

Abstract
Bradykinin (BK) is an inflammatory mediator and one of the most potent endogenous pain-inducing substances. When released at sites of tissue damage or inflammation, or applied exogenously, BK produces acute spontaneous pain and causes hyperalgesia (increased sensitivity to potentially painful stimuli). The mechanisms underlying spontaneous pain induced by BK are poorly understood. Here we report that in small nociceptive neurons from rat dorsal root ganglia, BK, acting through its B2 receptors, PLC, and release of calcium from intracellular stores, robustly inhibits M-type K+ channels and opens Ca2+-activated Cl– channels (CaCCs) encoded by Tmem16a (also known as Ano1). Summation of these two effects accounted for the depolarization and increase in AP firing induced by BK in DRG neurons. Local injection of inhibitors of CaCC and specific M-channel openers both strongly attenuated the nociceptive effect of local injections of BK in rats. These results provide a framework for understanding spontaneous inflammatory pain and may suggest new drug targets for treatment of such pain.
Introduction
Bradykinin (BK) has long been known as one of the most potent endogenous pain-inducing (algogenic) substances (1, 2), and it is a major inflammatory mediator (3). BK and related short peptides (kinins) are produced de novo at sites of injury or inflammation from their precursors, kininogens, by serine proteases such as tissue kallikrein or by proteases released from mast cells and basophils (4). Cellular effects of BK are mediated by specific receptors, B1 and B2. B1 receptors are usually absent from noninflamed tissues and have a low affinity for BK; thus, it is believed that the majority of the acute effects of BK are mediated by B2 receptors (B2Rs), which are G protein–coupled receptors acting via Gq/11 signaling cascades (5).
When applied exogenously to a human skin blister or injected into arteries supplying viscera in laboratory animals (1), BK induces two major effects: (a) severe spontaneous pain and (b) increased sensitivity to painful (hyperalgesia) and nonpainful (allodynia) thermal and mechanical stimuli (2, 5). The latter effect has attracted considerable attention and is now relatively well understood. It is believed that B2R activation induces sensitization of sensory ion channels, including TRPV1 (6) and TRPA1 (7). Additional mechanisms for BK-induced hyperalgesia are mediated by PKCε (8), which in turn also causes TRPV1 sensitization (9). Unfortunately, the understanding of the molecular events underlying acute pain directly induced by BK is not that advanced and is mostly based on the electrophysiological observations of BK-induced depolarization (10, 11), increased action potential (AP) firing (12), and inward current induction (10, 12). BK has been referred to as “the most potent endogenous algogenic substance known” (2); thus, the elucidation of its modes and mechanisms of action should improve our ability to understand and treat spontaneous inflammatory pain.
The aim of this study was to investigate the mechanisms of acute nociceptive signaling induced by BK. We characterized the major membrane currents directly affected by BK in nociceptive sensory neurons of dorsal root ganglia (DRG), investigated their molecular identities, the signaling cascades that regulate them, and their contribution to neuronal excitability and to BK-induced nocifensive behavior in rats. Our findings indicate that the major effects of BK on the excitability of nociceptors are mediated by PLC- and Ca2+-dependent inhibition of M-type K+ channels and by simultaneous opening of Ca2+-activated Cl– channels (CaCCs) encoded by transmembrane protein 16A (Tmem16a). Both these effects contribute to membrane depolarization in primary sensory afferents, resulting in the generation of ascending nociceptive signals. Thus, this study presents a step-by-step reconstitution of the molecular events of the acute nociceptive signal induced in the peripheral sensory system by BK. Our findings also put forward new candidate drug targets for the treatment of inflammatory pain.
Results
Signaling pathways triggered in DRG neurons by BK.
It is well established that the most abundant BK receptors, B2, couple to Gq/11 and activate PLC (5). PLC hydrolyzes the plasma membrane phospholipid phosphatidylinositol 4,5-bisphosphate (PIP2) and releases inositol 1,4,5-trisphosphate (IP3) into the cytosol; the latter can then trigger exit of Ca2+ from IP3-sensitive intracellular stores. Both PIP2 depletion and Ca2+ release are powerful signaling events capable of regulating neuronal ion channels. However, Ca2+ release after PLC activation does not always occur; likewise, localized hydrolysis of PIP2 does not always lead to a global drop in the membrane PIP2 level (13, 14). Our initial approach was therefore to examine these first steps of the BK signaling cascade in DRG.
In the first series of experiments, we transfected DRG neurons with PLCδ-PH-GFP, a fluorescent probe containing the PIP2-binding domain from PLCδ fused to GFP. This probe binds to PIP2 (15) and IP3 (for which the probe has higher affinity; refs. 16, 17). As shown in the confocal images in Figure 1A, under basal conditions, the PLCδ-PH-GFP probe localized predominantly to the plasma membrane. Application of 200 nM BK produced a strong, reversible translocation of the probe to the cytosol (manifested by a 1.6 ± 0.08–fold rise in cytosolic fluorescence, n = 8; Figure 1A) in 8 of 14 small neurons (soma diameter, 20–30 μm; typical neurons selected for confocal and electrophysiological experiments are shown in Supplemental Figure 1A; supplemental material available online with this article; doi: 10.1172/JCI41084DS1). This result suggests that PLC is activated and PIP2 is hydrolyzed upon BK stimulation in DRG neurons. However, since PLCδ-PH-GFP has a higher affinity for IP3 than for PIP2, translocation of the probe does not necessarily indicate a significant drop in membrane PIP2 levels, as even small amounts of hydrolyzed PIP2 may produce enough IP3 to cause the probe to translocate (13, 18). Thus, we used a new PIP2 probe (YFP-tubby, R332H mutated), which does not bind IP3 (19), to probe whether there was a significant depletion of the membrane PIP2 pool. Like PLCδ-PH-GFP, the YFP-tubby also localized predominantly to the plasma membrane (Figure 1B); however, unlike PLCδ-PH-GFP, YFP-tubby failed to translocate to the cytosol after BK application in 11 of 12 neurons tested (see also Supplemental Figure 2). BK, however, induced a robust translocation of the YFP-tubby in DRG neurons cotransfected with the probe and exogenous B2R (Figure 1C), suggesting that this probe is capable of reporting BK-induced PIP2 depletion when it is strong enough. These results suggest that although BK clearly causes PIP2 hydrolysis and IP3 generation in DRG neurons (Figure 1A), membrane PIP2 levels do not drop to a significant enough degree to induce translocation of the YFP-tubby probe, possibly because endogenous PIP2 levels are high and/or well buffered in DRG neurons.
Figure 1. Characterization of BK-induced signaling in DRG neurons.

(A) BK-induced translocation of the IP3/PIP2 probe PLCδ-PH-GFP, overexpressed in DRG neurons. Top panels show confocal micrographs of the transfected neuron before (Control), during (BK), and after (Wash) the application of 200 nM BK. Lower panels show the corresponding surface intensity plots. Color scale is absolute pixel intensity (×102). The plot on the right shows mean (±SEM) relative increase in cytosolic fluorescence in 8 DRG neurons during BK application (indicated by the bar). (B) As in A, but using YFP-tubby probe; n = 11. (C) As in B, but neurons were cotransfected with B2R cDNA (F/F0). Scale bars in A–C: 10 μm. (D and E) BK-induced Ca2+ signals in DRG neurons. (D) [Ca2+]i was measured in small DRG neurons loaded with fura-2AM as normalized ratio (R/R0) of fluorescence at 340 and 380 nm. BK (200 nM) and CAP (1 μM) were applied during periods indicated by the bars. (E) Experiment similar to that shown in D, but Ca2+-free extracellular solution was applied (as indicated by the shaded areas) before and during the BK application.
We then performed Ca2+ imaging experiments to test whether BK application produces Ca2+ signals in DRG neurons. In fura-2AM–loaded neurons (preidentified by their response to bath application of 30 mM KCl), 200 nM BK induced prominent Ca2+ signals in 155 of 252 neurons tested (Figure 1D). These signals were usually biphasic, with a sharp transient rise followed by a slower and smaller secondary elevation (Figure 1D). Removal of extracellular Ca2+ abolished the secondary responses but not the initial transient one (Figure 1E), suggesting that the first, transient phase of the signal was predominantly due to Ca2+ release from intracellular stores, while the second, slower phase was due to Ca2+ influx pathways (see below). In all Ca2+ imaging experiments, we applied the TRPV1 agonist capsaicin (CAP, 1 μM) at the end to identify TRPV1-positive nociceptors; 174 of 252 neurons were CAP positive; 107 of 155 BK-responsive neurons were CAP positive.
BK inhibits M current in small DRG neurons.
Previous studies revealed that (a) BK induces membrane depolarization in cultured small DRG neurons (10–12); and (b) these neurons express several M-type K+ channel subunits (Kv7.2, Kv7.3, and Kv7.5; ref. 20), whose inhibition induces depolarization and increases AP firing (21, 22). Since M channels can be inhibited via PLC pathways (reviewed in ref. 23), we investigated whether BK affects M current in cultured nociceptive DRG neurons. We identified nociceptors by size and morphology (Supplemental Figure 1A) and by their sensitivity to 1 μM CAP. M currents were recorded by amphotericin B–perforated patch-clamp using a voltage protocol depicted in Figure 2A (inset) and measured as the deactivation tail current amplitude when the membrane voltage was stepped from –20 to –60 mV. Bath application of BK (10 nM–1 μM) caused a concentration-dependent inhibition of M current (Figure 2B) in the majority of the tested neurons, with an EC50 of 60.0 ± 16.3 nM. The mean inhibition of M current by 200 nM BK amounted to 43.2% ± 4.6% (n = 15) and could be partially reversed upon washout (Figure 2A). We used a specific M channel blocker, XE991 (3 μM), to verify the identity of the deactivation current (Figure 2A and thereafter); XE991 abolished the deactivation tail currents by 96.5% ± 2.1% (n = 56). In a representative population of 47 small neurons recorded under the same conditions, 28 of 47 were CAP positive, 44 of 47 were BK positive, and 26 of 28 CAP-positive neurons were BK positive. The larger proportion of BK-positive cells seen in the patch-clamp versus Ca2+ imaging experiments is likely due to the different selection procedure: small “peanut-shaped” neurons (Supplemental Figure 1A) were selected for patch clamp, while imaging was carried out with total neurons in the optical field.
Figure 2. BK inhibits M current in DRG neurons.

(A) Time course for the effect of 200 nM BK on M current amplitude (Ideac, the amplitude of the deactivating tail current at –60 mV; see Methods). A specific M channel blocker, XE991 (3 μM), was applied at the end of the recording. Drugs were applied during the periods indicated by the bars. The lower inset shows the current traces recorded at the times indicated by the letters; the dotted line indicates a zero current level. The voltage protocol is depicted in the inset above. (B) Concentration dependency of the inhibitory effect of BK on M current. The curve represents the fit of data to a logistic equation (see Methods). The n number for each data point is given in parentheses. (C–E) As in A, but neurons were pretreated with (C) the specific B2R antagonist Hoe-140 (100 nM), (D) PLC inhibitor edelfosine (10 μM), or (E) IP3 receptor blocker xestospongin C (Xe-C, 1 μM). (F) Summary of the pharmacology of BK-induced M current inhibition. Bars show percentage M current inhibition by 200 nM BK (Control); BK + 100 nM Hoe-140; BK + 10 μM edelfosine; BK + 1 μM Xe-C; BK + 2 μM thapsigargin (Thap); BK + high BAPTA pipette solution; BK + 100 nM bisindolylmaleimide (BIM); and BK + 10 μM quinacrine (Quin). The number of experiments is given above each bar; **P ≤ 0.01.
It has been reported that some BK effects in DRG cultures can be mediated by non-neuronal satellite cells (24); thus, we tested whether the action of BK on M current would also require non-neuronal cells. We prepared purified DRG cultures (Supplemental Figure 3 and Supplemental Methods) and recorded from neurons that were not in physical contact with any surrounding cells (Supplemental Figure 3B). 200 nM BK still induced inhibition of M current by 46.0% ± 3.0% (n = 8; versus inhibition under normal culture, 45.1% ± 4.5%, n = 14; P > 0.05; Supplemental Figure 3A). Thus, the action of BK on M current was due to a primary effect on DRG neurons.
Next we tested the significance of the main steps in the B2R signaling pathway for the BK-induced M current inhibition in DRG nociceptors. Pretreatment of neurons with the B2R antagonist Hoe-140 (100 nM) and the PLC inhibitor edelfosine (10 μM) abolished M current inhibition (6.3% ± 1.6% and 10.4% ± 1.6% inhibition of M current, n = 9; Figure 2, C, D, and F). BK-induced M current inhibition was also reduced by disruption of intracellular Ca2+ signaling: xestospongin C (Xe-C, a membrane-permeable IP3 receptor antagonist), thapsigargin (a sarco-/endoplasmic reticulum Ca2+-ATPase inhibitor), and chelation of intracellular Ca2+ with the high-affinity Ca2+ buffer BAPTA (20 mM; 12-minute dialysis in “fast” whole-cell mode) significantly (P ≤ 0.01) reduced M current inhibition (Figure 2, E and F). The PLA2 inhibitor quinacrine (10 μM) and the PKC inhibitor bisindolylmaleimide (100 nM) were without effect (Figure 2F). In all, the above results suggest that BK-induced M current inhibition depends on the PLC- and IP3-mediated intracellular Ca2+ rises.
BK activates TMEM16A-dependent Cl– channels.
In all our patch-clamp experiments, BK-induced inhibition of M current was always accompanied by the simultaneous activation of an inward current at –60 mV (e.g., Figure 2A, inset). The kinetics of development of inward current very closely followed that of inhibition and recovery of M current (Supplemental Figure 4); the times for activation and decay of the inward current were 93 ± 4 seconds and 275 ± 16 seconds (n = 24), respectively, and the times for maximal inhibition and recovery of the M current were 95 ± 6 seconds and 272 ± 18 seconds (n = 24), respectively (Supplemental Figure 4). However, inhibition of M current and activation of an inward current were indeed distinct phenomena, as robust inward currents were still seen when BK was applied in the presence of XE991 (Supplemental Figure 5A). We reasoned that this significant inward current is likely to have a depolarizing effect on the membrane potential of neurons and thus may contribute to the excitatory effect of BK. In order to identify this current, we first tested TRP channel involvement, since the sensitizing effect of BK on different sensory neuron–specific TRP channels (e.g., TRPV1, ref. 6; and TRPA1, ref. 7) is well documented. In small DRG neurons voltage-clamped at –60 mV, 200 nM BK evoked inward currents with an average amplitude of –231.1 ± 27.9 pA (Figure 3, A and G, n = 27); 1 μM CAP also induced inward currents of comparable amplitude (–212.7 ± 37.7 pA, n = 22; Figure 3, C and G). The effect of CAP was totally abolished by the broad-spectrum Ca2+ channel blocker ruthenium red (RR), as the CAP-evoked current in the presence of 10 μM RR amounted to only –14.2 ± 1.5 pA (n = 12, P ≤ 0.01 compared with CAP alone). In a separate imaging experiment, 10 μM RR also completely abolished Ca2+ transients induced by the TRPA1 agonist mustard oil (100 μM, Supplemental Figure 6). In contrast, RR had no effect on the BK-induced inward current (–302.8 ± 41.1 pA in the presence of RR, n = 11; Figure 3, B, D, and G). Thus, we concluded that the BK-evoked inward current was not conducted by RR-sensitive TRP channels. The inward current was also not affected by the hyperpolarization-activated cyclic nucleotide–gated cation channel (HCN) blocker ZD7288 (10 μM; –231.2 ± 74.9 pA, n = 5; Figure 3, E and G). However, lowering intracellular Cl– ([Cl–]i) by using a K acetate–based pipette solution (see Methods) almost completely abolished BK-induced inward current (–25.4 ± 4.0 pA, n = 15, P ≤ 0.01 compared with control). The broad-spectrum chloride channel blocker 4,4ι-diisothiocyanatostilbene-2, 2ι-disulphonic acid (DIDS, 100 μM) also abolished BK-induced inward current (–26.0 ± 4.9 pA, n = 17, P ≤ 0.01 compared with control; Figure 3, F and G). Other Cl– channel blockers, niflumic acid (NFA, 100 μM; Supplemental Figure 5B) and 5-nitro-2-(3-phenylpropylamino)benzoic acid (NPPB, 100 μM; data not shown), also effectively blocked BK-induced inward currents. The above results suggest that the inward current evoked by BK was almost exclusively due to chloride channel opening.
Figure 3. BK induces Ca2+ activated CaCC in DRG neurons.

(A–F) Representative current traces showing inward currents induced by BK (200 nM) and CAP (1 μM). Traces were obtained by continuous recording at a holding potential of –60 mV. Dotted lines indicate zero current level. Timing of the BK and CAP application is indicated by black bars; white bars indicate pretreatment with 10 μM Ca2+ channel blocker RR (B and D), 10 μM HCN blocker ZD7288 (E), and 100 μM Cl– channel blocker DIDS (F). (G) Summary for the pharmacology of the BK-induced inward current in DRG. Bars show the peak inward current amplitude induced by the following substances: 1 μM CAP; CAP + 10 μM RR; 200 nM BK; BK + 10 μM RR; BK + 10 μM ZD7288; BK + 100 nM Hoe-140; BK + 10 μM edelfosine; BK + 1 μM Xe-C; BK + 2 μM thapsigargin; BK + high BAPTA pipette solution; BK + low [Cl–]i pipette solution; BK + 100 μM DIDS. The number of experiments is given above each bar in parentheses; **P ≤ 0.01.
CaCCs have been reported in DRG neurons (25–27); therefore, we tested whether the BK-induced inward current in DRG neurons was mediated by the same B2R/PLC/Ca2+ pathways as the M current inhibition. The B2R antagonist Hoe-140 and PLC blocker edelfosine both prevented the inward current responses (–47.9 ± 11.7 pA, n = 6 and –45.7 ± 11.4 pA, n = 7; both P ≤ 0.01 compared with control; Figure 3G). The BK-induced inward current was also significantly attenuated by treatment with either Xe-C (–57.5 ± 4.4 pA, n = 6, P ≤ 0.01 compared with control; Figure 3G), thapsigargin (–26.2 ± 8.1 pA, n = 9, P ≤ 0.01 compared with control; Figure 3G) or 20 mM BAPTA in fast whole-cell mode (–19.8 ± 7.5 pA, n = 4, P ≤ 0.01 compared with control; Figure 3G), further suggesting that this inward current is conducted by CaCCs activated via the PLC/IP3/Ca2+ pathway.
In the experiments described above, we varied [Cl–]i in neurons using perforated patch recording with the Cl–-permeable pore-forming agent amphotericin B. In order to evaluate the endogenous electrochemical driving force for Cl–, we used gramicidin in the patch pipette, which forms Cl–-impermeable pores (28). First we confirmed that in gramicidin-perforated patch BK still induces inward currents at –60 mV (–65.3 ± 11.1 pA, n = 8; Figure 4, A and B); the BK-induced inward current recorded in gramicidin-perforated patch in combination with “low [Cl–]i” pipette solution was smaller compared with that recorded with amphotericin B with “high [Cl–]i” pipette solution (Figure 4B) but still substantial. Accordingly, the low [Cl–]i pipette solution abolished BK-induced inward current when recordings were made with amphotericin B but not with gramicidin (Figure 4B). These data suggest that [Cl–]i in DRG neurons is high enough to maintain a driving force for inward current at –60 mV. To determine the endogenous reversal potential for Cl– currents in small DRG neurons, we studied GABA-induced currents in gramicidin-perforated patch. In these experiments, NaCl in the extracellular solution and KCl in the pipette solution were substituted with equimolar tetraethylammonium chloride (TEA-Cl) and CsCl, respectively. In accordance with previously published data (29), 200 μM GABA induced Cl– currents that reversed at –37.4 ± 2.7 mV; importantly, BK-induced currents reversed at the same potentials (–34.2 ± 4.3 mV; Figure 4, C and D), further supporting our assumption that BK-induced inward currents in small DRG neurons are in fact Cl– currents.
Figure 4. Characterization of BK-induced inward currents using gramicidin-perforated patch.

(A) Representative current trace showing BK-induced (200 nM) inward current in gramicidin-perforated patch using low [Cl–]i pipette solution. (B) Summary statistics for the BK-induced inward currents recorded with amphotericin B (high [Cl–]i and low [Cl–]i pipette solutions) and gramicidin (low [Cl–]i pipette solution). **P ≤ 0.01 compared with low [Cl–]i amphotericin B. (C) Representative current traces induced in small DRG neuron by the voltage ramp from –80 mV to +40 mV (depicted above the traces) applied on the peak of BK- (200 nM) or GABA-induced (200 μM) currents. (D) Mean reversal potentials for BK- and GABA-induced currents recorded as in C. The number of experiments is given below each bar.
The gene encoding an essential CaCC subunit (Tmem16a) has recently been identified (30–32). Thus, we used siRNA against Tmem16a to test whether it is a molecular correlate of the BK-induced Cl– current in small DRG neurons. Transfection (using a Nucleofector device, Amaxa) of siRNA significantly reduced levels of Tmem16a mRNA (Figure 5F; Supplemental Figure 7, C and D) and protein (Figure 5G and Supplemental Figure 7, A and B), as compared with the levels in acutely dissociated cells and cultures from P7 and adult rats and also as compared with the cultures transfected with the scrambled oligo control. A TMEM16A antibody stained neurons coexpressing TRPV1 and the M channel subunit Kv7.2 but not satellite glial cells (Supplemental Figure 1B and Supplemental Figure 7A). In cells transfected with Tmem16a siRNA, TMEM16A but not TRPV1 immunoreactivity was reduced (Supplemental Figure 7A). Tmem16a siRNA did not affect expression of Tmem16b, which was also expressed in DRG but at a much lower level than Tmem16a (Supplemental Figure 7, C and D).
Figure 5. CaCC current in DRG neurons depends on expression of TMEM16A.

(A) Current at –60 mV induced by BK (200 nM) in small DRG neurons is attenuated by Tmem16a siRNA (right trace) but not by the scrambled siRNA control (left trace). (B) Mean peak inward current amplitudes for experiments as in A, for cells transfected with 2 different scrambled oligos (scrambled 1 and 2), mock-transfected cells, and cells transfected with Tmem16a siRNA. *P ≤ 0.05; numbers of experiments are shown in the bars. (C–E) Tmem16a siRNA does not affect BK-induced inhibition of M current. Application of 200 nM BK to the neuron transfected with Tmem16a siRNA induced robust inhibition of M current, which was recorded by the voltage pulse from –30 mV to –60 mV (inset above the current traces in D). (E) Mean M current inhibition by BK in neurons transfected with scrambled oligos or Tmem16a siRNA. (F) Tmem16a mRNA levels (normalized to the housekeeping gene U6) in DRG cultures transfected with scrambled oligo (scrambled 1) or Tmem16a siRNA, as well as in untransfected culture and acutely dissociated DRG prep from an adult (8 weeks) rat; **P ≤ 0.01. (G) Western blot showing decrease in TMEM16A protein abundance (band at 116 kDa) in whole cell lysates of DRG cells transfected with Tmem16a siRNA (as compared with the scrambled oligo control 1) and whole protein lysate from acutely extracted DRG (no cell culture) of P7 rat; loading control was β-actin.
In small DRG neurons transfected with 2 different scrambled oligos, BK induced a robust inward current with a mean amplitude of –90.5 ± 18.6 pA (scrambled 1, n = 10; Figure 6, A and B) and of –115.6 ± 22.5 pA (scrambled 2, n = 5; Figure 6B); the amplitude of the BK-induced inward current in mock-transfected neurons (cells were electroporated but without any RNA) was –103.4 ± 27.6 pA (n = 5; Figure 5B). In contrast, neurons transfected with Tmem16a siRNA responded to BK with much smaller currents (–43.8 ± 15.0 pA, or 48% of the value for the scrambled 1 oligo control, n = 12, P < 0.05; Figure 5, A and B). The inward current amplitudes in all variants of transfected controls were smaller than that recorded in nontransfected cells (Figure 3), suggesting some nonspecific effects of electroporation; nevertheless, the effect of the siRNA was specific. Furthermore, in neurons with Tmem16a knockdown, BK still induced robust inhibition of M current (Figure 5, C–E).
Figure 6. Characterization of BK-induced inward currents and Ca2+ transients at 37°C.

(A–C) Representative current traces showing inward currents at 37°C induced by 200 nM BK and 1 μM CAP. (A) In 13 of 20 CAP-positive neurons, BK did not induce inward current during recording with low [Cl–]i pipette solution. (B) In 7 of 20 neurons, BK induced small inward currents (low [Cl–]i pipette solution). (C) In 4 of 4 CAP-positive neurons, BK induced large inward currents during recording with high [Cl–]i solution. (D) Summary for experiments in A–C. (E) Exemplary Ca2+ imaging experiments (similar to that in Figure 1, D and E) at 37°C. BK (1 μM) was applied either alone (Control) or after 5 minutes preincubation with (and in the presence of) 10 μM capsazepine (CZP) or a VGCC-blocking cocktail (1 μM ω-conotoxin GVIA, 100 nM agatoxin TK, 1 μM nifedipine, 50 μM NNC 55-0396). (F) Pharmacological characterization of the second-phase Ca2+ rises induced by BK. The y axis is the relative area/peak parameter and is a measure of the second-phase Ca2+ rise (see Methods and explanation in Results). Bars represent mean area/peak parameter for Ca2+ transients induced by BK only (Control) or by BK in the presence of 10 μM RR, 10 μM CZP, VGCC-blocking cocktail, 1 mM NiCl2 (Ni2+), 100 μM NFA or VGSC blockers (1 μM TTX and 100 nM A-803467). (G) Bars represent mean normalized peak of ratiometric BK-induced Ca2+ signal (R/R0); all labeling as in F. Only CAP- and BK-positive neurons were included in E and F. The number of cells is given in parentheses. **P ≤ 0.01.
Our data strongly suggest that the BK-induced inward current in small DRG neurons is CaCC; however, BK was also shown to sensitize several sensory TRP channels such as TRPV1 (6) and TRPA1 (7) that, when open, conduct inward cationic currents at negative voltages. Particularly, sensitization of TRPV1 by BK results in a leftward shift of the temperature response curve such that the threshold for channel activation shifts closer toward body temperature (33). We thus performed further patch-clamp and imaging experiments to test the involvement of TRP channels in the acute effects of BK on DRG nociceptors at 37°C. In the majority (13 of 20) of small CAP-positive neurons, BK-induced inward currents were not seen when recorded in amphotericin B–perforated patch mode in combination with low [Cl–]i pipette solution (Figure 6, A and D). In only 7 of 20 CAP-positive neurons, BK induced small non-Cl– inward currents of –48.9 ± 7.5 pA (Figure 6, B and D); these currents were much smaller than those recorded with high [Cl–]i pipette solution (–190.2 ± 69.9 pA, 4 of 4 neurons; Figure 6, C and D). We also tested the effect of BK on DRG neurons overexpressing exogenous B2Rs. Even with B2R overexpression, application of BK induced only very small inward currents of –11.24 ± 2.23 pA (n = 8; recordings were made with low [Cl–]i pipette solution in the presence of 3 μM XE991 at room temperature). As a control for these sets of experiments, we tested whether we could detect BK-induced sensitization of TRPV1 to CAP, and indeed we found that when 2 successive applications of 50 nM CAP were separated by 5 minutes of treatment with 200 nM BK, in 6 of 9 CAP-positive neurons, the second CAP application produced 1.7 ± 0.2–fold larger current than the first CAP application (data not shown). Together, these findings suggest that coupling of endogenous B2R to sensory TRP channels in small DRG neurons is such that it provides for sensitization but less so for activation of these channels.
We also performed detailed characterization of the BK-induced Ca2+ rises at 37°C in imaging experiments (Figure 6, E–G). In order to quantify the contribution of a second-phase response to the total BK-induced Ca2+ signal, we calculated the total area under the curve of the ratiometric Ca2+ signal and divided it by the peak amplitude of Ca2+ signal (see Figure 6F, inset, and Methods); the larger the contribution of the second-phase Ca2+ rise to the total Ca2+ signal, the higher the value of this area/peak parameter. The dotted line in Figure 6F indicates the predicted value for the area/peak parameter for a single Gaussian peak of amplitude (R/R0 = 2.25) and time to peak (15 seconds), characteristic of the mean (n = 34) BK-induced Ca2+ transient in small CAP-positive DRG neurons (i.e., the value for the mean BK-induced Ca2+ peak with no second phase).
RR had no effect on either the peak Ca2+ rise or area/peak parameter (Figure 6, F and G), while the more specific TRPV1 blocker capsazepine (10 μM) had a small but significant effect on the second-phase Ca2+ rise (but not on the peak rise; Figure 6, E–G). In contrast, the nonspecific voltage-gated Ca2+ channel (VGCC) blocker Ni2+, as well as a cocktail of specific N-, P/Q-, T-, and L-type Ca2+ channel blockers and toxins (see Methods), significantly reduced both the peak and the second phase of the BK-induced Ca2+ rise (with a proportionally greater effect on the second phase; Figure 6, E–G). NFA (100 μM) also substantially reduced the second-phase and peak Ca2+ rises, suggesting that Cl– channel–mediated depolarization may contribute to these rises. These results suggest that VGCCs significantly contribute to the BK-induced Ca2+ rises, particularly to the second phase. We also tested whether AP firing is necessary for VGCC activation during the second phase of the Ca2+ response by treating neurons with a combination of tetrodotoxin (TTX, 1 μM) and A-803467 (100 nM) to block TTX-sensitive and -insensitive voltage-gated sodium channels (VGSCs) (34). This combination of VGSC inhibitors effectively blocked AP firing in small DRG neurons (data not shown); however, it did not significantly affect either the peak or the second phase of the Ca2+ signals (Figure 6G). Taken together, the results of Figure 1, D and E, and of Figure 6 suggest that the BK-induced Ca2+ signal is complex in nature and that the primary event is the release of Ca2+ from intracellular stores, which triggers a secondary Ca2+ influx, mainly through VGCCs that are activated by depolarization caused by the primary Ca2+ release. AP firing does not seem to be necessary for this VGCC activation. The contribution of TRP channels to acute BK-induced Ca2+ signals is small even at 37°C; however, the involvement of other RR-insensitive cationic channels cannot be excluded.
Since VGCCs in small DRG neurons are activated during the second phase of BK-induced Ca2+ signals, we tested whether Ca2+ influx through VGCCs can in turn activate CaCCs. Andre and colleagues have reported that in medium and large mouse DRG neurons, CaCCs indeed can be activated by VGCC-mediated Ca2+ influx; however, in small neurons, VGCC-coupled CaCC current was not found (26). In our study (and also in ref. 35), CaCC current in CAP-positive nociceptors was activated by BK-induced Ca2+ transients; we thus asked whether this CaCC current can also be activated by VGCC-mediated Ca2+ influx. We performed fast whole-cell recordings using solutions and voltage protocols as in ref. 26. Consistent with the previous study, we found that in the majority (34 of 44) of small CAP-positive neurons, VGCC activation did not result in CaCC activation (Supplemental Figure 8A). In only 10 of 44 neurons, variable inward “tail” currents developed after the VGCC-activating pre-pulse (Supplemental Figure 8, B and C); these tail currents were sensitive to 100 μM NFA (data not shown). We concluded that in most small DRG neurons (in contrast to the medium/large ones), the coupling between VGCC and CaCC is minimal, which may suggest either poor spatial colocalization of CaCCs and VGCCs or a different molecular identity of CaCCs in small compared with large DRG neurons. Of note is also the fact that our voltage protocol for measuring M current involves repetitive stepping from –20 to –60 mV, a protocol that is likely to activate VGCCs; however, in perforated patch recordings M currents usually do not exhibit noticeable rundown, suggesting that Ca2+ influx through VGCCs in DRG neurons is not capable of producing noticeable inhibition of M channels either.
All experiments on M current inhibition and CaCC activation described above were performed on cultured neurons from P7 rats. Could we apply our findings to native neurons from adult animals? Robust M currents were reported in DRG neurons from P17 (20) and from 6-week-old adult rats (21); moreover, we recently reported that M current can be recorded from acute DRG slices, and this current can be inhibited by the activation of PLC (22). However, no such data exist for TMEM16A/CaCC (although strong TMEM16A immunoreactivity in small DRG neurons of adult mice has been reported; ref. 32). Thus, we tested whether BK-induced inward current can be recorded in acutely dissociated DRG neurons from adult rats. Consistent with the data shown in Figure 3, BK induced robust Cl–-selective inward currents in small DRG neurons from 8-week-old adult rats just 4 hours after dissociation (Supplemental Figure 5B). In addition, the levels of Tmem16a transcript were similar in DRG cultures and acutely dissociated DRGs from adult rats (Figure 5F).
M current inhibition and CaCC activation contribute to and can account for the excitatory effect of BK.
We examined how the effects of BK on M channels and CaCCs affect the excitability of small DRG neurons. In control neurons under current-clamp conditions (amphotericin B–perforated patch recording with regular, high [Cl–]i pipette solution), injection of 200 pA depolarizing current usually elicited only 1 or 2 APs (Figure 7A and Supplemental Figure 9, A and B). AP firing frequency was remarkably increased by 200 nM BK from 2.0 ± 0.6 AP/500 ms to 6.6 ± 1.3 AP/500 ms (n = 7, P ≤ 0.01; Supplemental Figure 9A). BK also substantially depolarized the membrane potential from –56.8 ± 2.3 mV to –44.0 ± 3.0 mV (n = 9, P ≤ 0.01; Supplemental Figure 9C). The specific M channel blocker XE991 (3 μM) also significantly increased the AP firing frequency from 1.4 ± 0.3 AP/500 ms to 4.1 ± 0.7 AP/500 ms (n = 11, P ≤ 0.01; Supplemental Figure 9B) and induced depolarization from –55.7 ± 2.4 mV to –42.7 ± 2.5 mV (n = 15, P ≤ 0.01; Supplemental Figure 9D). However, the number of APs elicited by the application of XE991 was smaller than that induced by BK (Supplemental Figure 9, A–C) or when BK and XE991 were coapplied after the first application of XE991 only (3.0 ± 0.4 AP/500 ms in the presence of XE991 only versus 8.8 ± 2.0 AP/500 ms in the presence of BK plus XE991, n = 5, P ≤ 0.05; Figure 7A). These data suggest that both BK and XE991 can depolarize DRG neurons and increase AP firing frequency; however, under symmetrical Cl– conditions, the effect of BK is stronger and contains a component that cannot be accounted for by the inhibition of M channels.
Figure 7. M channel inhibition and CaCC activation account for the BK-induced excitability in small DRG neurons.

Current clamp recordings from small DRG neurons; APs were elicited by 200 pA current injection for a 500-ms period. (A) Under symmetrical Cl– conditions (high [Cl–]i pipette solution), XE991 (3 μM) increased the number of APs elicited by the depolarizing current injection; subsequent application of 200 nM BK (together with XE991) further increased firing frequency; n = 5. (B) Experiment similar to A, but performed with low [Cl–]i pipette solution. Under such conditions, application of BK does not cause any additional increase in XE991-induced AP firing; n = 6. (C) Another experiment with low [Cl–]i pipette solution; this time, BK was applied first. In this case, BK was able to produce some excitation; subsequent application of XE991 (in the presence of BK) produced an additional increase in firing; n = 4. Bar charts at the bottom summarize the effects shown above; *P ≤ 0.05, **P ≤ 0.01.
To test whether CaCC activation contributes to the excitatory effect of BK, we used a low [Cl–]i pipette solution, which abolished the BK-induced inward current. Under such conditions, the ability of XE991 (3 μM) to increase AP firing was unchanged (an increase from 1.4 ± 0.4 AP/500 ms to 5.0 ± 1.0 AP/500 ms, n = 6, P ≤ 0.05; Figure 7B), while subsequent coapplication of BK plus XE991 failed to produce further excitation (4.6 ± 1.1 AP/500 ms, n = 6; Figure 7B). Moreover, at low [Cl–]i, BK (which inhibits some 50% of M current), when applied alone, was less potent in increasing AP firing than BK plus XE991 (with XE991 inhibiting M current almost completely; 2.8 ± 0.3 AP/500 ms in the presence of BK only versus 4.8 ± 0.8 AP/500 ms in the presence of BK plus XE991, n = 4, P ≤ 0.05; Figure 7C). Taken together, these experiments demonstrate that both M current inhibition and CaCC activation contribute to the acute excitatory action of BK on sensory neurons; moreover, the combination of both effects can satisfactorily account for such action.
M channel openers and CaCC blockers can reduce nocifensive behavior induced by BK.
We next tested whether pharmacological targeting of M channels and CaCC can influence BK-induced nocifensive behavior in rats. We evaluated nocifensive behavior (time spent licking, biting, and flinching the affected paw) after hind paw injection of 50 μl of saline containing the relevant compounds. Consistent with previous literature (2), intraplantar injection of BK (10 nmol/site) into the rat hind paw produced strong nocifensive behavior (quantified within the first 30 minutes after injection), which was not observed in rats injected with saline (Figure 8A). In order to investigate whether M channel inhibition or CaCC activation contributes to BK-induced nocifensive behavior, we preinjected the M channel opener retigabine or different CaCC blockers into the same plantar region of the hind paw where 5 minutes later BK was injected. This protocol limits central effects of the drugs and ensures that the preinjected drugs act on the same sensory neuron terminals as BK. We first tested whether the M channel opener retigabine can reduce BK-induced nocifensive behavior. We reasoned that M channels inhibited by BK might be “rescued” with retigabine. Retigabine (5 nmol/site) indeed significantly attenuated BK-induced pain responses (Figure 8A). Accordingly, in patch-clamp experiments, application of retigabine effectively reversed the inhibitory effect of BK on M current at –60 mV (Supplemental Figure 5E).
Figure 8. M channel openers and CaCC blockers attenuate BK-induced nocifensive behavior.

(A) Cl– channel blockers NPPB (10 nmol/site), NFA (50 nmol/site), DIDS (50 nmol/site), or M channel opener retigabine (RTG, 5 nmol/site) or vehicles (DMSO, Veh1; NaHCO3, Veh2) were dissolved in saline and injected into the rat hind paw 5 minutes before the injection of BK (10 nmol/site) or saline in 50 μl (second injection was done into the same site). Nocifensive behavior was quantified as the time spent licking, biting, and flinching during 30 minutes. n = 7 for each group. (B) RR attenuated CAP- but not BK-induced nocifensive behavior. RR (50 nmol/site) or vehicle were preinjected 5 minutes prior to CAP or BK injection; n = 8 for each group. (C) Injection of M channel blocker XE991 (30 nmol/site) induced nocifensive behavior and attenuated BK-induced nociception. XE991 or vehicle control were injected into the plantar surface of the right hind paw either on their own or as pretreatment prior to BK injection; n = 7 for each group. *P ≤ 0.05; **P ≤ 0.01.
We then tested whether pharmacological inhibition of CaCC would also have an anti-nocifensive effect. Preinjection of the Cl– channel blockers DIDS (50 nmol/site) and NPPB (10 nmol/site) indeed significantly attenuated the BK-induced nocifensive behavior; NFA (50 nmol/site) also had a tendency to reduce the effect of BK (Figure 8A). We also tested the potential antinociceptive effect of RR. Intraplantar preinjection of RR (50 nmol/site) had no effect on BK-induced nociception (Figure 8B); however, as expected, RR did attenuate the nocifensive behavior produced by intraplantar injection of CAP (3 μg/site). Finally, we tested the nociceptive effect of peripheral M channel inhibition. In accordance with our previous study (22), intraplantar injections of XE991 (30 nmol/site) alone induced moderate nocifensive behavior (Figure 8C). Coinjection of XE991 with BK did not result in additional nocifensive responses; rather, there appeared to be a reduction in the pain response, which possibly may have resulted from desensitization of peripheral terminals caused by XE991-mediated sustained depolarization. These data suggest that M channel inhibition does not add any additional component to BK-induced nociception.
Discussion
M channels and BK-induced excitability in sensory neurons.
M channels (Kv7 or KCNQ) conduct slow-kinetic, non-inactivating K+ currents with a threshold for activation below –60 mV, allowing a fraction of M channels to remain open at or near the resting membrane potential of a neuron. It is therefore suggested that neuronal M current serves as an intrinsic “voltage clamp” mechanism that controls the resting membrane potential, threshold for AP firing, and accommodation within AP trains (23). Accordingly, M channel inhibition depolarizes neurons, making them more excitable, while M current augmentation has the opposite effect (20, 36–38). Growing evidence suggests that functional M channels are expressed in peripheral sensory fibers and their activity strongly contributes to fiber excitability in vivo (22, 39–42). Here we show that BK inhibits M current in nociceptors, that M current inhibition is excitatory and causes nociception, and that the nociceptive effect of BK could be attenuated by the pre-application of a specific M channel opener (Figure 8).
Gq/11-coupled receptors can inhibit M currents via PLC-induced PIP2 depletion (as M channels require PIP2 for their activity; refs. 43–45), IP3-mediated Ca2+ rises (as M channels are inhibited by Ca2+/calmodulin; refs. 46, 47), or a combination of both (48). Our data suggest that in DRG neurons, the Ca2+-mediated pathway prevails. Indeed, pharmacological inhibition of Ca2+ release from IP3-sensitive stores or chelating intracellular Ca2+ with BAPTA (Figure 2) were almost as effective in preventing BK-induced M current inhibition as direct PLC inhibition. Imaging experiments (Figure 1) also suggest that despite activation of PLC by B2R, the overall membrane PIP2 level in most DRG neurons may not change dramatically, since YFP-tubby (PIP2 reporter that does not bind IP3; ref. 19) did not translocate in response to BK application (until B2Rs were overexpressed).
New role of CaCCs in nociceptors.
CaCCs are a prominent group of ion channels robustly expressed in many mammalian cell types, including neurons, smooth muscle and epithelial cells (49). In the nervous system, functional CaCCs are best characterized in neurons mediating different sensory inputs, such as olfactory neurons (50, 51) or photosensitive rods and cones (52, 53), where these channels amplify excitatory inputs (e.g., the response to odorants mediated by Ca2+ influx through opening of CNG channels; ref. 54).
A critical prerequisite for Cl– channels to conduct a depolarizing inward current is an equilibrium potential for Cl– (ECl) that is more positive than the resting membrane potential of a neuron (–80 to –55 mV). In an archetypal CNS neuron, [Cl–]i is low and ECl is more negative than the resting membrane potential, so the activation of Cl– channels (e.g., GABA) has a hyperpolarizing (inhibitory) effect. This, however, may not hold true for many types of sensory neurons, and there is evidence that there is a higher tonic [Cl–]i in peripheral sensory neurons, leading to ECl values in the range of –50 to –10 mV (refs. 55, 56 and the present study), which is well above the resting membrane potential. In our experiments on small nociceptive DRG neurons, both GABA and CaCC currents reversed at –35 mV to –40 mV (Figure 4). A recent report suggests that even further [Cl–]i accumulation in DRG neurons occurs in response to inflammation (55). In current clamp experiments (Figure 7 and Supplemental Figure 9), we clearly observed that under high [Cl–]i conditions, CaCC activation can amplify the excitatory effect of BK. Accordingly, intraplantar preinjection of CaCC blockers attenuated BK-induced nocifensive behavior in rats (Figure 8).
The molecular identity of CaCC remained controversial until very recently, when 3 groups independently identified a member of the transmembrane protein 16 (TMEM16 or anoctamin) protein family as a CaCC subunit (30–32). Most recently, another member of this family, TMEM16B was also shown to have CaCC activity (57). We still know little about the structural basis for the Cl– conductance and Ca2+ sensitivity of TMEM16A, but its identification as a molecular correlate of CaCC makes it possible to alter CaCCs in native cells via genetic manipulation. In this study, we detected TMEM16A immunoreactivity in TRPV1-positive DRG neurons (Supplemental Figures 1 and 7) and used the siRNA approach to show that Tmem16a knockdown reduces CaCC currents in these cells (Figure 5). We also detected Tmem16b expression in total DRG culture extracts, but its level of expression was approximately 100 times lower than that of Tmem16a.
An apparently different type of endogenous CaCC current has been reported in medium/large DRG neurons. This CaCC current was reported to couple to VGCC activation (26), while in the case of CaCC in small DRG neurons, such coupling is weak (the present study). Such a lack of coupling between CaCC and VGCC in nociceptors may be required in order to avoid a self-maintaining positive feedback loop, since if Ca2+ influx through VGCC would significantly activate CaCC (and inhibit M channels), this would then cause depolarization and Ca2+ influx through VGCC and further amplification of the cycle. Interestingly, Tmem16a is highly expressed in the majority of small DRG neurons but not in large neurons (32); moreover, it was recently suggested that Best1 underlies CaCC in large neurons (58). Taken together, these two observations may indicate that CaCC in different types of neurons consist of distinct molecular components.
Contribution of other currents to the acute effect of BK.
In our experiments, the inhibition of M current and activation of CaCC were the most prominent effects of BK on ionic currents in DRG neurons; moreover, these effects satisfactorily accounted for the observed excitatory action of BK. Our data suggest that the contribution of TRP channels to the acute effects of BK in nociceptors is not large, as the non-Cl– component of the BK-induced inward currents was small even at 37°C (Figure 6); overexpression of B2Rs in DRG neurons also did not result in large non-Cl– BK-activated currents; finally, TRP channel blockers had little effect on BK-induced Ca2+ transients (Figure 6) or nociceptive behavior (Figure 8). Our results seem at odds with publications suggesting that BK can activate TRPV1 (59, 60) and TRPA1 (61). Nevertheless we confirmed the sensitizing effect of BK on TRPV1 via endogenous B2Rs; moreover, small non-Cl– inward currents in response to BK recorded at 37°C suggest that some degree of sensory TRP channel activation by BK may happen in vivo. We believe that in our experimental conditions, B2R indeed produced some sensitization of sensory TRP channels, but the overall contribution of this effect to the acute BK-induced excitability and nociception was small. A recent report suggested that acute BK-induced nocifensive behavior in Trpv1-knockout mice was affected only marginally (62). The study tested two doses of BK, 0.1 and 1 nmol/site; the behavioral responses to BK did not differ between wild-type and Trpv1–/– animals at a higher BK dose, and at a lower dose the percentage of BK-responsive animals was smaller in the Trpv1–/– group, but the amount of distress experienced by the responsive animals did not change significantly. Moreover, BK-induced excitation of cutaneous C fibers was similar in wild-type and Trpv1–/– mice at a range of BK concentrations. Thus, we suggest that B2R-TRP coupling may have a stronger role in the BK-induced hyperalgesia/allodynia than in the acute nociception produced by BK.
General principles of spontaneous inflammatory nociception.
Inflammatory mediators (such as BK, proteases, histamine, prostaglandins, IL1-β, ATP, NGF, etc.) often act via G protein–coupled receptors or ligand-gated ion channels to acutely depolarize the plasma membrane of sensory fibers, reduce the threshold for AP firing, and increase firing frequency (3). Our data as well as emerging work from many other research groups allow us to propose certain common principles underlying the “spontaneous” excitation of peripheral sensory neurons in inflammation: (a) weakening of intrinsic “voltage clamp” — e.g., through M channel inhibition (ref. 22 and the present study); (b) depolarizing input — e.g., through activation of cationic channels such as P2X (63) and TRP (61, 64) or anionic channels such as CaCCs (the present study); (c) amplification of the depolarizing input — e.g., by the upregulation of VGSCs (65, 66) or by positive feedback activation of CaCCs.
Methods
The use of animals in this study was approved by the University of Leeds Ethical Review Committee and by the Animal Care and Ethical Committee of Hebei Medical University.
Rat DRG cell culture.
DRGs were extracted from all spinal levels of 7-day-old Wistar rats, and neurons were dissociated and cultured as described previously (22, 67). For preparation of purified DRG culture, see Supplemental Methods.
Electrophysiology.
Unless stated otherwise, recordings were made at room temperature. Patch electrodes were pulled from borosilicate glass and fire-polished to a final resistance of 2–4 MΩ when filled with internal solution. An EPC-10 patch clamp amplifier in combination with Patchmaster version 2.2 software (HEKA) was used for voltage and current clamp experiments. Most recordings were done using either amphotericin B– (250 μg/ml; Sigma-Aldrich) or gramicidin-perforated (100 μg/ml; Sigma-Aldrich) patch technique. Regular (high [Cl–]i) internal pipette solution contained (in mM): 150 KCl, 5 MgCl2, 10 HEPES, pH 7.4. In the low [Cl–]i pipette solution, KCl was replaced by equimolar K-acetate. Calcium clamping experiments were done using fast whole-cell recording using the following “high BAPTA” internal solution (in mM): 115 KCl, 5 MgCl2, 5 HEPES, 20 BAPTA, 3 K2ATP, 0.1 NaGTP, and 10 CaCl2, pH 7.4. The external solution contained (in mM) 160 NaCl, 2.5 KCl, 5 CaCl2, 1 MgCl2, 10 HEPES, and 8 glucose, pH 7.4. A low-profile perfusion chamber fed by a gravity perfusion system was used for solution exchange (2 ml/min, bath exchange time of ~15 seconds). M current amplitude was measured from the deactivation current elicited by a 500-ms square voltage pulse to –60 mV from a holding potential of –20 mV or –30 mV, calculated as the difference between the average of a 10-ms segment, taken 20–30 ms into the hyperpolarizing step, and the average during the last 50 ms of that step. Additional details are provided in Supplemental Methods.
Fluorescence imaging.
Ca2+ imaging was performed as described previously (46); briefly, neurons were loaded with fura-2AM (2 μM) in the presence of Pluronic F-127 (0.02%) and imaged using a Nikon TE-2000 microscope (10× objective) equipped with a T.I.L.L. Photonics Ca2+ imaging system (Polychrome V monochromator and IMAGO CCD camera). Cells were excited alternatively at 340 and 380 nm and images analyzed with TILLvisION 4.5.56. Only neurons responding with a greater than 10% rise (peak) in F340/F380 were counted as BK positive. The area/peak parameter in Figure 6F was calculated according to the following equation:
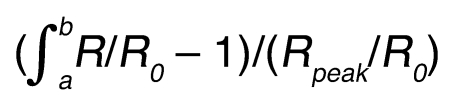
(Equation 1)
where a and b are the start and end points of the Ca2+ transient (Figure 6F, inset).
Translocation of PIP2 probes was recorded using a LiveScan Swept Field Confocal System (Nikon) as in ref. 22, and data were analyzed using NIS Elements imaging software (Nikon).
DNA constructs, siRNA, and transfections.
Freshly isolated DRG neurons were transfected with PLCδ-PH-GFP (gift from Tobias Meyer, Stanford University, California) or YFP-tubby (tubby-R332H-cYFP; gift from Andrew Tinker, UCL, London, United Kingdom) with or without B2R (gift from Mark Shapiro, UT Health Science Center San Antonio, Texas) using a Nucleofector device (Amaxa) and used 3–5 days after transfection. siRNA oligonucleotide sequences against Tmem16a (with matching scrambled controls) were designed using BLOCK-iT RNAi designer (Invitrogen): siRNA sense, CCUGGUCAGGAAAUACUUU[dT][dT], antisense, AAAGUAUUUCCUGACCAGG[dT][dT]; scrambled 1 sense, CCUACUAGGUAACAGGUUU[dT][dT], antisense, AAACCUGUUACCUAGUAGG[dT][dT]; scrambled 2 sense, GCAGUAUUCCGAUAGACUU[dT][dT], antisense, AAGUCUAUCGGAAUACUGC[dT][dT]. Amaxa’s Nucleofector device was used for siRNA delivery; the efficiency of Tmem16a knockdown was checked by quantitative RT-PCR (Bio-Rad), immunocytochemistry, and Western blot analysis (see Supplemental Methods). In patch-clamp experiments, RNA oligos were cotransfected with EGFP cDNA, and neurons with green fluorescence were analyzed.
Behavioral studies.
Sprague-Dawley rats (body weight, 190–240 g) were randomly grouped and allowed to acclimatize for at least 20 minutes in a transparent observation chamber before the experiment. The right hind paw of the animal received an intraplantar injection (50 μl) of BK (10 nmol/site) or vehicle, and the nocifensive responses (licking, biting, lifting, and flinching) were recorded using a video camera for 30 minutes. The videos were analyzed by an observer unaware of treatment allocations. In the experiments with M channel openers and CaCC or TRP blockers, these drugs were preinjected in the same plantar of the hind paw where 5 minutes later BK was injected. Drugs were dissolved in saline (pH 7.4) from stock solutions and applied in a volume of 50 μl at the following doses: retigabine, 5 nmol/site; DIDS, 50 nmol/site; NPPB, 10 nmol/site; NFA, 50 nmol/site; CAP, 3 μg/site; RR, 50 nmol/site; XE991, 30 nmol/site.
Statistics.
The concentration-response curve was fitted with logistic equation: y = A2 + (A1 – A2)/[1 + (x/x0)p], where y is the response; A1 and A2 are the maximum and minimum response, respectively, x is the drug concentration, and p is the Hill coefficient. All data are given as mean ± SEM. Differences between groups were assessed by Student’s t test or 1-way ANOVA. The differences were considered significant at P ≤ 0.05.
Chemicals.
Edelfosine was from Calbiochem; XE991 and ZD7288, from Tocris; Xe-C, from Cayman Europe; and fura-2AM, from Invitrogen. Retigabine was a gift from Kewei Wang (Peking University, Beijing, China); all other chemicals were from Sigma-Aldrich.
Supplementary Material
Acknowledgments
We thank Hannah Kirton and Katarzyna Marszalek for expert technical assistance and Mark Shapiro for helpful discussions. This work was supported by grants from the Wellcome Trust (080593/Z06/Z, 070740 and 080833/Z/06/Z) and the MRC (grant 82507) to N. Gamper and by the National Natural Science Foundation of China (30730031 to H. Zhang and 30500112 to X. Du) and the National Basic Research Program (grant 2007CB512100) to H. Zhang. H. Zhang was a recipient of a grant from the National Science Fund for Distinguished Young Scholars of China (no. 30325038). N. Gamper and H. Zhang received an international joint project grant from the Royal Society (JP080524) and a National Science Foundation of China grant (no. 30911130137). B. Liu received a fellowship grant from the China-UK Joint Laboratory in Membrane Biology.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2010;120(4):1240–1252. doi:10.1172/JCI41084.
Boyi Liu’s present address is: Department of Pharmacology, Yale University School of Medicine, New Haven, Connecticut.
References
- 1.Keele CA. The chemistry of pain production. Proc R Soc Med. 1967;60(4):419–422. doi: 10.1177/003591576706000434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dray A, Perkins M. Bradykinin and inflammatory pain. Trends Neurosci. 1993;16(3):99–104. doi: 10.1016/0166-2236(93)90133-7. [DOI] [PubMed] [Google Scholar]
- 3. McMahon SB, Bennet DLH, Bevan S. Inflammatory mediators and modulators. In: McMahon SB, Koltzenburg M, eds.Wall and Melzack’s Textbook of Pain. Edinburgh, United Kingdom: Elsevier Churchill Livingstone. 2006:49–72. [Google Scholar]
- 4.Raidoo DM, Bhoola KD. Pathophysiology of the kallikrein-kinin system in mammalian nervous tissue. Pharmacol Ther. 1998;79(2):105–127. doi: 10.1016/S0163-7258(98)00011-4. [DOI] [PubMed] [Google Scholar]
- 5.Couture R, Harrisson M, Vianna RM, Cloutier F. Kinin receptors in pain and inflammation. Eur J Pharmacol. 2001;429(1–3):161–176. doi: 10.1016/S0014-2999(01)01318-8. [DOI] [PubMed] [Google Scholar]
- 6.Chuang HH, et al. Bradykinin and nerve growth factor release the capsaicin receptor from PtdIns(4,5)P2-mediated inhibition. . Nature. 2001;411(6840):957–962. doi: 10.1038/35082088. [DOI] [PubMed] [Google Scholar]
- 7.Wang S, et al. Phospholipase C and protein kinase A mediate bradykinin sensitization of TRPA1: a molecular mechanism of inflammatory pain. Brain. 2008;131(Pt 5):1241–1251. doi: 10.1093/brain/awn060. [DOI] [PubMed] [Google Scholar]
- 8.Cesare P, Dekker LV, Sardini A, Parker PJ, McNaughton PA. Specific involvement of PKC-epsilon in sensitization of the neuronal response to painful heat. Neuron. 1999;23(3):617–624. doi: 10.1016/S0896-6273(00)80813-2. [DOI] [PubMed] [Google Scholar]
- 9.Mandadi S, et al. Increased sensitivity of desensitized TRPV1 by PMA occurs through PKCepsilon-mediated phosphorylation at S800. Pain. 2006;123(1–2):106–116. doi: 10.1016/j.pain.2006.02.016. [DOI] [PubMed] [Google Scholar]
- 10.Burgess GM, Mullaney I, McNeill M, Dunn PM, Rang HP. Second messengers involved in the mechanism of action of bradykinin in sensory neurons in culture. J Neurosci. 1989;9(9):3314–3325. doi: 10.1523/JNEUROSCI.09-09-03314.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dray A, Patel IA, Perkins MN, Rueff A. Bradykinin-induced activation of nociceptors: receptor and mechanistic studies on the neonatal rat spinal cord-tail preparation in vitro. Br J Pharmacol. 1992;107(4):1129–1134. doi: 10.1111/j.1476-5381.1992.tb13418.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oh EJ, Weinreich D. Bradykinin decreases K+ and increases Cl– conductances in vagal afferent neurones of the guinea pig. . J Physiol. 2004;558(Pt 2):513–526. doi: 10.1113/jphysiol.2004.066381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gamper N, Shapiro MS. Target-specific PIP2 signaling: how might it work? . J Physiol. 2007;582(Pt 3):967–975. doi: 10.1113/jphysiol.2007.132787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gamper N, Shapiro MS. Regulation of ion transport proteins by membrane phosphoinositides. Nat Rev Neurosci. 2007;8(12):921–934. doi: 10.1038/nrn2257. [DOI] [PubMed] [Google Scholar]
- 15.Stauffer TP, Ahn S, Meyer T. Receptor-induced transient reduction in plasma membrane PtdIns(4,5)P2 concentration monitored in living cells. . Curr Biol. 1998;8(6):343–346. doi: 10.1016/S0960-9822(98)70135-6. [DOI] [PubMed] [Google Scholar]
- 16.Hirose K, Kadowaki S, Tanabe M, Takeshima H, Iino M. Spatiotemporal dynamics of inositol 1,4,5-trisphosphate that underlies complex Ca2+ mobilization patterns. . Science. 1999;284(5419):1527–1530. doi: 10.1126/science.284.5419.1527. [DOI] [PubMed] [Google Scholar]
- 17.Varnai P, Balla T. Visualization of phosphoinositides that bind pleckstrin homology domains: calcium- and agonist-induced dynamic changes and relationship to myo-[3H]inositol-labeled phosphoinositide pools. J Cell Biol. 1998;143(2):501–510. doi: 10.1083/jcb.143.2.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gamper N, Reznikov V, Yamada Y, Yang J, Shapiro MS. Phosphatidylinositol 4,5-bisphosphate signals underlie receptor-specific Gq/11-mediated modulation of N-type Ca2+ channels. . J Neurosci. 2004;24(48):10980–10992. doi: 10.1523/JNEUROSCI.3869-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Quinn KV, Behe P, Tinker A. Monitoring changes in membrane phosphatidylinositol 4,5-bisphosphate in living cells using a domain from the transcription factor tubby. J Physiol. 2008;586(Pt 12):2855–2871. doi: 10.1113/jphysiol.2008.153791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Passmore GM, et al. KCNQ/M currents in sensory neurons: significance for pain therapy. J Neurosci. 2003;23(18):7227–7236. doi: 10.1523/JNEUROSCI.23-18-07227.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Crozier RA, Ajit SK, Kaftan EJ, Pausch MH. MrgD activation inhibits KCNQ/M-currents and contributes to enhanced neuronal excitability. J Neurosci. 2007;27(16):4492–4496. doi: 10.1523/JNEUROSCI.4932-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Linley JE, Rose K, Patil M, Robertson B, Akopian AN, Gamper N. Inhibition of M current in sensory neurons by exogenous proteases: a signaling pathway mediating inflammatory nociception. J Neurosci. 2008;28(44):11240–11249. doi: 10.1523/JNEUROSCI.2297-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Delmas P, Brown DA. Pathways modulating neural KCNQ/M (Kv7) potassium channels. Nat Rev Neurosci. 2005;6(11):850–862. doi: 10.1038/nrn1785. [DOI] [PubMed] [Google Scholar]
- 24.Heblich F, England S, Docherty RJ. Indirect actions of bradykinin on neonatal rat dorsal root ganglion neurones: a role for non-neuronal cells as nociceptors. J Physiol. 2001;536(Pt 1):111–121. doi: 10.1111/j.1469-7793.2001.00111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mayer ML. A calcium-activated chloride current generates the after-depolarization of rat sensory neurones in culture. J Physiol. 1985;364:217–239. doi: 10.1113/jphysiol.1985.sp015740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Andre S, et al. Axotomy-induced expression of calcium-activated chloride current in subpopulations of mouse dorsal root ganglion neurons. J Neurophysiol. 2003;90(6):3764–3773. doi: 10.1152/jn.00449.2003. [DOI] [PubMed] [Google Scholar]
- 27.Ward SM, Kenyon JL. The spatial relationship between Ca2+ channels and Ca2+-activated channels and the function of Ca2+-buffering in avian sensory neurons. . Cell Calcium. 2000;28(4):233–246. doi: 10.1054/ceca.2000.0151. [DOI] [PubMed] [Google Scholar]
- 28.Ebihara S, Shirato K, Harata N, Akaike N. Gramicidin-perforated patch recording: GABA response in mammalian neurones with intact intracellular chloride. J Physiol. 1995;484(Pt 1):77–86. doi: 10.1113/jphysiol.1995.sp020649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chabwine JN, et al. Differential contribution of the Na+-K+-2Cl– cotransporter NKCC1 to chloride handling in rat embryonic dorsal root ganglion neurons and motor neurons. . FASEB J. 2009;23(4):1168–1176. doi: 10.1096/fj.08-116012. [DOI] [PubMed] [Google Scholar]
- 30.Caputo A, et al. TMEM16A, a membrane protein associated with calcium-dependent chloride channel activity. Science. 2008;322(5901):590–594. doi: 10.1126/science.1163518. [DOI] [PubMed] [Google Scholar]
- 31.Schroeder BC, Cheng T, Jan YN, Jan LY. Expression cloning of TMEM16A as a calcium-activated chloride channel subunit. Cell. 2008;134(6):1019–1029. doi: 10.1016/j.cell.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang YD, et al. TMEM16A confers receptor-activated calcium-dependent chloride conductance. Nature. 2008;455(7217):1210–1215. doi: 10.1038/nature07313. [DOI] [PubMed] [Google Scholar]
- 33.Sugiura T, Tominaga M, Katsuya H, Mizumura K. Bradykinin lowers the threshold temperature for heat activation of vanilloid receptor 1. J Neurophysiol. 2002;88(1):544–548. doi: 10.1152/jn.2002.88.1.544. [DOI] [PubMed] [Google Scholar]
- 34.Jarvis MF, et al. A-803467, a potent and selective Nav1.8 sodium channel blocker, attenuates neuropathic and inflammatory pain in the rat. Proc Natl Acad Sci U S A. 2007;104(20):8520–8525. doi: 10.1073/pnas.0611364104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee MG, Macglashan DW, Jr, Undem BJ. Role of chloride channels in bradykinin-induced guinea pig airway vagal C-fibre activation. J Physiol. 2005;566(Pt 1):205–212. doi: 10.1113/jphysiol.2005.087577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gamper N, et al. Oxidative modification of M-type K+ channels as a mechanism of cytoprotective neuronal silencing. . EMBO J. 2006;25(20):4996–5004. doi: 10.1038/sj.emboj.7601374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jones S, Brown DA, Milligan G, Willer E, Buckley NJ, Caulfield MP. Bradykinin excites rat sympathetic neurons by inhibition of M current through a mechanism involving B2 receptors and G alpha q/11. Neuron. 1995;14(2):399–405. doi: 10.1016/0896-6273(95)90295-3. [DOI] [PubMed] [Google Scholar]
- 38.Shah MM, Migliore M, Valencia I, Cooper EC, Brown DA. Functional significance of axonal Kv7 channels in hippocampal pyramidal neurons. Proc Natl Acad Sci U S A. 2008;105(22):7869–7874. doi: 10.1073/pnas.0802805105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Blackburn-Munro G, Jensen BS. The anticonvulsant retigabine attenuates nociceptive behaviours in rat models of persistent and neuropathic pain. Eur J Pharmacol. 2003;460(2-3):109–116. doi: 10.1016/S0014-2999(02)02924-2. [DOI] [PubMed] [Google Scholar]
- 40.Hirano K, Kuratani K, Fujiyoshi M, Tashiro N, Hayashi E, Kinoshita M. Kv7.2-7.5 voltage-gated potassium channel (KCNQ2-5) opener, retigabine, reduces capsaicin-induced visceral pain in mice. Neurosci Lett. 2007;413(2):159–162. doi: 10.1016/j.neulet.2006.11.043. [DOI] [PubMed] [Google Scholar]
- 41.Lang PM, Fleckenstein J, Passmore GM, Brown DA, Grafe P. Retigabine reduces the excitability of unmyelinated peripheral human axons. Neuropharmacology. 2008;54(8):1271–1278. doi: 10.1016/j.neuropharm.2008.04.006. [DOI] [PubMed] [Google Scholar]
- 42.Roza C, Lopez-Garcia JA. Retigabine, the specific KCNQ channel opener, blocks ectopic discharges in axotomized sensory fibres. Pain. 2008;138(3):537–545. doi: 10.1016/j.pain.2008.01.031. [DOI] [PubMed] [Google Scholar]
- 43.Li Y, Gamper N, Hilgemann DW, Shapiro MS. Regulation of Kv7 (KCNQ) K+ channel open probability by phosphatidylinositol (4,5)-bisphosphate. . J Neurosci. 2005;25(43):9825–9835. doi: 10.1523/JNEUROSCI.2597-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Suh B, Hille B. Recovery from muscarinic modulation of M current channels requires phosphatidylinositol 4,5-bisphosphate synthesis. Neuron. 2002;35(3):507–520. doi: 10.1016/S0896-6273(02)00790-0. [DOI] [PubMed] [Google Scholar]
- 45.Zhang H, et al. PIP2 activates KCNQ channels, and its hydrolysis underlies receptor-mediated inhibition of M currents. . Neuron. 2003;37(6):963–975. doi: 10.1016/S0896-6273(03)00125-9. [DOI] [PubMed] [Google Scholar]
- 46.Gamper N, Shapiro MS. Calmodulin mediates Ca2+-dependent modulation of M-type K+ channels. . J Gen Physiol. 2003;122(1):17–31. doi: 10.1085/jgp.200208783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Selyanko AA, Brown DA. Intracellular calcium directly inhibits potassium M channels in excised membrane patches from rat sympathetic neurons. Neuron. 1996;16(1):151–162. doi: 10.1016/S0896-6273(00)80032-X. [DOI] [PubMed] [Google Scholar]
- 48.Brown DA, Hughes SA, Marsh SJ, Tinker A. Regulation of M(Kv7.2/7.3) channels in neurons by PIP2 and products of PIP2 hydrolysis: significance for receptor-mediated inhibition. . J Physiol. 2007;582(Pt 3):917–925. doi: 10.1113/jphysiol.2007.132498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Frings S, Reuter D, Kleene SJ. Neuronal Ca2+ -activated Cl– channels — homing in on an elusive channel species. . Prog Neurobiol. 2000;60(3):247–289. doi: 10.1016/S0301-0082(99)00027-1. [DOI] [PubMed] [Google Scholar]
- 50.Kleene SJ, Gesteland RC. Calcium-activated chloride conductance in frog olfactory cilia. J Neurosci. 1991;11(11):3624–3629. doi: 10.1523/JNEUROSCI.11-11-03624.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lowe G, Gold GH. Nonlinear amplification by calcium-dependent chloride channels in olfactory receptor cells. Nature. 1993;366(6452):283–286. doi: 10.1038/366283a0. [DOI] [PubMed] [Google Scholar]
- 52.Bader CR, Bertrand D, Schwartz EA. Voltage-activated and calcium-activated currents studied in solitary rod inner segments from the salamander retina. J Physiol. 1982;331:253–284. doi: 10.1113/jphysiol.1982.sp014372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Barnes S, Hille B. Ionic channels of the inner segment of tiger salamander cone photoreceptors. J Gen Physiol. 1989;94(4):719–743. doi: 10.1085/jgp.94.4.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Menini A. Calcium signalling and regulation in olfactory neurons. Curr Opin Neurobiol. 1999;9(4):419–426. doi: 10.1016/S0959-4388(99)80063-4. [DOI] [PubMed] [Google Scholar]
- 55.Funk K, Woitecki A, Franjic-Wurtz C, Gensch T, Mohrlen F, Frings S. Modulation of chloride homeostasis by inflammatory mediators in dorsal root ganglion neurons. Mol Pain. 2008;4:32. doi: 10.1186/1744-8069-4-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gilbert D, Franjic-Wurtz C, Funk K, Gensch T, Frings S, Mohrlen F. Differential maturation of chloride homeostasis in primary afferent neurons of the somatosensory system. Int J Dev Neurosci. 2007;25(7):479–489. doi: 10.1016/j.ijdevneu.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 57.Stohr H, et al. TMEM16B, a novel protein with calcium-dependent chloride channel activity, associates with a presynaptic protein complex in photoreceptor terminals. J Neurosci. 2009;29(21):6809–6818. doi: 10.1523/JNEUROSCI.5546-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Boudes M, et al. Best1 is a gene regulated by nerve injury and required for Ca2+-activated Cl– current expression in axotomized sensory neurons. . J Neurosci. 2009;29(32):10063–10071. doi: 10.1523/JNEUROSCI.1312-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Premkumar LS, Ahern GP. Induction of vanilloid receptor channel activity by protein kinase C. Nature. 2000;408(6815):985–990. doi: 10.1038/35050121. [DOI] [PubMed] [Google Scholar]
- 60.Shin J, et al. Bradykinin-12-lipoxygenase-VR1 signaling pathway for inflammatory hyperalgesia. Proc Natl Acad Sci U S A. 2002;99(15):10150–10155. doi: 10.1073/pnas.152002699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bandell M, et al. Noxious cold ion channel TRPA1 is activated by pungent compounds and bradykinin. Neuron. 2004;41(6):849–857. doi: 10.1016/S0896-6273(04)00150-3. [DOI] [PubMed] [Google Scholar]
- 62.Katanosaka K, et al. Contribution of TRPV1 to the bradykinin-evoked nociceptive behavior and excitation of cutaneous sensory neurons. Neurosci Res. 2008;62(3):168–175. doi: 10.1016/j.neures.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 63.Xu GY, Huang LY. Peripheral inflammation sensitizes P2X receptor-mediated responses in rat dorsal root ganglion neurons. J Neurosci. 2002;22(1):93–102. doi: 10.1523/JNEUROSCI.22-01-00093.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dai Y, et al. Proteinase-activated receptor 2-mediated potentiation of transient receptor potential vanilloid subfamily 1 activity reveals a mechanism for proteinase-induced inflammatory pain. J Neurosci. 2004;24(18):4293–4299. doi: 10.1523/JNEUROSCI.0454-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Maingret F, et al. Inflammatory mediators increase Nav1.9 current and excitability in nociceptors through a coincident detection mechanism. J Gen Physiol. 2008;131(3):211–225. doi: 10.1085/jgp.200709935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Binshtok AM, et al. Nociceptors are interleukin-1beta sensors. J Neurosci. 2008;28(52):14062–14073. doi: 10.1523/JNEUROSCI.3795-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liu B, Liang H, Liu L, Zhang H. Phosphatidylinositol 4,5-bisphosphate hydrolysis mediates histamine-induced KCNQ/M current inhibition. Am J Physiol Cell Physiol. 2008;295(1):C81–C91. doi: 10.1152/ajpcell.00028.2008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.