

Abstract
Organofluorine compounds play an important role in medicinal chemistry where they are responsible for up to 15% of the pharmaceutical products on the market. While natural products are valuable sources of new chemical entities, natural fluorinated molecules are extremely rare and the pharmaceutical industry has not benefited from a microbial source of this class of compounds. Streptomyces cattleya is an unusual bacterium in that it elaborates fluoroacetate and the amino acid 4-fluorothreonine. The discovery in 2002 of the fluorination enzyme FlA responsible for C-F bond formation in S. cattleya, and its subsequent characterization, opened up for the first time the prospect of genetically engineering fluorometabolite production from fluoride ion in host organisms. As a proof of principle, we report here the induced production of fluorosalinosporamide by replacing the chlorinase gene salL from Salinispora tropica with the fluorinase gene flA.
Fluorinated natural products are extremely rare, with only a handful of such metabolites identified. Fluoroacetate is the most common, found in a variety of plants, however despite an increased sophistication in high throughput screening technology and analytical methods aimed at natural product identification, it is over 20 years since the last fluorinated microbial products were identified. These were fluoroacetate and 4-fluorothreonine from the soil bacterium Streptomyces cattleya.1 This is in stark contrast to metabolites containing the other halogens.2,3 While numerous biochemical methods have evolved for the activation and incorporation of inorganic chloride, bromide, and iodide into organo-halo compounds, the unique physical properties of fluoride prevent oxidative activation to F+. Fluoride ion is also a poor nucleophile in water. The process of enzymatic fluorination in S. cattleya has been characterized and it has been shown that fluoride ion acts as a nucleophile and reacts with S-adenosyl-l-methionine (SAM) to generate the C–F bond in 5′-fluorodeoxyadenosine (5′-FDA). 5′-FDA is subsequently metabolized into the mammalian toxin fluoroacetate and the antibiotic 4-fluorothreonine (Figure 1).4,5 We now report the first example whereby the fluorinase gene has been incorporated into a host organism by genetic engineering, such that the heterologous host has a new capacity to generate a fluorinated natural product when grown in the presence of inorganic fluoride.
Figure 1.

Biosynthesis of fluorometabolites by Streptomyces cattleya21,22 (top), of salinosporamide A by Salinispora tropica10 (bottom) and fluorosalinosporamide engineering (middle).
Organofluorine compounds represent an important class of drugs in which the halogen can impart favorable pharmacokinetic properties.6,7 While fluorinated molecules are routinely synthesized in the laboratory, the incorporation of fluorine into structurally complex natural products can prove problematic, as most fluorination reagents are non-selective, often noxious and difficult to handle. To circumvent these problems, precursor-directed biosynthesis and mutasynthesis have contributed to the generation of a host of fluorinated natural product derivatives in which simple, synthetic organofluorine precursors are metabolized into more complex biosynthetic products.8 The discovery of fluorinase (5′-FDA synthase) and the characterization of its initiating role in the biosynthesis of fluorinated metabolites in S. cattleya4,5 has provided an opportunity to reprogram biosynthesis in a heterologous host for the assembly of an engineered organofluorine metabolite. We thus selected the marine bacterium Salinispora tropica as a suitable test organism since it uniquely biosynthesizes the potent anticancer agent salinosporamide A from a related halogenation pathway involving chlorine assimilation via 5′-ClDA in a reaction catalyzed by the chlorinase SalL (Figure 1).9,10
Results and Discussion
We previously prepared fluorosalinosporamide by a standard mutasynthesis approach in which the chlorinase gene salL was inactivated and the ensuing mutant was chemically complemented with synthetic 5′-FDA, 5-fluororibose11 or 4-fluorocrotonic acid.12 In order to engineer fluorosalinosporamide biosynthesis from inorganic fluoride, we generated a S. tropica mutant in which the salL chlorinase gene was chromosomally replaced with the S. cattleya flA fluorinase gene by λ-Red mediated recombination13 so that flA was placed under the control of the native salL promoter and ribosome binding site (Figure 2A). The correct gene replacement was confirmed by PCR (Figure 2B) and sequencing.
Figure 2.
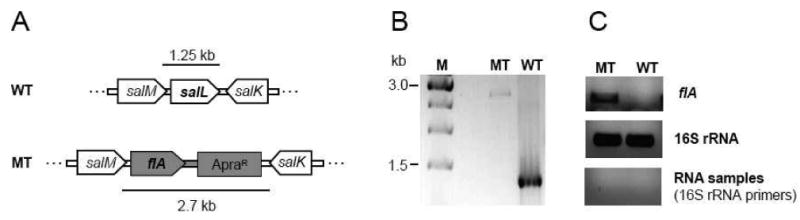
Fluorinase expression in S. tropica. (A) The chlorinase gene salL was chromosomally replaced with fluorinase flA and the apramycin resistance/oriT cassette from pIJ773 (ApraR) using PCR targeting.10,13 flA is thus under control of the natural salL promoter and ribosome binding site. The targeted locus of the wild-type (WT) and mutant (MT) chromosomes is shown for comparison. (B) The authenticity of mutants was confirmed by PCR. (C) flA transcription was analyzed by semi-quantitative RT-PCR. 16S rRNA was used as control of cDNA quality and integrity. The negative control with RNA instead of cDNA samples confirms the absence of genomic DNA contamination.
Cultivation of the salL- flA+ mutant in seawater-based medium initially showed no detectable salinosporamide A nor fluorosalinosporamide production (Figure 3) despite flA transcription being established in the mutant by reverse transcription PCR (RT-PCR) (Figure 2C). Previously it was reported that fluorinase can also function as a chlorinase in vitro when coupled to a l-amino acid oxidase or to an adenylic acid deaminase in order to drive the equilibrium of the reaction towards chlorinated products.14 Although the rate of the reaction is at least two orders of magnitude lower with chloride as a substrate compared to fluoride, we did expect at least some salinosporamide A production in the mutant. Upon semi-purification of a large-scale fermentation extract followed by LC/APCI-MS analysis, we ultimately detected salinosporamide A. Yet, the amount produced (35 μg/L) was three orders of magnitude lower compared to the wild-type S. tropica (50 mg/L), reflecting the differential specialization of SalL and FlA as chlorinase and fluorinase, respectively.
Figure 3.
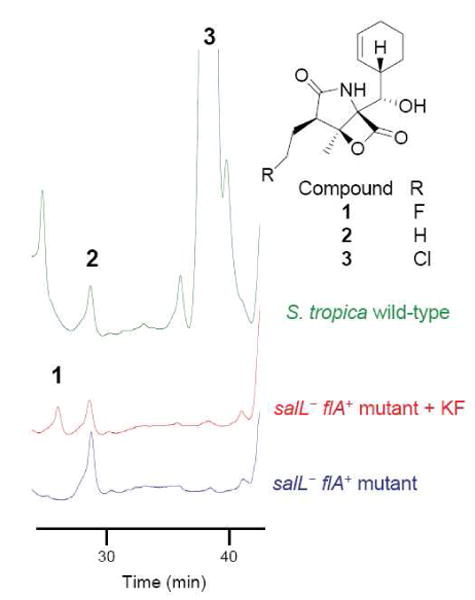
HPLC-MS analysis of culture extracts of S. tropica wild-type and the flA+ salL- mutant grown in A1 seawater-based media, and of the mutant grown in A1 seawater-based media pH 6.0 to which 2 mM potassium fluoride was added at day 2 (+ KF). HPLC was monitored at 210 nm.
The inability to biosynthesize fluorosalinosporamide, on the other hand, was likely due to the low concentration of fluoride in seawater (1.3 ppm). Cultivation of the S. tropica salL- flA+ mutant in seawater-based medium supplemented with 2 mM potassium fluoride as reported for S. cattleya15 caused growth inhibition preventing the detection of fluorosalinosporamide in a timely manner. To circumvent the fluoride toxicity issue, potassium fluoride was not added until the culture reached an early to mid exponential phase. Under these conditions, fluorosalinosporamide was clearly detectable in the culture broth by LC/(ESI)MS (Figure 3) and 19F-NMR (Figures 4 and 5).
Figure 4.
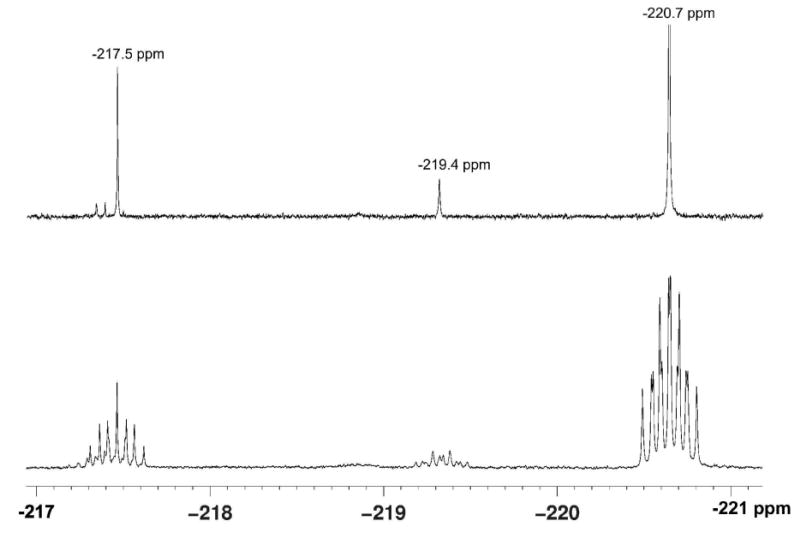
19F-NMR analysis of S. tropica salL- flA+ culture extracts. The upper trace shows the decoupled 19F{1H}-NMR spectrum of the organic fraction of the extract, whereas the lower trace is the proton coupled 19F-NMR spectrum. In all, five fluorometabolites were detected with the major product as fluorosalinosporamide (-220.7 ppm).
Figure 5.
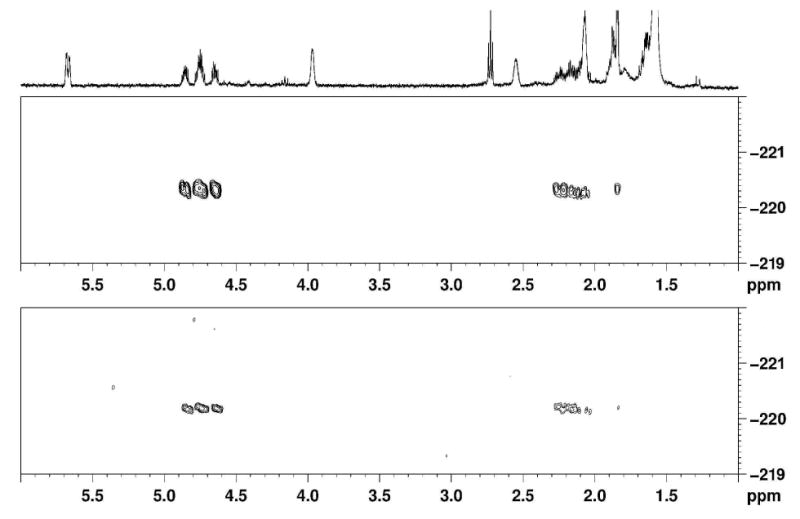
1H/19F-HMBC (500 MHz, CDCl3) correlation spectra. The upper trace is the spectrum of a reference sample of fluorosalinosporamide. The lower spectrum is a focused section illustrating fluorosalinosporamide in the S. tropica salL- flA+ extract grown in limiting fluoride. The fluorine atom is coupling to three sets of protons with chemical shifts consistent with the FCH2CH2CH-R fragment.
Fluoride uptake and fluorometabolite production by S. cattleya has been shown to be pH-dependent, with maximum rates at pH 6.0 and a sharp decline at higher and lower pH values.15 Likewise, maximum yields of fluorosalinosporamide production were detected at pH 6.0 (4 mg/L), while lower pH values (5.5 was tested) inhibited growth, preventing fluorometabolite production, and higher pH values (7-8) led to on average a 5-fold and 12-fold decrease in yields, respectively (Table 1).
Table 1.
Effect of pH on fluorosalinosporamide yields
| pH of culture broth | Fluorosalinosporamide yield (%) |
|---|---|
| 5.5 | --- (poor growth) |
| 6.0 | 100 ± 8* |
| 7.0 | 18 ± 5 |
| 8.0 | 8.5 ± 2 |
100% is equivalent to 4 mg/L.
The fluorosalinosporamide production yields obtained at optimum pH were comparable to those attained through mutasynthesis,11 which however was still one order of magnitude lower than the amount of salinosporamide A produced by the parent strain. In order to verify if there were any significant competing pathways diverting fluorinated intermediates into other side products, 19F-NMR analyses of the culture extracts were carried out. Figure 4 shows both 19F{1H}- and 19F-spectra revealing that at least five fluorinated metabolites were produced. The major fluorinated component in the S. tropica salL- flA+ extract is fluorosalinosporamide (at -220.7 ppm), which was confirmed by NMR comparison to a reference sample generated from previous mutasynthesis studies.11 In particular, the 2D 1H/19F-HMBC experiment comparing the extract with a reference sample of fluorosalinosporamide showed identical chemical shifts and correlations between 19F and 1H signals in the two samples (Figure 5).
Although the structures of the other fluorinated compounds have not yet been elucidated, the minor metabolites corresponding to 19F-NMR signals at -217.5 ppm and -219.4 ppm have very similar 1H/19F-NMR coupling patterns as observed with fluorosalinosporamide (Figure 4). Hence they are most likely close structural analogs. Natural structural modifications reported for salinosporamide A include the C-2 epimer salinosporamide F, the C-3 ethyl derivative salinosporamide I, the C-5 deoxy analog salinosporamide J, and the C-5 isopropyl derivative antiprotealide.16-18 The presence of salinosporamide degradation products in culture extracts has also been described which include the hydrolyzed β-lactone and decarboxylated products.19,20
Because 19F-NMR analysis showed that fluorosalinosporamide is indeed the major fluorinated metabolite produced by S. tropica salL- flA+, an explanation for the lower yield of fluorosalinosporamide produced by the mutant compared to salinosporamide A accumulated by the wild-type is not consistent with parallel pathways diverting fluorinated intermediates. Rather fluoride toxicity and the differential preference for chlorinated versus fluorinated substrates by downstream pathway enzymes in the salinosporamide biosynthetic pathway likely account for the observed disparate yields, especially considering that the mutasynthesis experiment gave similar results.
In conclusion, we have presented an example for the biosynthesis of fluorinated natural product analogs from fluoride ion by introducing the fluorinase gene from S. cattleya into a suitable heterologous host. Broader applicability of fluorometabolite engineering in different organisms may come from the concomitant expression of other pathway enzymes involved in the production of polyketide and/or non-ribosomal peptide building blocks such as fluoroacetyl-CoA, fluorothreonine or fluoroethylmalonyl-CoA.10,12,21,22
Experimental Section
General Experimental Procedures
Salinispora tropica CNB-44023 was cultured in A1 sea water-based medium as described.24 The pH of the medium was set up using 50 mM MES buffer (pH 5.5 to 7.0) or phosphate buffer (pH 8.0). The REDIRECT technology kit for PCR targeting13 was obtained from Plant Bioscience Limited (Norwich, UK). pCC1FOS-based (Epicentre) fosmid BHXS1782 was obtained from the Joint Genome Institute. Apramycin (200 μg/mL for S. tropica; 50 μg/mL for E. coli), chloramphenicol (12-25 μg/mL), carbenicillin (100 μg/mL), and nalidixic acid (100 μg/mL) were used for selection of recombinant strains. DNA manipulation and PCR were performed according to standard procedures.25,26 Details of gene replacement in S. tropica including a conjugation protocol can be found in references 9 and 10.
Replacement of salL by flA in S. tropica
The cassette for replacement of salL with flA was generated by a two-step PCR strategy in which flA was fused to the apramycin resistance cassette from pIJ773.13 In the first step, flA and the apramycin resistance cassette were amplified in two parallel reactions termed here “PCR 1” and “PCR 2”, respectively. Primers used in PCR 1 were P1_flAki (GTC GGT TTC CGA CCG ATA AAA CGG AGG TCA CTC ACC ATG GCT GCC AAC AGC ACA CG) and P2_flAki (TT CGA ACT GCA GGT CGA CGG ATC CCC GGA AT GGT ACG TCG TCG CGC GTT C) and in PCR 2, P3_flAki (ATT CCG GGG ATC CGT CGA CC) and P4_flAki (GAC AGG AGC TAC CCA GCA GCA TGC CGC CCC CTG GGT CAG TGT AGG CTG GAG CTG CTT C) in which priming sites are underlined. PCR 1 was subject to 30 cycles of 94 °C (30 s), 58 °C (45 s) and 72 °C (90 s) using Pfu Turbo DNA polymerase (Stratagene) and FlA_HT plasmid27 as template. PCR 2 was carried out using the High Fidelity PCR system (Roche) as reported.13 PCR products were digested with DpnI to eliminate template plasmid, purified using the QIAQuick PCR purification kit (Qiagen) and mixed to approximately equimolar concentrations for the second round which was carried out using the High Fidelity PCR system (Roche) and primers P1_flAki and P4_flAki. Cycle conditions consisted of 1 cycle of 94 °C (2 min), followed by a cycle of 94 °C (45 s) and 72 °C (5 min), 10 cycles of 94 °C (45 s), 60 °C (45 s), 72 °C (2.5 min), 15 cycles of 94 °C (45 s), 65 °C (45 s), 72 °C (2.5 min) and a final extension step at 72 °C (10 min). The desired fragment was then gel purified using the QIAQuick gel extraction kit (Qiagen) and used for replacement of salL in fosmid BHXS1782 by PCR targeting as previously described.9,13 Gene replacement in the fosmid was confirmed by PCR and sequencing using primers PflAseq_f (GAG AAG CCT GCG GTT CGT G) and (PflAseq_r GA ACT GCA GGT CGA CGG ATC). S. tropica mutants were confirmed by PCR.
RT-PCR analysis
RNA was isolated from cells collected from a second generation S. tropica A1 liquid culture grown for 67 h (late exponential phase) using the RiboPure-Bacteria kit (Ambion). cDNA was synthesized using the SuperScript III First-Strand Synthesis System for RT-PCR (Invitrogen) as previously described.10 Primers used in the PCR step to interrogate flA transcription were PflArt_f (GAG ATC GTC CGC TTC AAC C) and PflArt_r (TGA GGT AGA TGG CGA TGT TG). Absence of genomic DNA contamination in the RNA samples and cDNA quality were assessed using 16S rRNA primers.10
Analysis of fluorosalinosporamide production
Best yields of fluorosalinosporamide production were obtained by culturing the S. tropica salL- flA+ mutant in A1 pH 6.0 for two days (early exponential phase) before adding 2 mM of potassium fluoride. XAD 7 resin (Sigma) was added at the time of fluoride supplementation and extracted with acetone after two weeks. The dried extract was then subject to EtOAc-H2O partition and the organic phase analyzed by HPLC with a Phenomenex C18 column (150 × 4.6 mm; 5 μm particle size) at flow rate of 0.7 mL min−1, using MeCN (B) in H2O (A) as mobile phase (0 to 30% B in 5 min, then 30% B for 20 min, 30 to 35% B in 5 min, 35% B for 8 min, 35 to 100% B in 5 min, 100% B for 5 min) and monitoring at 210 nm. Fluorosalinosporamide identity was confirmed with an authentic standard by LC/(+)ESI-MS ([M+H]+: m/z 298).11
Salinosporamide A detection in the S. tropica salL- flA+mutant
A dried crude extract of the S. tropica salL- flA+ mutant grown in A1 seawater-based medium (no fluoride added) was fractioned by silica gel flash chromatography using increasing concentrations of acetone in CH2Cl2 as mobile phase. Fractions containing salinosporamides (eluted at 90:10, CH2Cl2:acetone) were combined, dried in vacuo and analyzed by LC/(APCI)-MS using a Michrom Magic C-18 column (150 × 1.0 mm, 5 μm particle size, 200Å pore size) and a gradient of 7.5 to 97.5% MeCN in H2O (50 μL/min flow rate) as mobile phase (salinosporamide A retention time of 18.5 min, m/z 314 and 316, 3:1, [M+H]+).
19F-NMR analysis
The dried acetone extracts of S. tropica salL- flA+ were resuspended directly in CDCl3 and 19F-NMR (and HMBC) spectra were run on a Bruker AV-500MHz instrument (19F at 470.33 MHz). 19F-NMR spectra were recorded with and without proton decoupling. 19F-NMR chemical shifts were calculated relative to CFCl3. Figure 6 shows that the experimental 19F-NMR fluorosalinosporamide spectrum matches well a DAISY simulation28 of the spectrum (-220.7 ppm; 2JH, F= 47.0 Hz, 2 equivalent H, and 3JH, F = 28.8 and 24.3 Hz, 2 non-equivalent diastereotopic H).
Figure 6.
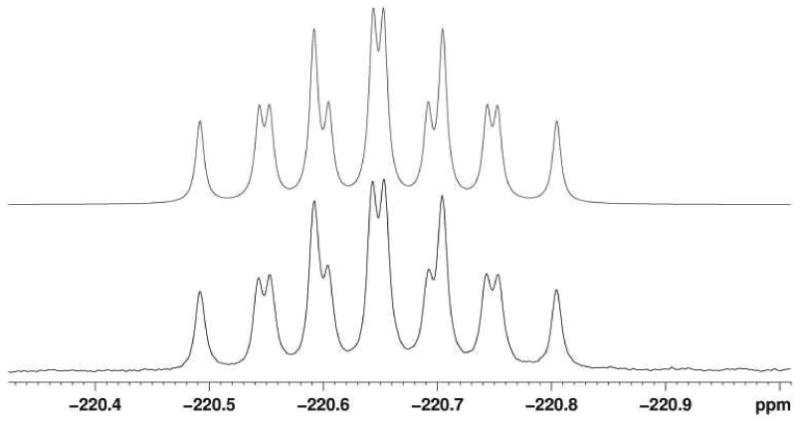
Simulation of the 19F-NMR spectrum of fluorosalinosporamide (upper trace) compared to the experimental spectrum (lower trace). 2J(1H,19F) = 47.0 Hz (2 equivalent H), 3J(1H,19F) = 28.8 and 24.3 Hz (2 non-equivalent diastereotopic H).
Acknowledgments
We kindly thank W. Fenical and P. R. Jensen (Scripps Institution of Oceanography) for the S. tropica wild-type strain, T. Lebl (University of St. Andrews) for assistance in running the 19F-NMR spectra, B. Gust (University of Tübingen, Germany) and Plant Bioscience Limited for providing the REDIRECT technology kit for PCR-targeting, and the UCSD Chemistry and Biochemistry Mass Spectrometry Facility for LC/APCI-MS analysis for salinosporamide A detection. This research was supported by NIH grant CA127622 to B.S.M and a BBSRC grant BB/F007426/1 to D.O'H. A.S.E. is a Tularik postdoctoral fellow of the Life Sciences Research Foundation.
Footnotes
Dedicated to the late Richard E. Moore of the University of Hawaii at Manoa for his pioneering work on bioactive natural products
References and Notes
- 1.Harper DB, O'Hagan D. Nat Prod Rep. 1994;11:123–133. doi: 10.1039/np9941100123. [DOI] [PubMed] [Google Scholar]
- 2.Neumann CS, Fujimori DG, Walsh CT. Chem Biol. 2008;15:99–109. doi: 10.1016/j.chembiol.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 3.Blasiak LC, Drennan CL. Acc Chem Res. 2009;42:147–155. doi: 10.1021/ar800088r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dong C, Huang F, Deng H, Schaffrath C, Spencer JB, O'Hagan D, Naismith JH. Nature. 2004;427:561–565. doi: 10.1038/nature02280. [DOI] [PubMed] [Google Scholar]
- 5.O'Hagan D, Schaffrath C, Cobb SL, Hamilton JT, Murphy CD. Nature. 2002;416:279. doi: 10.1038/416279a. [DOI] [PubMed] [Google Scholar]
- 6.Müller K, Faeh C, Diederich F. Science. 2007;317:1881–1886. doi: 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]
- 7.Hagmann WK. J Med Chem. 2008;51:4359–4369. doi: 10.1021/jm800219f. [DOI] [PubMed] [Google Scholar]
- 8.Murphy CD, Clark BR, Amadio J. Appl Microbiol Biotechnol. 2009;84:617–629. doi: 10.1007/s00253-009-2127-0. [DOI] [PubMed] [Google Scholar]
- 9.Eustáquio AS, Pojer F, Noel JP, Moore BS. Nat Chem Biol. 2008;4:69–74. doi: 10.1038/nchembio.2007.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eustáquio AS, McGlinchey RP, Liu Y, Hazzard C, Beer LL, Florova G, Alhamadsheh MM, Lechner A, Kale AJ, Kobayashi Y, Reynolds KA, Moore BS. Proc Natl Acad Sci U S A. 2009;106:12295–12300. doi: 10.1073/pnas.0901237106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eustáquio AS, Moore BS. Angew Chem Int Ed Engl. 2008;47:3936–3938. doi: 10.1002/anie.200800177. [DOI] [PubMed] [Google Scholar]
- 12.Liu Y, Hazzard C, Eustáquio AS, Reynolds KA, Moore BS. J Am Chem Soc. 2009;131:10376–10377. doi: 10.1021/ja9042824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gust B, Challis GL, Fowler K, Kieser T, Chater KF. Proc Natl Acad Sci U S A. 2003;100:1541–1546. doi: 10.1073/pnas.0337542100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deng H, Cobb SL, McEwan AR, McGlinchey RP, Naismith JH, O'Hagan D, Robinson DA, Spencer JB. Angew Chem Int Ed Engl. 2006;45:759–762. doi: 10.1002/anie.200503582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reid KA, Hamilton JT, Bowden RD, O'Hagan D, Dasaradhi L, Amin MR, Harper DB. Microbiology. 1995;141:1385–1393. doi: 10.1099/13500872-141-6-1385. [DOI] [PubMed] [Google Scholar]
- 16.Reed KA, Manam RR, Mitchell SS, Xu J, Teisan S, Chao TH, Deyanat-Yazdi G, Neuteboom ST, Lam KS, Potts BC. J Nat Prod. 2007;70:269–276. doi: 10.1021/np0603471. [DOI] [PubMed] [Google Scholar]
- 17.McGlinchey RP, Nett M, Eustáquio AS, Asolkar RN, Fenical W, Moore BS. J Am Chem Soc. 2008;130:7822–7823. doi: 10.1021/ja8029398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Manam RR, Macherla VR, Tsueng G, Dring CW, Weiss J, Neuteboom ST, Lam KS, Potts BC. J Nat Prod. 2009;72:295–297. doi: 10.1021/np800578e. [DOI] [PubMed] [Google Scholar]
- 19.Williams PG, Buchanan GO, Feling RH, Kauffman CA, Jensen PR, Fenical W. J Org Chem. 2005;70:6196–6203. doi: 10.1021/jo050511+. [DOI] [PubMed] [Google Scholar]
- 20.Denora N, Potts BC, Stella VJ. J Pharm Sci. 2007;96:2037–2047. doi: 10.1002/jps.20835. [DOI] [PubMed] [Google Scholar]
- 21.Deng H, Cross SM, McGlinchey RP, Hamilton JT, O'Hagan D. Chem Biol. 2008;15:1268–1276. doi: 10.1016/j.chembiol.2008.10.012. [DOI] [PubMed] [Google Scholar]
- 22.Huang F, Haydock SF, Spiteller D, Mironenko T, Li TL, O'Hagan D, Leadlay PF, Spencer JB. Chem Biol. 2006;13:475–484. doi: 10.1016/j.chembiol.2006.02.014. [DOI] [PubMed] [Google Scholar]
- 23.Udwary DW, Zeigler L, Asolkar R, Singan V, Lapidus A, Fenical W, Jensen PR, Moore BS. Proc Natl Acad Sci U S A. 2007;104:10376–10381. doi: 10.1073/pnas.0700962104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beer LL, Moore BS. Org Lett. 2007;9:845–848. doi: 10.1021/ol063102o. [DOI] [PubMed] [Google Scholar]
- 25.Sambrook J, Russell DW. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press; New York: 2001. [Google Scholar]
- 26.Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA. Practical Streptomyces Genetics. John Innes Foundation; Norwich, UK: 2000. [Google Scholar]
- 27.Zhu X, Robinson DA, McEwan AR, O'Hagan D, Naismith JH. J Am Chem Soc. 2007;129:14597–14604. doi: 10.1021/ja0731569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zinin VN, Ilyasov AV, Weber U, Hägele G, Thiele H. J Fluorine Chem. 1995;70:289–292. [Google Scholar]