronutrient (fat/carbohydrate/protein) composition of energy intake of RTH adults and children was not significantly different from that of control subjects (data not shown).
Figure 3. Energy intake in children with RTH.

Ad libitum energy intake (expressed per unit LBM) assessed by a test meal in children with RTH (n = 12; black circles) and healthy controls (n = 16; gray circles). Horizontal lines indicate the mean.
Muscle substrate oxidation — TCA cycle flux and ATP synthesis in RTH.
A subset of adults with RTH (n = 5) underwent 13C/31P MRS studies to assess mitochondrial energy metabolism in skeletal muscle. Thyroid function, as reflected by T4 levels, was 2-fold increased, and REE was approximately 15% increased in these individuals, as compared with age- and BMI-matched control subjects (Table 3), with these changes being representative of the RTH group as a whole (Table 1 and Figure 1A). Rates of substrate oxidation via the TCA cycle were 75% higher in the muscle of RTH individuals compared with control subjects (120.7 ± 17.2 vs. 68.9 ± 5.1 nmol/(g × min); P = 0.0006; Figure 4A). Resting rates of muscle ATP synthesis, as assessed by 31P MRS, tended to be slightly lower in RTH patients compared with control subjects (P = 0.10; Figure 4B), resulting in a significant increase (~60%) in mitochondrial energy uncoupling, as assessed as the ratio between TCA cycle flux and rates of ATP synthase flux, in the RTH group as compared with the control subjects (P = 0.0056; Figure 4C).
Table 3 .
Thyroid status, ISI, and energy expenditure in adult patients with RTH and control subjects undergoing MRS

Figure 4. Mitochondrial function in patients with RTH.

(A) Rates of muscle TCA cycle flux assessed by 13C MRS in control and RTH subjects. (B) Rates of muscle ATP synthase flux assessed by 31P MRS in control and RTH subjects. (C) Muscle mitochondrial energy uncoupling as determined by the ratio of muscle TCA cycle flux to ATP synthesis in control and RTH subjects. Mean ± SEM.
Assessments of insulin sensitivity.
The homeostatic model assessment of insulin resistance (HOMA-IR) was significantly higher in the RTH patients than control subjects (P < 0.05), indicating reduced whole-body insulin sensitivity (Table 1). A subgroup of 5 RTH patients underwent an oral glucose tolerance test (OGTT) in order to assess glucose tolerance and insulin sensitivity, based on the insulin sensitivity index (ISI). Concordant with the HOMA results, the ISI (3.12 ± 0.58) tended to be lower (P = 0.11) than in control subjects and was also in the lowest ISI quartile of a normal reference population (22). These 5 RTH patients also underwent 1H MRS measurements of intramyocellular lipid (IMCL) content, and, as has been observed in other human populations, the reduced ISI in RTH patients (n = 5) was associated with 2.7-fold higher IMCL content (P = 0.0019) than control subjects (Figure 5). There is also a strong correlation between fasting plasma FFA concentrations and insulin resistance (23, 24), and our observation that plasma FFA concentrations were approximately 30% higher in RTH subjects compared with controls (P < 0.01; Table 1) may also be relevant in this context. In addition, subjects with RTH showed a fasting lipid profile (slightly elevated triglycerides and significantly reduced HDL cholesterol levels; P < 0.01; Table 1) congruent with systemic insulin resistance.
Figure 5. IMCL content in patients with RTH.

IMCL content in the soleus muscle in control and RTH subjects assessed by 1H MRS. Mean ± SEM.
Discussion
We have assessed energy balance in a cohort of adults and children with RTH, harboring THRB gene mutations, and documented significantly increased, elevated REE and BMR, using both ventilated hood and chamber calorimetry, respectively. Furthermore, the raised energy expenditure was intermediate between euthyroid control and thyrotoxic subjects. Consistent with the known role of skeletal muscle as a major contributor to whole-body energy expenditure (13), REE was highly correlated with LBM in all 3 groups. Energy expenditure did correlate with thyroid hormone levels, albeit less strongly in RTH than thyrotoxicosis; however, REE was also positively correlated with resting heart rate, and this correlation was quite similar in both groups. To investigate the role of skeletal muscle in the observed increased energy expenditure, 13C/31P MRS techniques were used to assess muscle mitochondrial energy metabolism (TCA cycle flux and ATP synthesis) in patients with RTH and age- and BMI-matched healthy control subjects. Whereas rates of muscle mitochondrial TCA cycle flux, as measured by 13C MRS, were increased by 75% (P = 0.0006) in RTH, muscle-specific ATP synthesis was slightly (nonsignificantly; P = 0.10) diminished in this group, leading overall to an approximately 60% increase in mitochondrial energy uncoupling in the RTH versus control subjects (P = 0.0056). First, these findings show that thyroid hormone excess is indeed associated with a reduction in coupling between TCA cycle activity and ATP synthesis in vivo (25), and second, they represent the first demonstration to our knowledge that chronic, endogenous thyroid hormone excess can cause these bioenergetic changes, similar to effects of short-term exogenous T3 administration to healthy volunteers (26). These observations also support the notion that skeletal muscle is less refractory to thyroid hormone action than the hypothalamic-pituitary axis in RTH. With 3 receptor subtypes (TRα1, TRβ1, and TRβ2) mediating thyroid hormone action, the relative predominance of receptor isoforms in different tissues might explain our findings. The pituitary and hypothalamus express mainly TRβ2 (1), and RTH mutations also involve this isoform, leading to defective feedback regulation in the hypothalamic-pituitary-thyroid axis and elevated thyroid hormone levels. Conversely, TRα isoform expression predominates in murine (27) and human skeletal muscle (28); GC-24, a TRβ selective agonist, is 10-fold less potent than T3 (an equipotent TRα and TRβ agonist) at inducing target gene (Serca1) expression in rodent skeletal muscle (29). We suggest that such predominance of the α receptor isoform in skeletal muscle may explain retention of sensitivity to thyroid hormone by this target tissue. We have previously described similar retention of cardiac responsiveness to elevated thyroid hormone levels in RTH subjects, possibly mediated by a preponderance of TRα expression in myocardium (30). In this context, our observation that mean heart rate and REE correlate to a similar degree in both RTH and thyrotoxicosis (Figure 1E) suggests equivalent thyroid hormone responsiveness of these tissues in the 2 states. Collectively, these observations suggest that TRα-rich organs (skeletal muscle, myocardium) are major determinants of thyroid hormone–mediated changes in whole-body energy expenditure in humans, and other lines of evidence in animals are consistent with this. GC-1 and KB-141, TRβ-selective hormone analogs, are much less potent than T3 at increasing metabolic rate in rodents (31, 32); mice lacking TRα1 but not TRβ1 exhibit lower core body temperature (33, 34); elevation of heart rate and energy expenditure by T3 is unaffected in TRβ-null mice (35) but is significantly blunted in TRα-null animals (32). However, we do not suggest that skeletal and cardiac muscle are the sole tissues mediating T3-dependent energy expenditure. At similar thyroid hormone levels, energy expenditure was more elevated in thyrotoxicosis than RTH (Figure 1, C and D). Lower SHBG levels, signifying liver thyroid hormone resistance (Table 1) and consequent reduced hepatic contribution to whole-body REE, could account for the blunted energy expenditure response in RTH. Other tissues may also be contributing to this differential response.
The molecular basis of changes in skeletal muscle energy metabolism we observed by MRS can be extrapolated from analyses of gene expression in skeletal muscle after short-term T3 treatment of human volunteers (36). Consistent with the increased TCA cycle activity observed in RTH or T3-treated subjects (26), T3 administration induces several muscle genes encoding enzymes (pyruvate dehydrogenase complex [PDK4], succinate-CoA ligase α subunit, glutamate dehydrogenase 1, 3-oxoacid CoA transferase) involved in oxidative phosphorylation (36); likewise, the muscle mitochondrial uncoupling we observed in vivo could be mediated by adenine nucleotide translocase-1 and -2 and uncoupling protein 3 (UCP3), whose expression is upregulated by T3 (36).
We observed an increase in ad libitum energy intake in RTH adults, and this may reflect either a compensatory change in food intake to maintain energy balance and/or direct effects of thyroid hormones on central pathways regulating appetitive behavior, as suggested by abnormal hunger and satiety ratings (Figure 2, B and C) in affected subjects. Children with RTH showed marked hyperphagia, comparable with levels seen in monogenic syndromes with disordered appetite, with their energy intake being disproportionately raised compared with the elevation in REE. However, RTH is known to be associated with attention-deficit hyperactivity disorder (ADHD) (15), and this behavioral abnormality was present in approximately 50% of our childhood RTH group. Physical activity is a significant component of total, free-living energy expenditure (37), and we suggest that hyperkinetic activity due to ADHD could increase this, providing an additional stimulus to enhance energy intake. The mechanisms underlying hyperphagia in hyperthyroidism, including the relative contribution of central (hypothalamic) versus peripheral pathways (e.g., adipokine, gut peptide), is unknown. Neither thyrotoxicosis nor experimental, T3-mediated hyperthyroidism are associated with consistent changes in serum leptin (38, 39), and indeed circulating leptin levels were not reduced in our RTH subjects (data not shown), arguing against a role for this peptide. Unlike T3, the administration of the TRβ-specific agonist GC-1 has no effect on food intake in rats, suggesting that the orexigenic effects of thyroid hormone may also be mediated by the TRα (40), and it is tempting to speculate that hyperphagia in RTH is mediated via elevated thyroid hormone acting centrally via this receptor isoform.
RTH subjects also exhibited insulin resistance as reflected by HOMA-IR. The ISI also tended to be lower in a smaller subset of RTH subjects, in whom we observed a 2.7-fold increase in IMCL content. Previous studies have demonstrated a strong relationship between IMCL content and insulin resistance in sedentary individuals, and it is possible that the increased IMCL content may be a contributing factor to their insulin resistance (23, 41, 42). However, the combination of increased IMCL content and muscle mitochondrial uncoupling we observed in RTH subjects is surprising, because in previous studies, transgenic mice with UCP3 overexpression in skeletal muscle were shown to be protected from insulin resistance due to decreased intramuscular diacylglycerol (DAG) concentrations associated with increased fat oxidation (43). One possible explanation for these discrepant results is that besides stimulating muscle fat oxidation and mitochondrial energy uncoupling in skeletal muscle, thyroid hormone also promotes myocellular lipogenesis by upregulation of malic enzyme gene expression, as has been documented in the liver (44–46). It is therefore possible that the rate of intramuscular lipogenesis exceeds the rate of oxidation, resulting in net accumulation of IMCL and DAG. An alternative explanation for raised IMCL content in RTH subjects is that elevated circulating FFA levels in affected subjects (Table 1) and enhanced delivery of this substrate to skeletal muscle exceed the oxidative capacity of this tissue, leading to triglyceride and DAG accumulation. A further possibility is that the liver RTH action in RTH might influence hepatic insulin sensitivity. A recent study showing that T3-responsive genes account for most of the dysregulated transcripts in the livers of obese, insulin resistant subjects (47) supports this notion. Our studies of energy metabolism may also be relevant to the management of RTH. Although, our energy expenditure data did not suggest differential peripheral tissue thyroid hormone sensitivity in subjects with PRTH- versus GRTH-associated mutations, longitudinal measurements of energy expenditure in the subset of RTH individuals who experience hyperthyroid symptoms episodically may reveal an association with some genotypes (8). Energy expenditure measurements may also be a useful parameter in titrating thyroid hormone lowering therapy of this disorder. The reduction in whole-body insulin sensitivity and dyslipidemia we have documented in this study, together with increased arterial stiffness and raised LDL cholesterol seen in another RTH group (48), suggests heightened cardiometabolic risk in this disorder, and prospective studies are needed to evaluate this further. Finally, these observations highlight the use of RTH as a human genetic model to map the relative contribution of thyroid hormone receptor isoforms in mediating hormone action in different target tissues. Such knowledge may be of particular relevance to ongoing drug development programs, which seek to develop receptor isoform and tissue-selective thyromimetics (49).
Methods
Subject recruitment.
Adults (n = 54) and children (n = 13) with RTH, harbored diverse, heterozygous mutations in the THRB gene (adults, R243W [n = 4], G251R [n = 2], A268G [n = 1], A279V [n = 1], M310T [n = 1], M310V [n = 4], A317V [n = 1], R320C [n = 5], R320H [n = 1], Y321S [n = 2], D322A [n = 1], R338W [n = 1], G347E [n = 1], D351G [n = 1], I353M [n = 1], R383C [n = 7], R383H [n = 1], R438C [n = 4], R438H [n = 5], F451S [n = 1], P453S [n = 2], P453T [n = 2], V458A [n = 1], E460G [n = 2], and E460K [n = 2]; children, R243W [n = 1], V264D [n = 1], M310T [n = 1], A317V [n = 3], D322A [n = 2], R338W [n = 3], F459L [n = 1], and P453S [n = 1]), identified as described previously (4). Subjects attended the clinical research facilities either in Cambridge, United Kingdom (n = 47) or Cardiff, United Kingdom (n = 7) for evaluation, which included their medical history, physical examination, measurement of body composition, ventilated hood indirect calorimetric measurement of REE, telemetric recording of sleeping heart rate (Actiheart, Cambridge Neurotechnology Ltd.), blood samples for measurement of biochemical, thyroid, and other metabolic parameters (Table 1) in the fasting (12-hour overnight) state and ad libitum test meal (n = 36). Adult REE data was compared with 45 healthy controls and 11 patients with conventional thyrotoxicosis (Table 1). A subset of RTH patients (n = 13) also consented to participate in measurement of BMR by chamber calorimetry, and observations were compared with age-, gender-, and BMI-matched controls (n = 22) (Figure 1B). Five adult RTH subjects (3 males and 2 females) and nineteen healthy, age-, body weight–, and BMI-matched controls (data from 15 control subjects have been reported previously; ref. 50) (Table 3) underwent MRS assessment of skeletal muscle metabolism, mitochondrial bioenergetics, and measurement of insulin resistance, using oral glucose tolerance testing (Yale University). Written consent was obtained from each subject after the purpose, nature, and potential complications of the studies were explained. The protocols were approved by the Cambridge Research Ethics Committee and the Yale University Human Investigation Committee.
Body composition.
Three-compartment body composition measurements were made by DEXA, using cross-calibrated instruments (Lunar Prodigy, GE Medical Systems for Cambridge subjects; Hologic QDR-4500/W densitometer, Hologic Inc. for Cardiff subjects), with total bone mass, LBM, and fat mass being recorded for each individual. In adults, the percentage of body fat was expressed as SDS scores, using manufacturer reference data adjusted for age, gender, BMI, and ethnicity.
Whole-body REE and BMR.
Participants were requested to refrain from strenuous exercise 24 hours prior to study and to avoid caffeinated drinks and nicotine during their research stay. REE measurement was made at thermoneutral ambient temperature, using a ventilated hood indirect calorimeter (GEM, GEMNutrition) the morning after an overnight fast of at least 12 hours and, if on any medication, 24 hours after cessation of this. During REE measurement, subjects were asked to remain awake, but relaxed and motionless for up to 20 minutes, and children were studied in the presence of a member of the study team to ensure compliance. Baseline measurements of composition of room air were made before and after each measurement period. Hood ventilation rates were adjusted to optimize measurements for each individual child or adult. BMR was measured as a component of total daily energy expenditure during whole-body indirect calorimetry, using a 24-m3 open circuit respiratory chamber. The chamber was constructed as a bed-sitting room and maintained at a constant ambient temperature of 23°C as described elsewhere (51, 52). Participants were fed to maintain energy balance (energy requirements were estimated from prior ventilated hood REE measurement) and served breakfast, lunch, and dinner of equal energy and macronutrient content. During chamber calorimetry, participants were asked to comply with a standardized timetable, incorporating both sedentary and exercise activities, and asked to quantify their perception of hunger and satiety using a linear 100-mm visual analogue scale (VAS) at specified intervals. BMR was measured between 7 am and 8 am on the 2 consecutive mornings within the calorimeter, after an overnight fast. Gas concentrations and air flow rates were recorded every 200 seconds throughout calorimetry and were retrospectively analyzed to yield macronutrient oxidation rates and energy expenditures for the specific periods of interest. Retrospective comparison of energy intake and expenditure during chamber calorimetry identified 2 underfed subjects (–0.59 MJ/d and –1.04 MJ/d), and data from these individuals was not included in VAS analyses. Energy balance for the remaining subjects averaged 0.008 ± 0.26 MJ/d. Measurements of fasting whole-body oxygen consumption and REE were repeated in 5 RTH subjects prior to them undergoing 1H/13C/31P MRS (Deltatrack II indirect calorimeter, Sensormedics).
Ad libitum energy intake.
Assessment of food intake in adults and children with RTH, controls, and subjects with appetite disorders was undertaken using an ad libitum, buffet-style breakfast meal of fixed size and macronutrient content (18 MJ; 52% carbohydrate, 30% fat, and 18% protein), which was provided between 8:00 AM and 9:00 AM after an overnight fast and carried out under standardized conditions (12). Subjects were asked to eat until they were “comfortably full.” Food was weighed before and after consumption and total energy intake and nutrient composition were calculated using standard tables (53).
OGTT.
After 12-hour overnight fast, whole-body insulin sensitivity was assessed with a 3-hour, 75 g OGTT, with data from this being used to calculate an ISI (50). Thirty minutes after insertion of an antecubital intravenous line, fasting blood samples were collected for determination of fasting plasma glucose and insulin concentrations. Blood samples were collected at 10, 20, 30, 60, 90, 120, 150, and 180 minutes after administration of dextrose load (Glucola, Curtin Matheson Scientific) for determination of plasma glucose and insulin concentrations. ISI (10–4 dl/min per μU/ml) was estimated from plasma glucose and insulin concentrations measured during the OGTT, by using the oral glucose minimal model as described below. This ISI measures overall effects of insulin to stimulate glucose disposal and inhibit glucose production (50).
Analytical methods.
For all subjects studied in the United Kingdom, thyroid biochemistry (serum fT4, fT3, and TSH) was measured using fluoroimmunometric and sensitive “second generation” assays, respectively (DELFIA, PerkinElmer Life Sciences). The intraassay and interassay coefficients of variation were less than 10% throughout. Fasting plasma glucose and serum lipid profile were measured using Siemens Dimension RXL, and fasting plasma insulin was measured using DELFIA assay (PerkinElmer Life Sciences). Subjects participating in MRS studies underwent an OGTT, with plasma glucose and insulin being measured using a YSI STAT 2700 Analyzer (YSI) and double-antibody radioimmunoassay kit (Linco), respectively.
Calculations.
Given the linear relationship between energy expenditure and metabolically active tissue mass, measured REE in adults was expressed as REE (MJ/d) per kilogram LBM (measured by DEXA) to allow comparison between individuals of different body weights and compositions. For RTH children, measured REE was compared with values derived from validated predictive equations (54–56), with results expressed as a percentage (measured/predicted REE).
Ad libitum food consumption was quantified as energy intake per unit LBM (measured by DEXA) to allow comparison between individuals of different ages and body weights, as described elsewhere (12). The homeostasis model assessment (HOMA-IR) is based on the assumption that normal-weight healthy subjects aged less than 35 years have an insulin resistance of 1 and β-cell function of 100%. HOMA-IR calculates insulin resistance and β-cell function from fasting glucose and insulin concentrations (22), using the following formula: HOMA-Ir = (FPG × FPI)/(22.5 × 18), where FPG indicates fasting plasma glucose (mg/dl) and FPI indicates fasting insulin concentrations (μU/ml).
The higher the HOMA-IR value, the more resistant an individual is to insulin. The ISI was calculated from the plasma glucose and insulin concentrations before and during the OGTT using the following formula, where FPG is the fasting plasma glucose concentration (mg/dl), FPI the fasting plasma insulin concentration (μU/ml), Ḡ is the average plasma glucose concentration during OGTT, and Ī is the average plasma insulin concentration during OGTT (22):
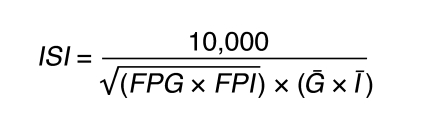
(Equation 1)
The ISI represents composite whole-body insulin sensitivity, reflecting both hepatic and peripheral tissue insulin sensitivity, with higher values reflecting increased sensitivity to insulin. Insulin resistance has been defined as an ISI in the lowest tertile of an entire population of young, lean, and healthy individuals (22).
MRS measurement.
On the day of their OGTT, 5 RTH patients and 19 control subjects were brought to the Yale University School of Medicine Magnetic Resonance Center for a measurement of the IMCL content in their right calf muscle. Within the time span of the study (from 2003–2005), the MR center acquired a new stronger magnet, and depending on the date they were performed, studies were either carried out using the 2.1T (14 controls) or the 4T magnet (5 RTH patients and 5 controls). On a different day, the RTH patients and the control subjects were brought back to the MR center for a measurement of their muscle mitochondrial function, assessed by the combined measurement of the rates of TCA cycle with 13C MRS and ATP synthase flux with 31P MRS. Examples of the MR spectra acquired are included as Supplemental Figures 1 and 2 (supplemental material available online with this article; doi: 10.1172/JCI38793DS1).
1H MRS of IMCL content.
IMCL content of the soleus muscle was measured using localized 1H MRS. Spectra were acquired on a Bruker Biospec system (Bruker BioSpin Corporation) at either 2.1T using a PRESS sequence or at 4T using a STEAM sequence. Voxel sizes were 1.5 × 1.5 × 1.5 cm3 and 1.0 × 1.0 × 1.0 cm3, respectively. Signal excitation/detection at both field strengths was achieved using twin, orthogonal 13-cm surface coils arrayed in quadrature and tuned to the appropriate 1H frequency. Detailed descriptions of these methods can be found in refs. 57 and 58. Due to methodological differences in acquiring the data using 2 different sequences at 2 different field strengths, a conversion factor was calculated from our entire database of IMCL measurements, and the data were corrected accordingly.
13C MRS measurement of TCA cycle flux.
Substrate oxidation via the TCA cycle was measured using 13C MRS to monitor the incorporation of 13C label into the muscle glutamate pool during a constant infusion of 13C labeled acetate. After an overnight fast and placement of 2 antecubital intravenous catheters, subjects were brought via wheelchair to the 2.1T or 4T MR system. Subjects lay supine in the magnet with the soleus/gastrocnemius muscle complex of the calf positioned directly over a 9-cm 13C surface coil and surrounded by a pair of 13-cm quadrature 1H surface coils, as described above, for imaging shimming and decoupling. 13C MR spectra were acquired on the 2.1T system using a nonlocalized, pulse-acquire sequence with nuclear overhauser enhancement, WALTZ16 decoupling, and suppression of the lipid signal at 34.7 ppm by T1-based nulling following an adiabatic inversion pulse (59, 60). Spectra were obtained with 10-minute time resolution at a repetition time (TR) of 1.4 seconds, corresponding to 424 averages. At 4T, spectra were acquired using a localized adiabatic 13C[1H] polarization-transfer sequence with WALTZ16 decoupling. Selection of a 90-cm3 volume within the calf muscles was achieved using 2-dimensional adiabatic outer-volume suppression. The residual lipid signal from IMCL content was suppressed using T1-selective nulling as described above (59, 60). Temporal resolution was 5 minutes, corresponding to 160 averages at a TR of 1.7 seconds. 13C MRS spectra were acquired for 20 minutes before and during an 120-minute [2-13C] acetate infusion (350 mmol × l–1 sodium salt 99% 13C enriched, Isotech) at an infusion rate of 2.9 mg × kg–1 × min–1. The increment in 13C4 glutamate enrichment during the infusion was determined by integration of the residual C4 glutamate peak in a difference spectrum, created by subtracting averaged baseline spectra from the 13C spectrum for each time point. This procedure eliminates the contribution of any residual lipid signal to the analysis. Fractional enrichments and plasma acetate were measured from blood samples collected at 10-minute intervals and analyzed on a Hewlett-Packard 5890 gas chromatography (HP-1 capillary column, 12 m × 0.2 mm × 0.33 mm film thickness), interfaced to a Hewlett-Packard 5971A mass selective detector, operating in the electron impact ionization mode (61).
TCA cycle flux was assessed by fitting the time course of C4 glutamate enrichment to a metabolic model of the TCA cycle using CWave software. The model consists of isotopic and mass balance equations that describe the metabolic fate of the plasma [2-13C]-acetate (59, 60). Briefly, following uptake by the myocyte [2-13C]-acetate is converted into acetyl CoA, which enters the TCA cycle and condenses with oxaloacetate to form [4-13C]–labeled citrate. Oxidation of citrate occurs with preservation of the 13C label, eventually forming [4-13C] α-ketoglutarate. Rapid equilibration between α-ketoglutarate and glutamate leads to the formation of [4-13C] glutamate. Entry of unlabeled substrates via acetyl CoA is incorporated into the model as a separate reaction. CWave determines the rate of total carbon flow from acetyl CoA to α-ketoglutarate using a nonlinear least-squares algorithm to fit the curve of C4-glutamate enrichment and the plasma [2-13C]-acetate concentration and enrichment as an input function (57). At the end of this study, the intravenous lines were removed and the subjects were given a 15 minute break, before the measurement of Pi → ATP flux by 31P MRS.
31P MRS measurement of Pi → ATP flux (ATP synthesis).
Unidirectional rates of Pi → ATP flux were measured using the saturation-transfer technique previously described at 2.1T (26) and reproduced at 4T. 31P MR spectra were acquired at 2.1T and 4T using a 9-cm diameter surface coil, with a concentric, outer 13-cm diameter 1H coil for scout imaging and shimming. The steady state magnetization of inorganic phosphate (Mz) was measured in the presence of a selective irradiation of the γ resonance of ATP and compared with the equilibrium magnetization of inorganic phosphate (M0) in a control spectrum without irradiation of γ-ATP. The apparent rate of longitudinal relaxation (T1*) was measured in an inversion-recovery experiment, and the inorganic phosphate concentration was calculated from a fully relaxed 31P spectrum. The total acquisition time for the 31P experiment was about 120 minutes.
Statistics.
All data are expressed as mean ± SEM. Two-tailed Student’s t tests or 1-way ANOVA with post-hoc Dunnett’s analyses were performed on data; P values of less than 0.05 were considered significant. Since each parameter in Table 1 had a specific hypothesis for testing, these data were not corrected for multiple comparisons.
Supplementary Material
Acknowledgments
The authors would like to thank Donna De’Eugenio, Gina Solomon, and the staff of the Wellcome Trust Clinical Research Facility, Cambridge, the Yale Center for Clinical Investigation, and the Clinical Research Unit, Cardiff, for expert technical assistance. This work was supported by Wellcome Trust funding (to K. Chatterjee, D. Savage, and S. Farooqi) and grants from the United States Public Health Service (R01 AG-23686, P01 DK-68229, M01 RR-00125) and the NIHR Cambridge Comprehensive Biomedical Research Centre. K. Falk Petersen and G.I. Shulman are recipients of Distinguished Clinical Scientist Awards from the American Diabetes Association. S. Dufour is a research associate and G.I. Shulman is an investigator of the Howard Hughes Medical Institute.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2010;120(4):1345–1354. doi:10.1172/JCI38793.
References
- 1.Lazar MA. Thyroid hormone receptors: multiple forms, multiple possibilities. Endocr Rev. 1993;14(2):184–193. doi: 10.1210/edrv-14-2-184. [DOI] [PubMed] [Google Scholar]
- 2.Refetoff S, Weiss RE, Usala SJ. The syndromes of resistance to thyroid hormone. Endocr Rev. 1993;14(3):348–399. doi: 10.1210/edrv-14-3-348. [DOI] [PubMed] [Google Scholar]
- 3.Parrilla R, Mixson AJ, McPherson JA, McClaskey JH, Weintraub BD. Characterization of seven novel mutations of the c-erbAb gene in unrelated kindreds with generalized thyroid hormone resistance: evidence for two “hot spot” regions of the ligand binding domain. J Clin Invest. 1991;88(6):2123–2130. doi: 10.1172/JCI115542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Adams M, et al. Genetic analysis of twenty-nine kindreds with generalised and pituitary resistance to thyroid hormone. J Clin Invest. 1994;94(2):506–515. doi: 10.1172/JCI117362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weiss RE, Weinberg M, Refetoff S. Identical mutations in unrelated families with generalized resistance to thyroid hormone occur in cytosine-guanine-rich areas of the thyroid hormone receptor b gene. J Clin Invest. 1993;91(6):2408–2415. doi: 10.1172/JCI116474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sakurai A, Miyamoto T, Refetoff S, De Groot LJ. Dominant negative transcriptional regulation by a mutant thyroid hormone receptor b in a family with generalised resistance to thyroid hormone. Mol Endocrinol. 1990;4(12):1988–1994. doi: 10.1210/mend-4-12-1988. [DOI] [PubMed] [Google Scholar]
- 7.Chatterjee VKK, et al. Thyroid hormone resistance syndrome: inhibition of normal receptor function by mutant thyroid receptors. J Clin Invest. 1991;87(6):1977–1984. doi: 10.1172/JCI115225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beck-Peccoz P, Chatterjee VK. The variable clinical phenotype in thyroid hormone resistance syndrome. Thyroid. 1994;4(2):225–232. doi: 10.1089/thy.1994.4.225. [DOI] [PubMed] [Google Scholar]
- 9.Kim B. Thyroid hormone as a determinant of energy expenditure and the basal metabolic rate. Thyroid. 2008;18(2):141–144. doi: 10.1089/thy.2007.0266. [DOI] [PubMed] [Google Scholar]
- 10.Al-Adsani H, Hoffer LJ, Silva JE. Resting energy expenditure is sensitive to small dose changes in patients on chronic thyroid hormone replacement. J Clin Endocrinol Metab. 1997;82(4):1118–1125. doi: 10.1210/jc.82.4.1118. [DOI] [PubMed] [Google Scholar]
- 11.Pijl H, et al. Food choice in hyperthyroidism: potential influence of the autonomic nervous system and brain serotonin precursor availability. J Clin Endocrinol Metab. 2001;86(12):5848–5853. doi: 10.1210/jc.86.12.5848. [DOI] [PubMed] [Google Scholar]
- 12.Farooqi IS, et al. Beneficial effects of leptin on obesity, T cell hyporesponsiveness, and neuroendocrine/metabolic dysfunction of human congenital leptin deficiency. J Clin Invest. 2002;110(8):1093–1103. doi: 10.1172/JCI15693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zurlo F, Larson K, Bogardus C, Ravussin E. Skeletal muscle metabolism is a major determinant of resting energy expenditure. J Clin Invest. 1990;86(5):1423–1427. doi: 10.1172/JCI114857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Petersen KF, et al. Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science. 2003;300(5622):1140–1142. doi: 10.1126/science.1082889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brucker-Davis F, et al. Genetic and clinical features of 42 kindreds with resistance to thyroid hormone. Ann Intern Med. 1995;123(8):572–583. doi: 10.7326/0003-4819-123-8-199510150-00002. [DOI] [PubMed] [Google Scholar]
- 16.Sarne DH, Refetoff S, Rosenfield RL, Farriaux JP. Sex hormone-binding globulin in the diagnosis of peripheral tissue resistance to thyroid hormone: the value of changes after short term triiodothyronine administration. J Clin Endocrinol Metab. 1988;66(4):740–746. doi: 10.1210/jcem-66-4-740. [DOI] [PubMed] [Google Scholar]
- 17.Hayashi Y, et al. Do clinical manifestations of resistance to thyroid hormone correlate with the functional alteration of the corresponding mutant thyroid hormone-beta receptors? J Clin Endocrinol Metab. 1995;80(11):3246–3256. doi: 10.1210/jc.80.11.3246. [DOI] [PubMed] [Google Scholar]
- 18.Mixson AJ, Renault JC, Ransom S, Bodenner DL, Weintraub BD. Identification of a novel mutation in the gene encoding the beta-triiodothyronine receptor in a patient with apparent selective pituitary resistance to thyroid hormone. Clin Endocrinol (Oxf). 1993;38(3):227–234. doi: 10.1111/j.1365-2265.1993.tb00999.x. [DOI] [PubMed] [Google Scholar]
- 19.Safer JD, et al. The thyroid hormone receptor-beta gene mutation R383H is associated with isolated central resistance to thyroid hormone. J Clin Endocrinol Metab. 1999;84(9):3099–3109. doi: 10.1210/jc.84.9.3099. [DOI] [PubMed] [Google Scholar]
- 20.Farooqi IS, Keogh JM, Yeo GS, Lank EJ, Cheetham T, O’Rahilly S. Clinical spectrum of obesity and mutations in the melanocortin 4 receptor gene. . N Engl J Med. 2003;348(12):1085–1095. doi: 10.1056/NEJMoa022050. [DOI] [PubMed] [Google Scholar]
- 21.Farooqi IS, et al. Clinical and molecular genetic spectrum of congenital deficiency of the leptin receptor. N Engl J Med. 2007;356(3):237–247. doi: 10.1056/NEJMoa063988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Petersen KF, et al. Increased prevalence of insulin resistance and nonalcoholic fatty liver disease in Asian-Indian men. Proc Natl Acad Sci U S A. 2006;103(48):18273–18277. doi: 10.1073/pnas.0608537103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krssak M, et al. Intramyocellular lipid concentrations are correlated with insulin sensitivity in humans: a 1H NMR spectroscopy study. Diabetologia. 1999;42(1):113–116. doi: 10.1007/s001250051123. [DOI] [PubMed] [Google Scholar]
- 24.Perseghin G, Ghosh S, Gerow K, Shulman GI. Metabolic defects in lean nondiabetic offspring of NIDDM parents: a cross-sectional study. Diabetes. 1997;46(6):1001–1009. doi: 10.2337/diabetes.46.6.1001. [DOI] [PubMed] [Google Scholar]
- 25.Harper ME, Brand MD. Use of top-down elasticity analysis to identify sites of thyroid hormone-induced thermogenesis. Proc Soc Exp Biol Med. 1995;208(3):228–237. doi: 10.3181/00379727-208-43852b. [DOI] [PubMed] [Google Scholar]
- 26.Lebon V, et al. Effect of triiodothyronine on mitochondrial energy coupling in human skeletal muscle. J Clin Invest. 2001;108(5):733–737. doi: 10.1172/JCI11775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bookout AL, Jeong Y, Downes M, Yu RT, Evans RM, Mangelsdorf DJ. Anatomical profiling of nuclear receptor expression reveals a hierarchical transcriptional network. Cell. 2006;126(4):789–799. doi: 10.1016/j.cell.2006.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nishimura M, Naito S, Yokoi T. Tissue-specific mRNA expression profiles of human nuclear receptor subfamilies. Drug Metab Pharmacokinet. 2004;19(2):135–149. doi: 10.2133/dmpk.19.135. [DOI] [PubMed] [Google Scholar]
- 29.Miyabara EH, et al. Thyroid hormone receptor-beta-selective agonist GC-24 spares skeletal muscle type I to II fiber shift. Cell Tissue Res. 2005;321(2):233–241. doi: 10.1007/s00441-005-1119-3. [DOI] [PubMed] [Google Scholar]
- 30.Kahaly GJ, Matthews CH, Mohr-Kahaly S, Richards CA, Chatterjee VK. Cardiac involvement in thyroid hormone resistance. J Clin Endocrinol Metab. 2002;87(1):204–212. doi: 10.1210/jcem.87.1.8170. [DOI] [PubMed] [Google Scholar]
- 31.Grover GJ, et al. Effects of the thyroid hormone receptor agonist GC-1 on metabolic rate and cholesterol in rats and primates: selective actions relative to 3,5,3′-triiodo-L-thyronine. Endocrinology. 2004;145(4):1656–1661. doi: 10.1210/en.2003-0973. [DOI] [PubMed] [Google Scholar]
- 32.Grover GJ, et al. Selective thyroid hormone receptor-beta activation: a strategy for reduction of weight, cholesterol, and lipoprotein (a) with reduced cardiovascular liability. Proc Natl Acad Sci U S A. 2003;100(17):10067–10072. doi: 10.1073/pnas.1633737100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wikstrom L, et al. Abnormal heart rate and body temperature in mice lacking thyroid hormone receptor a1. EMBO J. 1998;17(2):455–461. doi: 10.1093/emboj/17.2.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Johansson C, Gothe S, Forrest D, Vennstrom B, Thoren P. Cardiovascular phenotype and temperature control in mice lacking thyroid hormone receptor-beta or both alpha1 and beta. Am J Physiol. 1999;276(6 Pt 2):H2006–H2012. doi: 10.1152/ajpheart.1999.276.6.H2006. [DOI] [PubMed] [Google Scholar]
- 35.Weiss RE, Murata Y, Cua K, Hayashi Y, Seo H, Refetoff S. Thyroid hormone action on liver, heart and energy expenditure in thyroid hormone receptor b-deficient mice. Endocrinology. 1998;139(12):4945–4952. doi: 10.1210/en.139.12.4945. [DOI] [PubMed] [Google Scholar]
- 36.Clement K, et al. In vivo regulation of human skeletal muscle gene expression by thyroid hormone. Genome Res. 2002;12(2):281–291. doi: 10.1101/gr.207702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ravussin E, Lillioja S, Anderson TE, Christin L, Bogardus C. Determinants of 24-hour energy expenditure in man. Methods and results using a respiratory chamber. J Clin Invest. 1986;78(6):1568–1578. doi: 10.1172/JCI112749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Valcavi R, Zini M, Peino R, Casanueva FF, Dieguez C. Influence of thyroid status on serum immunoreactive leptin levels. J Clin Endocrinol Metab. 1997;82(5):1632–1634. doi: 10.1210/jcem.82.5.3954. [DOI] [PubMed] [Google Scholar]
- 39.Mantzoros CS, Rosen HN, Greenspan SL, Flier JS, Moses AC. Short-term hyperthyroidism has no effect on leptin levels in man. J Clin Endocrinol Metab. 1997;82(2):497–499. doi: 10.1210/jc.82.2.497. [DOI] [PubMed] [Google Scholar]
- 40.Villicev CM, et al. Thyroid hormone receptor beta-specific agonist GC-1 increases energy expenditure and prevents fat-mass accumulation in rats. J Endocrinol. 2007;193(1):21–29. doi: 10.1677/joe.1.07066. [DOI] [PubMed] [Google Scholar]
- 41.Sinha R, et al. Assessment of skeletal muscle triglyceride content by (1)H nuclear magnetic resonance spectroscopy in lean and obese adolescents: relationships to insulin sensitivity, total body fat, and central adiposity. Diabetes. 2002;51(4):1022–1027. doi: 10.2337/diabetes.51.4.1022. [DOI] [PubMed] [Google Scholar]
- 42.Hwang JH, Pan JW, Heydari S, Hetherington HP, Stein DT. Regional differences in intramyocellular lipids in humans observed by in vivo 1H-MR spectroscopic imaging. J Appl Physiol. 2001;90(4):1267–1274. doi: 10.1152/jappl.2001.90.4.1267. [DOI] [PubMed] [Google Scholar]
- 43.Choi CS, et al. Overexpression of uncoupling protein 3 in skeletal muscle protects against fat-induced insulin resistance. J Clin Invest. 2007;117(7):1995–2003. doi: 10.1172/JCI13579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Oppenheimer JH, Schwartz HL, Lane JT, Thompson MP. Functional relationship of thyroid hormone-induced lipogenesis, lipolysis, and thermogenesis in the rat. J Clin Invest. 1991;87(1):125–132. doi: 10.1172/JCI114961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Petersen KF, Blair JB, Shulman GI. Triiodothyronine treatment increases substrate cycling between pyruvate carboxylase and malic enzyme in perfused rat liver. Metabolism. 1995;44(11):1380–1383. doi: 10.1016/0026-0495(95)90133-7. [DOI] [PubMed] [Google Scholar]
- 46.Petersen KF, Cline GW, Blair JB, Shulman GI. Substrate cycling between pyruvate and oxaloacetate in awake normal and 3,3′-5-triiodo-L-thyronine-treated rats. Am J Physiol. 1994;267(2 Pt 1):E273–E277. doi: 10.1152/ajpendo.1994.267.2.E273. [DOI] [PubMed] [Google Scholar]
- 47.Pihlajamaki J, et al. Thyroid hormone-regulated regulation of gene expression in human fatty liver. J Clin Endocrinol Metab. 2009;94(9):3521–3529. doi: 10.1210/jc.2009-0212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Owen PJD, Chatterjee VKK, John R, Halsall D, Lazarus JH. Augmentation index in resistance to thyroid hormone. Clin Endocrinol (Oxf). 2009;70(4):650–654. doi: 10.1111/j.1365-2265.2008.03422.x. [DOI] [PubMed] [Google Scholar]
- 49.Berkenstam A, et al. The thyroid hormone mimetic compound KB2115 lowers plasma LDL cholesterol and stimulates bile acid synthesis without cardiac effects in humans. Proc Natl Acad Sci U S A. 2008;105(2):663–667. doi: 10.1073/pnas.0705286104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med. 2004;350(7):664–671. doi: 10.1056/NEJMoa031314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brown D, Cole TJ, Dauncey MJ, Marrs RW, Murgatroyd PR. Analysis of gaseous exchange in open-circuit indirect calorimetry. Med Biol Eng Comput. 1984;22(4):333–338. doi: 10.1007/BF02442102. [DOI] [PubMed] [Google Scholar]
- 52. Elia M, Livesey G. Energy expenditure and fuel selection in biological systems: the theory and practice of calculations based on indirect calorimetry and tracer methods. In:Control of Eating, Energy Expenditure and the Bioenergetics of Obesity . Simopoulos AP, ed. Basel, Switzerland: Karger; 1992:68–131. [DOI] [PubMed] [Google Scholar]
- 53. Department of Health.Dietary Reference Values for Food Energy and Nutrients for the United Kingdom: Report of the Panel on Dietary Reference Values of the Committee on Medical Aspects of Food Policy . London, United Kingdom: Stationery Office Books; 1991. [PubMed] [Google Scholar]
- 54.Schofield WN. Predicting basal metabolic rate, new standards and review of previous work. Hum Nutr Clin Nutr. 1985;39(Suppl 1):5–41. [PubMed] [Google Scholar]
- 55. Harris JA, Benedict FG.A biometric study of basal metabolism in man . Publication no. 279. Washington, DC: Carnegie Institute of Washington; 1919:40–44. [Google Scholar]
- 56.Molnar D, Jeges S, Erhardt E, Schutz Y. Measured and predicted resting metabolic rate in obese and nonobese adolescents. J Pediatr. 1995;127(4):571–577. doi: 10.1016/s0022-3476(95)70114-1. [DOI] [PubMed] [Google Scholar]
- 57.Mayerson AB, et al. The effects of rosiglitazone on insulin sensitivity, lipolysis, and hepatic and skeletal muscle triglyceride content in patients with type 2 diabetes. Diabetes. 2002;51(3):797–802. doi: 10.2337/diabetes.51.3.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Petersen KF, et al. The role of skeletal muscle insulin resistance in the pathogenesis of the metabolic syndrome. Proc Natl Acad Sci U S A. 2007;104(31):12587–12594. doi: 10.1073/pnas.0705408104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Befroy DE, Falk Petersen K, Rothman DL, Shulman GI. Assessment of in vivo mitochondrial metabolism by magnetic resonance spectroscopy. Methods Enzymol. 2009;457:373–393. doi: 10.1016/S0076-6879(09)05021-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Befroy DE, et al. Impaired mitochondrial substrate oxidation in muscle of insulin-resistant offspring of type 2 diabetic patients. Diabetes. 2007;56(5):1376–1381. doi: 10.2337/db06-0783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jucker BM, et al. Assessment of mitochondrial energy coupling in vivo by 13C/31P NMR. Proc Natl Acad Sci U S A. 2000;97(12):6880–6884. doi: 10.1073/pnas.120131997. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.