

Abstract
Two new ω-phenyl polyketide peroxides, plakinic acids K and L, were isolated from a two-sponge association of Plakortis halichondroides and Xestospongia deweerdtae. The absolute configurations of the remote dimethyl-branched stereocenters in plakinic acid K were assigned by degradation of plakinic acid K to a long-chain naphthamide and analysis by liposomal circular dichroism (L-CD) and comparison with synthetic standards.
The marine sponges of the genera Plakortis and Plakinastrella1 are prolific producers of cyclic polyketide peroxides that exhibit a broad spectrum of biological properties, including antifungal-1, antimalarial-2, antiprotozoal-3 and immunosuppressant4 activities. Sponge-derived cyclic peroxides may occur as 1,2-dioxane or 1,2-dioxolane ring systems.5 We recently reported the stereochemical assignments of two ω-phenyl polyketide peroxides, plakinic acids I (1) and J (2) from the symbiotic two-sponge association, Plakortis halichondroides–Xestospongia deweerdtae Lehnert & van Soest, 1999,6 which showed differential inhibition against haplodefficient lag1Δ/LAG1 strains of Saccharomyces cerevisiae.7 The remote methyl-branched C8 stereocenters of 1 and 2 were solved by the application of liposomal circular dichroism (L-CD)8 – a very sensitive technique (limit of detection ~16 nmol) that amplifies CD Cotton effects (CEs) through ordering of the long-chain lipids in highly-uniform, unilamellar liposomes.7. Here, we show a remarkable long-range perturbation of naphthamide chromophores by a single methyl branch, over eight bonds away, that discriminates between spectroscopically indistinguishable diastereomers. L-CD was used to assign configurations of both proximal C8 and distal C12 methyl-branched centers in two new ω-phenyl polyketide peroxides, plakinic acids K (3) and L (4).
P. halichondroides–X. deweerdtae, collected from the Bahamas, was extracted with MeOH:CH2Cl2 (1:1) to yield a dark-brown residue which was sequentially extracted with n-hexane, chloroform, n-butanol and water. Antifungal bioassay-guided fractionation revealed that the CHCl3-soluble extract exhibited potent activity against S. cerevisiae lag1Δ/LAG1 strain. This fraction was subjected to flash chromatography (silica, 0–100% MeOH-CHCl3, stepped gradient) and the active fraction further purified by HPLC (RP-C18 CH3CN/H2O) to yield known compounds 1, 2,7 new methyl-branched plakinic acids K (3), L (4) and plakinic acid M, a linear homolog of 1.9
Plakinic acid K (3) was isolated as an optically active colorless oil (4.74 × 10−3 % yield, w/w, wet weight), [α]D −137 (c 1.31, CHCl3) of molecular formula C26H44O4 as determined by HREIMS [M+] m/z 432.3239, with 14 mass units greater than 1. The UV, IR, 1H and 13C NMR spectra of 3 were almost identical to those of 1, except for an additional methyl group (δH 0.82, d, J = 6.4 Hz; δC 19.9, CH3). Inspection of the COSY and HMBC spectra established the position of the second methyl branch at C12 (see Supporting Information). Using a similar analysis, plakinic acid L (4, 7.5 × 10−3 % yield w/w wet weight, [α]D −26.2 (c 1.90, CHCl3) C25H40O4; HREIMS [M+] m/z 404.2918) was shown to be a homolog of 2.
The common origin of 1–4, similar specific rotations and 1H NMR suggest that the respective stereocenters at C3, C4 and C6 have the same configurations as those of 1 and 2.10 The configurations of the proximal C8/C7 stereocenters in 1 and 2 were assigned as R by interpretation of the intense CEs in L-CD of the derived 6-methoxy-2-naphthamide (in contrast, CD of the naphthamides in isotropic media, such as methanol solutions, showed only baseline).7 We had proposed7 that the strong, bi-signate CEs displayed in L-CD spectra arise from pair-wise intramolecular exciton coupling of naphthamides within the liposomal bilayers. Phospholipid bilayers, comprised of saturated long chains, promote hexagonal close packing of extended CH2 groups.
The dimensions of the problem expand significantly for assignment of the remote C12/C11 methyl-branched stereocenters in 3 and 4. We surmised the remote methyl branch in 3 and 4 may alter the chain packing and influence the magnitude and form of the naphthamide CEs in L-CD. This would constitute net transmission of stereochemical information from the remote C12 center to the naphthamide group, however, it was not certain the effect would be large enough to be readily observable in the CD spectrum.
In order to test this hypothesis, 311 was degraded (Scheme 1) by cleavage of the C6–C7 bond using Fe(II)-promoted reductive fragmentation (FeCl2, aq. CH3CN-H2O, N2-sparged) to give primary alkyl chloride 512, which was subsequently transformed by a three-step sequence7 to give naphthamide 7.
Scheme 1.

Degradation of 3 and conversion to 7
Compound 7 was formulated into a highly-uniform, unilamellar liposomes of distearoyl-sn-glycero-3-phosphocholine (DSPC) as previously described,13 (phospholipid: 7 ratio = 20:1) prior to measurement of the CD spectrum. Remarkably, no signal appeared in the CD spectrum of 7 (Figure 2a) immediately after formulation in liposomes, but over 24 hours intense, complex CEs gradually appeared that were stable (>30 days) and reproducible [λ 196 nm (Δε −11.6), 213 (+37.4), 226 (− 6.5), 255 (+5.6)]. Fitting the time-dependence of the CE at λ = 226 nm in L-CD spectrum of 7 (Figure 2c) to an exponential function gave t1/2 = 385 minutes. Evidently, the kinetics of lipid chain reorganization of liposomal 7 at room temperature are considerably slower than the corresponding naphthamides derived from 1 and 2 in which less complex CEs appeared essentially immediately.14 The assignment of configuration in 7 was completed by comparison with synthetic model compounds (2S,6R)-8 and (2R,6R)-9 which were prepared as follows.
Figure 2.
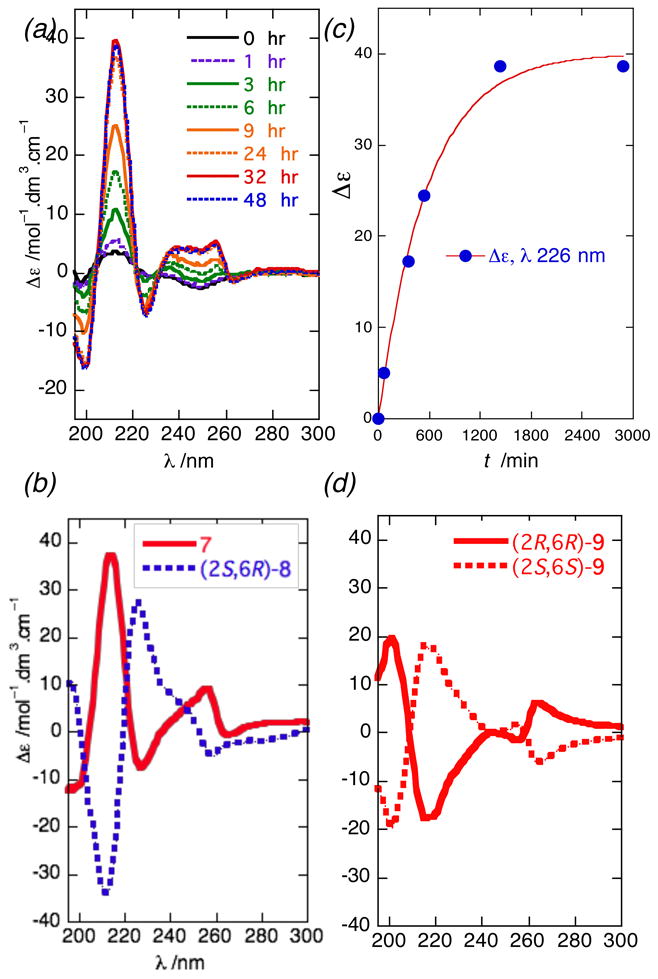
Liposomal circular dichroism (L-CD) spectra (T= 23 °C). (a) 7 from t = 0 to 48 hrs after formulation of liposomes (H2O, diastearoyl-sn-3-glycero-phosphocholine, 2 mg/mL; mole ratio of phospholipid:7 = 20:1). (b) 7, c = 2.24 × 10−4 M and (2S,6R)-8, c = 2.25 × 10−4 M. (c) Time-dependence of the Cotton effect of 7 (λ = 226nm), post-formulation. (d) (2R,6R)-9, c = 2.47 × 10−4 M and ent-9 (calculated).
Tosylation of (S)-(−)-citronellol15 10 gave 1116 in 87% yield (Scheme 2), which upon treatment with lithium phenylacetylide (reflux, THF), afforded enyne 12 in 92% yield.
Scheme 2.

Synthesis of an epimeric mixture of acids 17.
Two-step Johnson-Lemieux oxidation17 of 12 provided aldehyde 14 (Scheme 2), which was subjected to Horner-Wadsworth-Emmons olefination to give α,β-unsaturated ester 15 as an inseparable 1:1 mixture of E/Z isomers. Hydrogenation of enyne 15 (Pd/C) afforded saturated ester 16, which was saponified (LiOH, THF/water) to give acid 17 as a 1:1 mixture of diastereomers, epimeric at C2. Acid 17 was coupled with (S)-phenethylamine (HATU, i-Pr2NEt) (Scheme 3) to provide a mixture of diastereomeric amides (2S,6R)-18 and (2R,6R)-19, which were separated by silica gel chromatography.18 Amides 18 and 19 were individually treated with BH3•THF to afford secondary amines 20 and 21. Hydrogenolysis of 20 and 21 (Pd/C, CF3CH2OH) provided primary amines 22 and 23 in 92% and 72% yield, respectively. Separate acylation of 22 and 23 (6-methoxy-2-naphthoyl chloride, Et3N, DMAP) gave naphthamides (2S,6R)-8 and (2R,6R)-9.
Scheme 3.

Synthesis of standards, naphthamides 8 and 9.
Comparisons of the L-CD spectra of three stereoisomers are shown in Figure 2: 7 derived from 3 or 4, and synthetic compounds (2S,6R)-8 and (2R,6R)-9. The signs and magnitudes of the CEs are primarily dominated by perturbation of the naphthamide chromophore by the proximal stereocenter C2 within the first sphere of asymmetry. It is also evident that longer range perturbation from the remote methyl branch alters the forms of L-CD spectra of (2S,6R)-8 and (2S,6S)-9) as a function of the C6 configuration (Figures 2b and d). Whereas the diastereomers (2S,6R)-8 and (2R,6R)-9 were indistinguishable by NMR,19 they were clearly differentiated by the CEs and fine structure of their L-CD spectra. In order to evaluate the effect of the distal C6 stereocenter, the L-CD of (2S,6R)-8 was compared with the inverted L-CD of (2R,6R)-9 (Figure 2d) which corresponds to (2S,6S)-9. The CEs in 8 [λmax 196 (Δε +11.0), 213 (−34.0), 226 (+27.3), 258 (−4.9)] were changed in 9, particularly λ 217 nm (Δε +17.5) and λ 256 nm (Δε −6.2) (see Table S4 for complete data).
The L-CD of 7 did not match either enantiomer of 9: it was equal in magnitude and form, but opposite in sign to that of (2S,6R)-8 (Figure 2b). Therefore, 7 and (2S,6R)-8 are enantiomers and the complete configurations of 3 and 4 are 3S,4S,6R,8R,12S and 3R,5R,7R,11S, respectively.
Compounds 1–4 were assayed for antifungal activity against strains of Candida albicans, C. glabrata, C. krusei and Cryptococcus neoformans (Table 1). All compounds were exceedingly potent antifungal agents (MICs ≤ 0.5 μg/mL) against all seven strains. The monomethyl-branched ω-phenyl polyketide peroxides 1 and 2 were about twice as active as plakinic acids K (3) and L (4). Compound 2 showed the most potent activity (MIC< 0.12 μg/mL) against C. albicans 96–489, C. albicans UCD-FRI and C. glabrata suggesting that the 1,2-dioxolane ring and a monomethyl-branch in the ω-phenylalkyl side-chain are key determinants of antifungal activity.
Table 1.
Antifungal activities of plakinic acids I-L (1–4). Minimum inhibitory concentration, MIC (μg/mL)a.
| 1 | 2 | 3 | 4 | |
|---|---|---|---|---|
| C. albicans, ATCC 14503 | 0.25 | 0.25 | 0.50 | 0.50 |
| C. albicans 96–489b | 0.06 | <0.03 | 0.12 | 0.12 |
| C. albicans UCD-FR1b | 0.25 | 0.12 | 0.50 | 0.50 |
| C. glabrata | 0.12 | 0.06 | 0.12 | 0.12 |
| C. krusei | 0.06 | 0.12 | 0.12 | 0.12 |
| Cryptococcus neoformans var. gattii | 0.12 | 0.25 | 0.12 | 0.50 |
| C. neoformans var. grubii | 0.12 | 0.25 | 0.12 | 0.50 |
The in vitro susceptibilities were determined by the microbroth dilution method according to the guidelines of the National Committee for Clinical Laboratory Standards (NCCLS).
Fluconazole-resistant (MIC >64 (μg/mL).
In conclusion, liposomal CD differentiates long-chain methyl-branched naphthamides, with epimeric configurations at a stereocenter eight bonds removed from the chromophore. Comparison of the L-CD spectrum of the naphthamide, obtained by degradation of plakinic acid K (3), with those of diastereomeric synthetic models, allowed assignment of C8/C7 and C12/C11 of plakinic acids K (3) and L (4), respectively. This remarkable long-range propagation of stereochemical information is made possible by amplification of the Cotton effects through ordering of lipid chains within the liposomal bilayer. The current work also provides CD references for interrogation of remote double methyl-branched polyketides by L-CD. Additional applications of L-CD are the subject of ongoing investigations in our laboratories.
Supplementary Material
Figure 1.

Structures of plakinic acids I (1), J (2), K (3) and L (4).
Acknowledgments
We thank A. Marcus (University of Oregon) for helpful discussions, S. Zea (Universidad Nacional de Colombia) for identification of the sponges, and C. Green (Takasago International Corporation, U.S.A.) for a generous gift of (S)-(−)-citronellal. The authors are grateful to B. Morinaka and A. Jansma for assistance with NMR experiments, and J. Pawlik (University of North Carolina, Wilmington) and the crew of the RV Seward Johnson for logistics of sponge collection. EI HRMS data were provided by Y. Su. The 500 MHz NMR spectrometers were purchased with funds from NSF (CRIF, CHE0741968). Financial support of this work was provided by the National Institutes of Health (CA1225601 and AI039987).
Footnotes
Supporting Information Available. Full isolation and characterization of 3, 4, plakinic acid M (S1), degradation of 3 and 4, synthetic procedures, 1H and 13C NMR spectra of 3–23, and CD spectra of 7–9. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Phillipson DW, Rinehart KL., Jr J Am Chem Soc. 1983;105:7735–7736. [Google Scholar]; (b) Davidson BS. J Org Chem. 1991;56:6722–6724. [Google Scholar]; (c) Qureshi A, Salva J, Harper MK, Faulkner DJ. J Nat Prod. 1998;61:1539–1542. doi: 10.1021/np9802724. [DOI] [PubMed] [Google Scholar]; (d) Sandler JS, Colin PL, Hooper JNA, Faulkner DJ. J Nat Prod. 2002;65:1258–1261. doi: 10.1021/np020228v. [DOI] [PubMed] [Google Scholar]
- 2.Hu JF, Kelly M, Gao GF, Hamann MT. Tetrahedron. 2001;57:9379–9383. [Google Scholar]
- 3.Perry TP, Dickerson A, Khan AA, Kondru RK, Beratan DN, Wipf P, Kelly M, Hamann MT. Tetrahedron. 2001;57:1483–1487. [Google Scholar]
- 4.Costantino V, Fattorusso E, Mangoni A, Di Rosa M, Ianaro A. J Am Chem Soc. 1997;119:12465–12470. [Google Scholar]
- 5.Different polyketide peroxides have been found in tunicates. Reyes F, Rodríguez-Acebes R, Fernández R, Bueno S, Francesch A, Cuevas C. J Nat Prod. 2010;73:83–85. doi: 10.1021/np900700h.Fontana A, Gonzalez MC, Gavagnin M, Templado J, Cimino G. Tetrahedron Lett. 2000;41:429–432.Durán R, Zubía E, Ortega MJ, Naranjo S, Salvá J. Tetrahedron. 2000;56:6031–6037.
- 6.While n- and iso-alkyl cyclic peroxides are found in free-living P. halichondroides, we found only the P. halichondroides-X. deweerdtae association produces ω-phenyl-terminated polyketide peroxides.
- 7.Dalisay DS, Quach T, Nicholas GN, Molinski TF. Angew Chem Int Ed Engl. 2009;48:4367–71. doi: 10.1002/anie.200900888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) MacMillan JB, Molinski TF. J Am Chem Soc. 2004;126:9944–5. doi: 10.1021/ja047741a. [DOI] [PubMed] [Google Scholar]; (b) Macmillan JB, Linington RG, Andersen RJ, Molinski TF. Angew Chem Int Ed Engl. 2004;43:5946–51. doi: 10.1002/anie.200461158. [DOI] [PubMed] [Google Scholar]
- 9.See S1, Supporting Information for complete characterization.
- 10.The absolute configurations at stereocenters around the 1,2-dioxane ring in 1 were solved conventionally by integrated analysis of ROESY spectra and the modified Mosher’s ester method. The absolute configurations of C4 and C5 within the 1,2-dioxolane ring of 2 were assigned by comparison of the [α]D with those of synthetic ‘plakinates’ of defined configuration. Dai P, Trullinger TK, Liu X, Dussault PH. J Org Chem. 2005;71:2283–92. doi: 10.1021/jo0522254. and Ref. 7.
- 11.Plakinic acid L (4 ) also gave 5 under the same conditions.
- 12.Compound 5 arises from C6–C7 bond scission initiated by a ‘chloro-Fenton’ reaction that captures Cl from the Fe ligand sphere by radical rebound. Ref. 7 and Sawyer DT, Hage JP, Sobkowiak A. J Am Chem Soc. 1995;117:106–9.
- 13.The crude liposome suspension, prepared by shell-evaporation of a solution of naphthamides and DSPC in CHCl3 (2 mg/mL), followed by sonication in water and thermal annealing, was repeatedly extruded under pressure through a 100 nm pore polycarbonate membrane to give uniform diameter, unilamellar liposomes. Ref. 8.
- 14.The higher-order complexity of L-CD spectrum of 7 (which is the subject of our ongoing investigations) is indicative of delocalized intermolecular excitons, or J-arrays. Fidder H, Knoester J, Wiersma DA. J Chem Phys. 1993;98:6564–66.
- 15.(−)-Citronellol (10) obtained from TCI-EP [lot GM01] was of low optical purity (72% ee, [α]D20 −3.42 (neat); lit. [α]D20 −4.76° (neat) Rienäcker R, Ohloff G. Angew Chem. 1961;73:240.). Consequently, pure (−)-10 (98% ee, [α]D20 −5.18 (neat)) was obtained by reduction (NaBH4, MeOH), of (−)-citronellal (Takasago, 98%ee).
- 16.Mori K, Masuda S, Suguro T. Tetrahedron. 1981;37:1329–1340. [Google Scholar]
- 17.Pappo R, Allen DS, Jr, Lemieux RU, Johnson WS. J Org Chem. 1956;21:478–9. [Google Scholar]
- 18.The assignment of the C2 configurations in 18 and 19 followed from 1H NMR comparisons with analogs of known configuration. Nicholas GM, Molinski TF. Tetrahedron. 2000;56:2921–27.
- 19.13C NMR (CDCl3, 125 MHz): Σ [Δδ (8–9)]2 < 0.07 ppm.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.