

Abstract
Background
It has been proposed that elements of the renin angiotensin system expressed in the arterial wall are critical for the development of atherosclerosis. Angiotensin converting enzyme (ACE) is highly expressed by the endothelium and is responsible for a critical enzymatic step in the generation of angiotensin II. However, the functional contribution of ACE expression in the vascular wall in atherogenesis is unknown. Therefore, we made use of unique genetic models in which mice without expression of ACE in the vascular wall were crossed with apoE-/- mice in order to determine the contribution of tissue ACE expression to atherosclerotic lesion formation.
Methods and Results
Mice expressing either a soluble form of ACE (ACE 2/2) or mice with somatic ACE expression restricted to the liver and kidney (ACE 3/3) on an ApoE-/- background were placed on a standard chow or Western diet for 6 months. Atherosclerotic lesion area in the ACE 2/2 mice was significantly lower than that seen in the ACE 3/3 mice. However, these animals also had significantly lower blood pressure and reduced plasma ACE activity which precluded establishing a specific causal relationship between absent tissue ACE activity and decreased atherosclerotic lesion extent. Therefore, we studied the ACE 3/3 mice which are normotensive and lack vascular ACE expression. In the ACE 3/3 animals, atherosclerotic lesion area was no different from wild type controls despite reduced plasma ACE activity.
Conclusions
We concluded that under these experimental conditions, expression of ACE in the arterial wall is not required for atherosclerotic lesion formation.
Keywords: angiotensin, atherosclerosis, endothelium
Introduction
The traditional view of the renin angiotensin system is that of a systemic, hormonal regulatory pathway with a major functional role in hemodynamic homeostasis. This concept has now been superseded by a plethora of data defining important roles for angiotensin II that extend far beyond blood pressure control.1-3 The new paradigm for angiotensin II includes many functions such as roles for angiotensin II in cell growth and apoptosis, production of growth factors, generation of reactive oxygen species, thrombosis, immune responses, and tissue remodeling.1, 3-6 All of these processes are important in the normal physiology of many target organs and equally important in the pathophysiology of many disease processes. The target organs for angiotensin II include virtually every tissue in the body including the heart, brain, kidney, and the vascular wall.
A significant component of our new understanding of the pathophysiological role of angiotensin II comes from an appreciation of the fact that the renin-angiotensin system is not an exclusively “systemic” system at all.2, 7, 8 Virtually all of the enzymatic components of the renin angiotensin system have been identified in a variety of cell types throughout the body.2 Local production of angiotensin II may be of equal or greater importance in multiple pathologic scenarios.7 This is of particular relevance in the setting of atherosclerosis where localized generation of angiotensin II within the arterial wall has been implicated as a critical proximal step in atherogenesis.2 Thus, the concept that the combined synthetic functions of the liver, kidney and lung form the only source of angiotensin II has been abandoned in favor of an appreciation of the potentially more important local production of angiotensin II within specific tissues.2, 7
Angiotensin converting enzyme (ACE) is particular germane to atherogenesis not only because it is a primary enzymatic source of angiotensin II, but also due to the fact that ACE inhibition is a common therapeutic strategy that has been suggested to be useful in the treatment of atherosclerosis. In terms of local expression in the vascular wall, ACE has been identified in the vascular endothelium as well as macrophages.9, 10 Endothelial ACE has been proposed to be particularly relevant in the pathogenesis of atherosclerosis.11-13
Using the apoE-/- model of atherosclerosis, we sought to determine the functional significance of vascular wall ACE expression in atherosclerosis using genetically modified animals that lack ACE expression in the vascular wall. We used two previously characterized genetic mouse models of selective ACE expression. The ACE 2/2 mice only express the amino terminal potion of somatic ACE.14 The lack of the membrane anchoring portion of ACE in these animals results in ACE being “soluble” and not tissue associated. Thus, while ACE is synthesized in the same distribution as full length ACE, cells secrete the amino terminal domain of ACE as the enzyme is not be tethered. ACE is synthesized locally, but due to the lack of tethering, ACE is not retained in the vascular wall. We also used ACE 3/3 mice in which the ACE locus was genetically modified to disassociate ACE expression from the somatic ACE promoter and place it under the control of the albumin promoter.15, 16 In these mice there is also no ACE expression in the vascular wall as ACE expression is restricted to the liver and to a lesser degree the kidney. Using these two models, both of which exhibit a lack of ACE expression in the arterial wall, we studied the contributions of vascular wall ACE to atherosclerotic lesion formation.
Methods
Animals and Diets
All animal studies were approved by the Emory University Institutional Animal Care and Use Committee in accordance with the guidelines set forth by the NIH Guide for the Care and Use of Laboratory Animals. ACE-knockout mice type 2 (ACE 2/2)14 and type 3 (ACE 3/3)16 were provided by Dr. Ken Bernstein. ACE 2/2 and ACE 3/3 mice were backcrossed for 4 generations to C57BL/6 mice. All mice were bred as ACE heterozygotes, because ACE 2/2 mice are sterile. This breeding strategy also generates mice that are wild type littermates with respect to ACE 2/2 and ACE 3/3 (ACE 2/2WT and ACE 3/3WT) which are used as controls.
Female mice heterozygous for the ACE gene (ACE 2/2HZ and ACE 3/3HZ) were mated with male ApoE-/- mice on a C57BL/6 background that were purchased from The Jackson Laboratory (Bar Harbor, ME), generating F1 offspring that were either heterozygous or wild type with respect to the mutant ACE gene and heterozygous for the apoE gene (ApoE+/-). These F1 mice that were heterozygous with respect to the mutant ACE genes were then mated to produce F2 mice that were heterozygous with respect to the mutant ACE genes and homozygous for apoE deficiency (ApoE-/-) which were then mated to produce F3 offspring.
From the F3 offspring we acquired ACE 2/2/ApoE-/- and ACE 3/3/ApoE-/- mice for our experiments. Controls for the ACE 2/2/ApoE-/- and ACE 3/3/ApoE-/- mice were age-matched littermates from the same generation with ACE 2/2WT/ApoE-/- and ACE 3/3WT/ApoE-/- genotypes respectively. All mice were genotyped by polymerase chain reaction (PCR) from tail DNA, as previously described.14, 17
At 8 weeks of age, male mice from each of the four genotypes were placed on either the standard or Western diet for 6 months. The western-type diet (0.15% cholesterol, 42% fat) used in all experiments was purchased from Harlan Teklad, Inc (TD 88137, Indianapolis, Indiana). Systolic blood pressure was measured using a computerized, non-invasive, tail-cuff method (BP2000, Visitech) as previously described.18
Animals were euthanized by slow CO2 inhalation after 6 months of treatment. Blood was collected by cardiac puncture for lipid profiles and plasma ACE activity. Plasma lipid analyses were performed by Cardiovascular Specialty Labs (Atlanta, GA). Triglycerides and total cholesterol were determined by enzymatic methods on a CX5 chemistry analyzer (Beckman Coulter, Fullerton, CA) with Beckman reagents and controls. Low-density lipoprotein cholesterol and high-density lipoprotein cholesterol were estimated with the homogenous enzymatic kits from Equal Diagnostics (Exton, PA). VLDL was determined by calculation as total cholesterol minus the sum of Low-density lipoprotein cholesterol and high-density lipoprotein cholesterol. Plasma ACE activity was measured using a commercially available kit (Alpco, # 01-RK-ACD).
Evaluation of Atherosclerotic Lesions
The heart and aorta were pressure-perfused with 0.9% sodium chloride solution, followed by pressure fixation at approximately 100 mm Hg with a 10% formaldehyde solution. The extent of atherosclerotic lesion formation in the ascending and descending thoracic and abdominal aorta was analyzed as previously described.18 For cross-sectional analysis of lesion area in the ascending aorta hearts and ascending aortas were embedded in paraffin, and 5-μm thick serial sections were prepared for staining with hematoxylin and eosin. Digital images were captured from serial sections at the level of the sinus of valsalva and lesion area analyzed using NIH Image software. In addition, atherosclerotic cap thickness and necrotic core areas were measured using the digital calipers in the software.
Immunostaining for ACE was performed using a rabbit polyclonal, anti-mouse ACE antibody developed in Dr. Bernstein's laboratory.16 Samples were incubated with a biotinylated goat anti-rabbit secondary antibody from Vector Laboratories (1:200 dilution) followed by imaging with a streptavidin conjugated quantum dot (Q-Dot 605) from Invitrogen (1:100 dilution). All samples were counterstained with the nuclear counter stain, 4′-6-Diamidino-2-phenylindole (DAPI).
All data are presented as mean ± SEM. Statistical significance was determined by ANOVA. Post hoc analysis was performed using the Duncan New Multiple Range Test.
Results
Atherosclerotic lesion areas in ACE 2/2/ApoE-/- mice
Atherosclerotic lesion area in the aorta was quantified after 6 months of standard or Western diet. Panel A in figure 1 shows mean data for the analysis of the ascending aorta from the four treatment groups. The ACE 2/2/ApoE-/- mice demonstrated markedly reduced lesion area when compared to the wild type littermates for both diet treatment groups. In both the ACE 2/2/ApoE-/- mice and the wild type littermates, treatment with the western diet resulted in an increase in atherosclerotic lesion area. Similar trends were obtained for the en face analysis of the descending aorta (figure 1B/C).
Figure 1. Atherosclerosis in ACE 2/2/ApoE-/- mice.
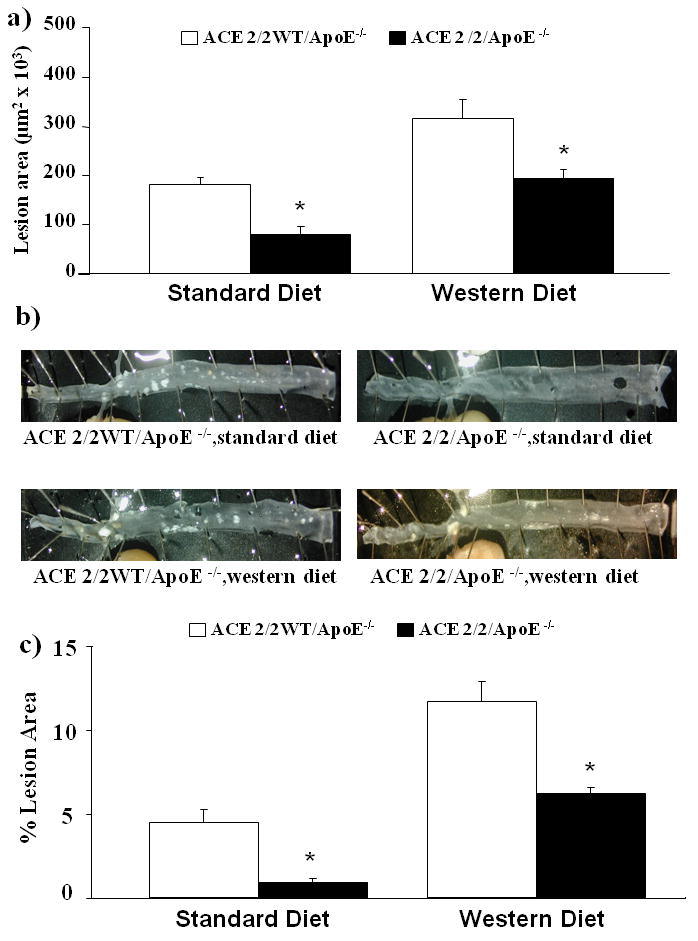
A) Atherosclerotic lesion area in the ascending aorta of ACE 2/2/ApoE-/- mice and their wild type litter mates (ACE 2/2WT/ApoE-/-) after treatment with either a standard diet or western diet for 6 months. B) Representative examples of en face preparations of the descending thoraco-abdominal aorta of ACE 2/2/ApoE-/- mice and their wild type littermates (ACE 2/2WT/ApoE-/-) after treatment with either a standard diet or western diet for 6 months. C) Mean data from en face analysis. (*= p<0.05 vs. ACE 2/2WT/ApoE-/-, n= 8-11/group). *=p<0.001
The ACE 2/2 mice have been previously shown to have a phenotype that is characterized by reduced blood pressure and reduced plasma ACE activity. 14, 16 We confirmed that this phenotype persisted when the ACE 2/2 mice were crossed onto the apoE-/- background. As shown in Figure 2, ACE 2/2 mice had a mean tail cuff blood pressure which was significantly lower than that observed in their wild type littermates. Plasma ACE activity in the ACE 2/2/ApoE-/- mice was 1.1±0.2 U/μg protein which was also significantly lower than that observed in the wild type mice (7.8±1.3 U/μg protein, p<0.001). Analysis of serum lipids revealed no significant effect of the ACE 2/2/ApoE-/- phenotype (Table 1). Immunostaining of the aorta of the ACE 2/2/ApoE-/- mice for ACE confirmed a lack of ACE expression in the vessel wall (figure 5).
Figure 2. Blood pressure in ACE 2/2/ApoE-/- mice.
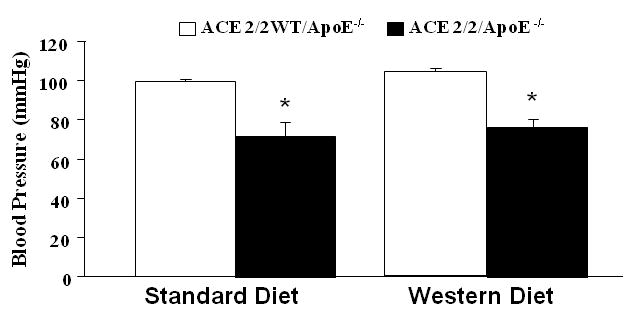
Blood pressure as measured using the tail cuff technique. (* p<0.001 vs. ACE 2/2WT/ApoE-/-). Shown are mean blood pressures measured after 6 months of treatment.
Table 1.
| CHOL mg/dl |
TG mg/dl |
LDLc mg/dl |
HDLc mg/dl |
VLDLc mg/dl |
|
|---|---|---|---|---|---|
| ACE 2/2WT/ApoE-/- Standard diet |
409.92±37.89 | 137.78±26.75 | 131.33±4.50 | 18.63±1.31 | 259.96±38.43 |
| ACE 2/2/ApoE-/- Standard diet |
441.29±52.74 | 159.97±46.63 | 131.94±19.91 | 25.19±2.93 | 284.16±39.15 |
| ACE 2/2WT/ApoE-/- Western diet |
883.20±72.08 | 209.91±28.44 | 445.80±47.16 | 24.03±2.99 | 413.37±30.52 |
| ACE 2/2/ApoE-/- Western diet |
1036.25±174.89 | 350.10±82.45 | 488.63±101.62 | 23.29±3.73 | 524.34±92.83 |
| ACE 3/3WT/ApoE-/- Standard diet |
404.22±27.87 | 235.22±65.96 | 126.11±9.64 | 24.92±2.35 | 253.19±25.58 |
| ACE 3/3/ApoE-/- Standard diet |
447.44±32.71 | 173.86±32.93 | 149.89±15.33 | 24.76±2.10 | 272.80±25.63 |
| ACE 3/3WT/ApoE-/- Western diet |
676.45±86.44 | 210.08±40.33 | 367.00±67.67 | 29.85±3.13 | 279.61±46.31 |
| ACE 3/3/ApoE-/- Western diet |
733.78±60.78 | 165.39±14.51 | 444.56±33.32 | 33.99±3.41 | 255.23±35.61 |
Values are mean±SEM.
TG indicates triglycerides; HDLc, high-density lipoprotein cholesterol; LDLc, low-density lipoprotein; VLDLc, very low-density lipoprotein cholesterol and CHOL, total cholesterol.
Figure 5. Immunostaining for ACE in ACE 2/2/ApoE-/- and ACE 3/3/ApoE-/- mice.
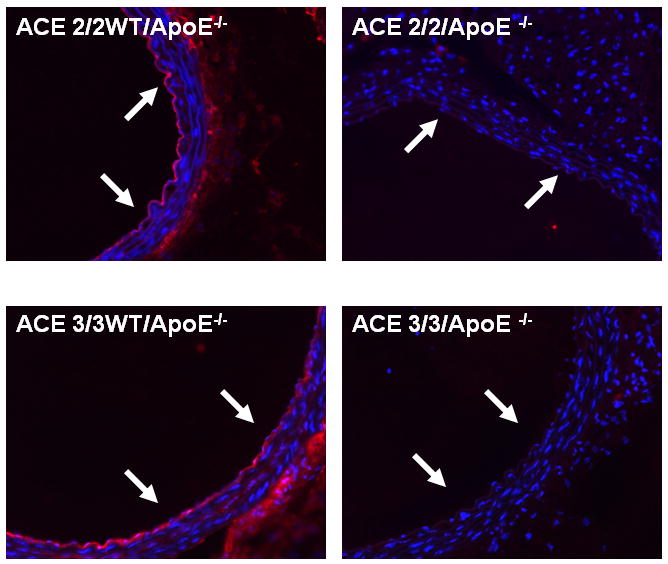
Representative sections from the descending aortas of non-atherosclerotic ACE 2/2/ApoE-/- and ACE 3/3/ApoE-/- mice and their wild type littermates immunostained for ACE. In wild type mice there is ACE staining in the endothelium and to a lesser degree in the adventitia. In the vessel wall of the ACE 2/2/ApoE-/- and ACE 3/3/ApoE-/- mice there is a total lack of ACE staining. Arrows indicate endothelium.
While the ACE 2/2 ApoE-/- mice exhibited a reduction in atherosclerotic lesion area, the mechanism remains unclear as the animals have reduced blood pressure, reduced plasma ACE activity and no ACE expression in the vessel wall. Therefore, we also examined atherogenesis in ACE 3/3 mice on the apoE-/- background. These mice have ACE expression restricted to the liver and to a lesser degree the kidney as expression is under control of the albumin promoter. Critical to the studies presented here is that despite a total lack of ACE expression in the vascular wall; these mice are normotensive thus allowing us to remove this potentially mitigating factor.19 Figure 3A shows the atherosclerotic lesion area in the ascending aorta of ACE 3/3/ApoE-/- mice as compared to their wild type littermates (ACE 3/3WT/ApoE-/-) for both diet treatment groups. For both the standard chow diet and the western diet treatment groups there was no difference in the lesion area between the ACE 3/3/ApoE-/- mice and their wild type littermates. Atherosclerotic lesion structure was not different between the two genotypes as evidenced similar mean fibrous cap thickness in the low fat (102,852±6197 vs. 96,432±2749 μm2) and Western diet (226,555±13,196 vs. 225,654±6627 μm2) of ACE 3/3/ApoE-/- vs. ACE 3/3 WT/ApoE-/- mice respectively. Similarly, cap thickness was not different between the two groups after low fat feeding (57±10 μm2 vs. 57±10 μm2 in high fat) or feeding with the Western diet (117±10 vs. 113±10 μm2) of ACE 3/3/ApoE-/- vs. ACE 3/3 WT/ApoE-/- mice respectively.
Figure 3. Atherosclerosis in ACE 3/3/ApoE-/- mice.
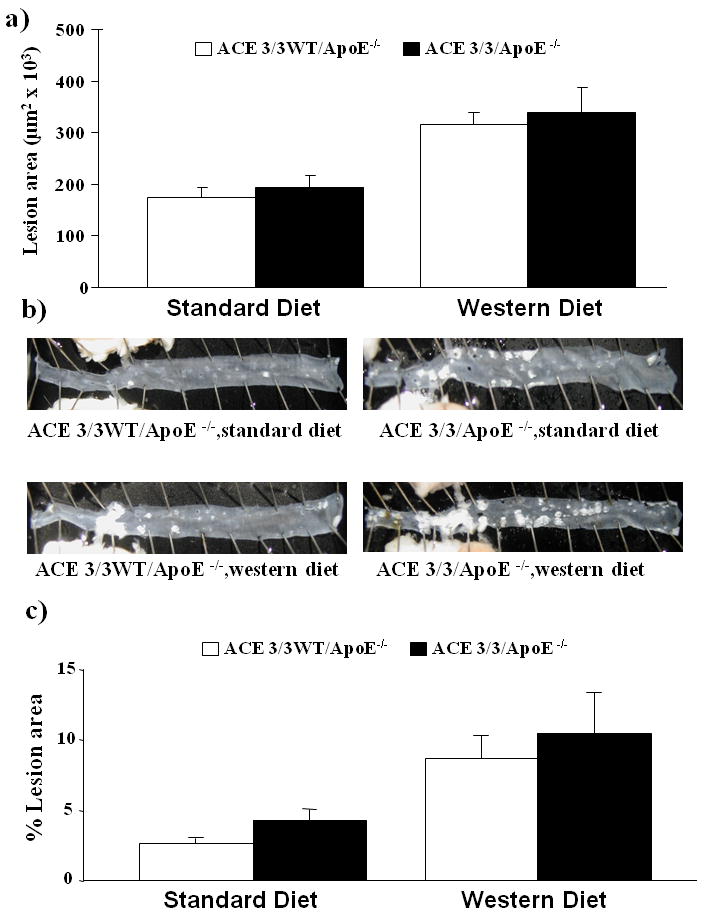
A) Mean atherosclerotic lesion area in the ascending aorta of ACE 3/3/ApoE-/- mice and their wild type litter mates (ACE 3/3WT/ApoE-/-) after treatment with either a standard diet or western diet for 6 months. B) Representative examples of en face preparations of the descending thoraco-abdominal aorta of ACE 3/3/ApoE-/- mice and their wild type littermates (ACE 3/3WT/ApoE-/-) after treatment with either a standard diet or western diet for 6 months. C) Mean data from en face analysis. There was no statistically significant effect of genotype on lesion area. Western diet significantly increased lesion area in all groups (p<0.05, n= 8-11/group)
Similar results were obtained for the en face lesion area of the descending aorta (figures 3B/C). The blood pressure of the ACE 3/3/ApoE-/- mice was not significantly different from their wild type littermates (Figure 4). Plasma ACE activity was lower in the ACE 3/3/ApoE-/- mice (3.5±0.5 U/μg protein) as compared to their wild type littermates (7.3±0.9 U/μg protein). There was no ACE expression the vascular wall as determined by immunostaining (figure 5). In addition, serum lipid profiles were similar for the two genotypes (Table 1).
Figure 4. Blood pressure in ACE 3/3/ApoE-/- mice.
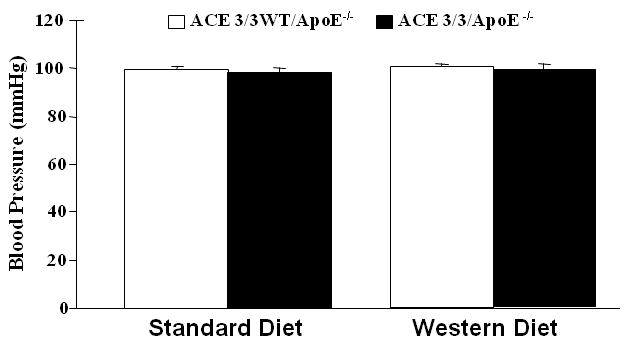
Blood pressure as measured using the tail cuff technique. Shown are mean blood pressures measured after 6 months of treatment. There were no significant differences between blood pressures measured in ACE 3/3/ApoE-/- mice and their wild type litter mates (ACE 3/3WT/ApoE-/-).
Discussion
It is well established that the renin angiotensin system is critically involved in the pathogenesis of atherosclerosis.6, 8, 20, 21 Studies in several different atherosclerotic models show that treatment with ACE inhibitors and angiotensin receptor blockers almost uniformly inhibit the formation of atherosclerotic lesions.6, 22-30 Wassman et al also showed that when AT1 receptor-deficient mice were crossed with apoE-/- mice, atherosclerotic lesion formation was reduced.31 Conversely, when angiotensin II is administered to apoE-/- or LDLR-/- mice, atherosclerotic lesion extent and complexity is dramatically increased.18, 32, 33 Thus, it is clear that angiotensin II promotes atherosclerosis. However, the cellular sources of angiotensin II remain uncertain.
Traditional views of the renin angiotensin system as a systemic mechanism for generating angiotensin II have been challenged by the concept that the local generation of angiotensin II within the arterial wall occurs independently of the systemic renin angiotensin system. Therefore, we employed two unique, genetic models to determine the contribution of the vascular wall tissue-based renin angiotensin system to atherosclerosis.
When we crossed ACE 2/2 mice with ApoE-/- mice to generate apoE mice with only the soluble form of ACE present, we found that atherosclerosis was dramatically reduced. However, these animals also had substantially lower blood pressure as compared to their apoE-/- littermates. Additionally, the plasma ACE activity in the ACE 2/2/ApoE-/- mice was approximately 20% of control levels. Thus, while it was obvious that there was a profound effect on atherosclerosis, it was not clear from these studies if the lack of tissue ACE expression, the lower overall ACE activity or the reduction in blood pressure was responsible for the reduction in atherosclerosis.
Our findings in the ACE2/2 mice are similar to those previously reported by another group which demonstrated a decrease in atherosclerotic lesion area.34 Note should be made that the previously study used only 2 mice that were homozygous for the modified ACE gene and that the major comparison was between wild type and heterozygous mice which yielded an intermediate result in terms of atherosclerotic lesion area. In addition, the previous publication reported no difference in blood pressure in the heterozygote mice whereas in our study with homozygous ACE2/2 mice, there was a significant decrease in blood pressure. Despite these differences, the overall finding that a deficiency in tissue ACE resulted in less atherosclerosis is consistent with our findings.
In order to further delineate the potential contribution of tissue ACE, we employed another unique mouse model, the ACE 3/3 mouse. In this mouse, ACE somatic expression is under control of the albumin promoter which results in restriction of somatic ACE expression to the liver with a modest degree of expression in the kidney. There is no ACE expression or activity in the endothelium, inflammatory cells or other tissues. The blood pressure of these animals is normal, as is the renal function and hematocrit. Plasma ACE activity is still reduced but the plasma levels of angiotensin II are normal. When we crossed the ACE 3/3 mice with ApoE-/- mice, we found that atherosclerosis was not affected. The lesion extent in the descending thoracic aorta and cross sectional area in the ascending aorta was not different. Furthermore, the average thickness of the fibrous cap and the area of the central, necrotic core was not different between the ACE 3/3/ApoE-/- mice and their littermate controls (ACE 3/3WT/ApoE-/- mice).
Taken together, these data demonstrate that ACE expression in the arterial wall is not necessary for the development of atherosclerosis. This does not completely exclude a potential role for vascular wall ACE. Recent work from our laboratory using the DOCA salt model of hypertension suggests in that particular model of hypertension in which there is low circulating renin activity, there is a strikingly important role for local generation of angiotensin II within the arterial wall.35 What we have shown here is that under normal conditions, tissue ACE expression is not an absolute requirement for atherosclerotic lesion formation.
It is also important to note that we have not performed the converse study with ACE expression limited to the endothelium as we do not have that genetic model available. Our data show that vascular wall is not necessary for atherosclerosis but they do not allow us to determine if vascular wall ACE is sufficient for atherosclerosis. One could argue that as the majority of ACE is expressed in the endothelium,7 the wild type state is largely reflective of endothelial ACE expression.
Our data do not discriminate between the different potential cellular sources of ACE in the arterial wall as all ACE expression in the arterial wall was absent. In the normal artery, the endothelium is the predominant source of ACE expression. In the setting of atherosclerosis and other vascular inflammatory states, macrophages9, 11 and potentially other inflammatory cells including T cells11, 36 infiltrating the vessel wall may be significant additional sources of ACE activity. In the late stages of atherosclerosis, it has been hypothesized that local ACE production by macrophages may contribute to advanced lesion progression and plaque destabilization. In our studies we did not identify any differences in lesion morphology between the ACE 3/3 mice and their littermates that were wild type with respect to ACE. However, lesion morphology in the apoE-/- mouse is not identical to human disease and plaque rupture is an infrequent or absent event in mice. Thus, we cannot rule out a role for vascular tissue ACE in more advanced disease states in human disease.
While these studies focused on the role of ACE expressed within the arterial wall, they do not address the potentially important roles of other components of the renin angiotensin system that are present in the different cellular components of the arterial wall. 2 Importantly, it has recently been shown that renin expression in macrophages is critical for the development of atherosclerosis.37 Thus, while these results argue against an obligate role for vascular wall ACE expression in atherosclerosis, they do not exclude important contributions by other components of the renin angiotensin system. ACE may not be the rate limiting step in angiotensin II production in vivo and thus more modest changes in ACE expression may not have similar effects on the development of atherosclerosis.
The finding that there was reduced atherosclerosis in the ACE 2/2 mice provides supportive data for the effect of blood pressure on atherosclerotic lesion formation. We have previously shown that elevated blood pressure alone can augment atherosclerosis in apoE-/- mice.18 While the finding of reduced atherosclerosis in the ACE 2/2 mice may be attributable to either a lower blood pressure or reduced plasma ACE activity in these animals, the finding that atherosclerosis was unaffected in the ACE 3/3 mice helps to dissect out these effects. The fact that the ACE 3/3 mice have reduced plasma ACE activity and are normotensive suggests that the lower blood pressure in the ACE 2/2 mice is largely responsible for the reduction in atherosclerotic lesion formation.
In summary, we have shown that the absence of tissue ACE expression does not inhibit the development of atherosclerosis in apoE-/- mice. In contrast to previous suggestions that endothelial ACE contributes significantly to atherosclerotic lesion formation, our data demonstrate that other sources of ACE are sufficient to sustain atherosclerosis.
Acknowledgments
Funding Sources
These studies were supported by NIH grants RO1 HL70531, P01 HL58000, RO1 HL090584, K99 HL088000, R01 DK039777, R01 DK051445 and VA Merit Funding.
Footnotes
Subject Codes: [134] Atherosclerosis Pathophysiology, [128] ACE/Angiotensin receptors, [145] Genetically altered mice
Disclosures
None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Bibliography
- 1.Daugherty A, Cassis L. Angiotensin II-mediated development of vascular diseases. Trends Cardiovasc Med. 2004;14(3):117–120. doi: 10.1016/j.tcm.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 2.Dzau VJ. Theodore Cooper Lecture: Tissue angiotensin and pathobiology of vascular disease: a unifying hypothesis. Hypertension. 2001;37(4):1047–1052. doi: 10.1161/01.hyp.37.4.1047. [DOI] [PubMed] [Google Scholar]
- 3.Gibbons GH. The pathophysiology of hypertension: the importance of angiotensin II in cardiovascular remodeling. Am J Hypertens. 1998;11(11 Pt 2):177S–181S. doi: 10.1016/s0895-7061(98)00198-8. [DOI] [PubMed] [Google Scholar]
- 4.Griendling KK, Ushio-Fukai M. Reactive oxygen species as mediators of angiotensin II signaling. Regul Pept. 2000;91(13):21–27. doi: 10.1016/s0167-0115(00)00136-1. [DOI] [PubMed] [Google Scholar]
- 5.Griendling KK, Ushio-Fukai M, Lassegue B, Alexander RW. Angiotensin II signaling in vascular smooth muscle. New concepts. Hypertension. 1997;29(1 Pt 2):366–373. doi: 10.1161/01.hyp.29.1.366. [DOI] [PubMed] [Google Scholar]
- 6.Weiss D, Sorescu D, Taylor WR. Angiotensin II and atherosclerosis. Am J Cardiol. 2001;87(8A):25C–32C. doi: 10.1016/s0002-9149(01)01539-9. [DOI] [PubMed] [Google Scholar]
- 7.Dzau VJ, Bernstein K, Celermajer D, Cohen J, Dahlof B, Deanfield J, Diez J, Drexler H, Ferrari R, van Gilst W, Hansson L, Hornig B, Husain A, Johnston C, Lazar H, Lonn E, Luscher T, Mancini J, Mimran A, Pepine C, Rabelink T, Remme W, Ruilope L, Ruzicka M, Schunkert H, Swedberg K, Unger T, Vaughan D, Weber M. The relevance of tissue angiotensin-converting enzyme: manifestations in mechanistic and endpoint data. Am J Cardiol. 2001;88(9 Suppl):1L–20L. doi: 10.1016/s0002-9149(01)01878-1. [DOI] [PubMed] [Google Scholar]
- 8.Lee MA, Bohm M, Paul M, Ganten D. Tissue renin-angiotensin systems. Their role in cardiovascular disease. Circulation. 1993;87(5 Suppl):IV7–13. [PubMed] [Google Scholar]
- 9.Potter DD, Sobey CG, Tompkins PK, Rossen JD, Heistad DD. Evidence that macrophages in atherosclerotic lesions contain angiotensin II. Circulation. 1998;98(8):800–807. doi: 10.1161/01.cir.98.8.800. [DOI] [PubMed] [Google Scholar]
- 10.Okamura A, Rakugi H, Ohishi M, Yanagitani Y, Takiuchi S, Moriguchi K, Fennessy PA, Higaki J, Ogihara T. Upregulation of renin-angiotensin system during differentiation of monocytes to macrophages. J Hypertens. 1999;17(4):537–545. doi: 10.1097/00004872-199917040-00012. [DOI] [PubMed] [Google Scholar]
- 11.Diet F, Pratt RE, Berry GJ, Momose N, Gibbons GH, Dzau VJ. Increased accumulation of tissue ACE in human atherosclerotic coronary artery disease. Circulation. 1996;94(11):2756–2767. doi: 10.1161/01.cir.94.11.2756. [DOI] [PubMed] [Google Scholar]
- 12.Dzau VJ, Bernstein K, Celermajer D, Cohen J, Dahlof B, Deanfield J, Diez J, Drexler H, Ferrari R, Van Gilst W, Hansson L, Hornig B, Husain A, Johnston C, Lazar H, Lonn E, Luscher T, Mancini J, Mimran A, Pepine C, Rabelink T, Remme W, Ruilope L, Ruzicka M, Schunkert H, Swedberg K, Unger T, Vaughan D, Weber M. Pathophysiologic and therapeutic importance of tissue ACE: a consensus report. Cardiovasc Drugs Ther. 2002;16(2):149–160. doi: 10.1023/a:1015709617405. [DOI] [PubMed] [Google Scholar]
- 13.Schiffrin EL. Beyond blood pressure: the endothelium and atherosclerosis progression. Am J Hypertens. 2002;15(10 Pt 2):115S–122S. doi: 10.1016/s0895-7061(02)03006-6. [DOI] [PubMed] [Google Scholar]
- 14.Esther CR, Marino EM, Howard TE, Machaud A, Corvol P, Capecchi MR, Bernstein KE. The critical role of tissue angiotensin-converting enzyme as revealed by gene targeting in mice. J Clin Invest. 1997;99(10):2375–2385. doi: 10.1172/JCI119419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shen XZ, Xiao HD, Li P, Lin CX, Billet S, Okwan-Duodu D, Adams JW, Bernstein EA, Xu Y, Fuchs S, Bernstein KE. New insights into the role of angiotensin-converting enzyme obtained from the analysis of genetically modified mice. J Mol Med. 2008;86(6):679–684. doi: 10.1007/s00109-008-0325-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cole J, Quach DL, Sundaram K, Corvol P, Capecchi MR, Bernstein KE. Mice lacking endothelial angiotensin-converting enzyme have a normal blood pressure. Circulation research. 2002;90(1):87–92. doi: 10.1161/hh0102.102360. [DOI] [PubMed] [Google Scholar]
- 17.Esther CR, Jr, Howard TE, Marino EM, Goddard JM, Capecchi MR, Bernstein KE. Mice lacking angiotensin-converting enzyme have low blood pressure, renal pathology, and reduced male fertility. Lab Invest. 1996;74(5):953–965. [PubMed] [Google Scholar]
- 18.Weiss D, Kools JJ, Taylor WR. Angiotensin II-induced hypertension accelerates the development of atherosclerosis in apoE-deficient mice. Circulation. 2001;103(3):448–454. doi: 10.1161/01.cir.103.3.448. [DOI] [PubMed] [Google Scholar]
- 19.Cole JM, Khokhlova N, Sutliff RL, Adams JW, Disher KM, Zhao H, Capecchi MR, Corvol P, Bernstein KE. Mice lacking endothelial ACE: normal blood pressure with elevated angiotensin II. Hypertension. 2003;41(2):313–321. doi: 10.1161/01.hyp.0000050650.52007.83. [DOI] [PubMed] [Google Scholar]
- 20.Kon V, Jabs K. Angiotensin in atherosclerosis. Curr Opin Nephrol Hypertens. 2004;13(3):291–297. doi: 10.1097/00041552-200405000-00005. [DOI] [PubMed] [Google Scholar]
- 21.Ferrario CM, Richmond RS, Smith R, Levy P, Strawn WB, Kivlighn S. Renin-angiotensin system as a therapeutic target in managing atherosclerosis. Am J Ther. 2004;11(1):44–53. doi: 10.1097/00045391-200401000-00010. [DOI] [PubMed] [Google Scholar]
- 22.Kowala MC, Grove RI, Aberg G. Inhibitors of angiotensin converting enzyme decrease early atherosclerosis in hyperlipidemic hamsters. Fosinopril reduces plasma cholesterol and captopril inhibits macrophage-foam cell accumulation independently of blood pressure and plasma lipids. Atherosclerosis. 1994;108(1):61–72. doi: 10.1016/0021-9150(94)90037-x. [DOI] [PubMed] [Google Scholar]
- 23.Hope S, Brecher P, Chobanian AV. Comparison of the effects of AT1 receptor blockade and angiotensin converting enzyme inhibition on atherosclerosis. Am J Hypertens. 1999;12(1 Pt 1):28–34. doi: 10.1016/s0895-7061(98)00203-9. [DOI] [PubMed] [Google Scholar]
- 24.Hayek T, Attias J, Coleman R, Brodsky S, Smith J, Breslow JL, Keidar S. The angiotensin-converting enzyme inhibitor, fosinopril, and the angiotensin II receptor antagonist, losartan, inhibit LDL oxidation and attenuate atherosclerosis independent of lowering blood pressure in apolipoprotein E deficient mice. Cardiovasc Res. 1999;44(3):579–587. doi: 10.1016/s0008-6363(99)00239-4. [DOI] [PubMed] [Google Scholar]
- 25.Sun YP, Zhu BQ, Browne AE, Pulukurthy S, Chou TM, Sudhir K, Glantz SA, Deedwania PC, Chatterjee K, Parmley WW. Comparative effects of ACE inhibitors and an angiotensin receptor blocker on atherosclerosis and vascular function. J Cardiovasc Pharmacol Ther. 2001;6(2):175–181. doi: 10.1177/107424840100600209. [DOI] [PubMed] [Google Scholar]
- 26.Candido R, Jandeleit-Dahm KA, Cao Z, Nesteroff SP, Burns WC, Twigg SM, Dilley RJ, Cooper ME, Allen TJ. Prevention of accelerated atherosclerosis by angiotensin-converting enzyme inhibition in diabetic apolipoprotein E-deficient mice. Circulation. 2002;106(2):246–253. doi: 10.1161/01.cir.0000021122.63813.32. [DOI] [PubMed] [Google Scholar]
- 27.Schuh JR, Blehm DJ, Frierdich GE, McMahon EG, Blaine EH. Differential effects of renin-angiotensin system blockade on atherogenesis in cholesterol-fed rabbits. J Clin Invest. 1993;91(4):1453–1458. doi: 10.1172/JCI116350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Keidar S, Attias J, Coleman R, Wirth K, Scholkens B, Hayek T. Attenuation of atherosclerosis in apolipoprotein E-deficient mice by ramipril is dissociated from its antihypertensive effect and from potentiation of bradykinin. J Cardiovasc Pharmacol. 2000;35(1):64–72. doi: 10.1097/00005344-200001000-00008. [DOI] [PubMed] [Google Scholar]
- 29.Hayek T, Attias J, Smith J, Breslow JL, Keidar S. Antiatherosclerotic and antioxidative effects of captopril in apolipoprotein E-deficient mice. J Cardiovasc Pharmacol. 1998;31(4):540–544. doi: 10.1097/00005344-199804000-00011. [DOI] [PubMed] [Google Scholar]
- 30.da Cunha V, Tham DM, Martin-McNulty B, Deng G, Ho JJ, Wilson DW, Rutledge JC, Vergona R, Sullivan ME, Wang YX. Enalapril attenuates angiotensin II-induced atherosclerosis and vascular inflammation. Atherosclerosis. 2005;178(1):9–17. doi: 10.1016/j.atherosclerosis.2004.08.023. [DOI] [PubMed] [Google Scholar]
- 31.Wassmann S, Czech T, van Eickels M, Fleming I, Bohm M, Nickenig G. Inhibition of diet-induced atherosclerosis and endothelial dysfunction in apolipoprotein E/angiotensin II type 1A receptor double-knockout mice. Circulation. 2004;110(19):3062–3067. doi: 10.1161/01.CIR.0000137970.47771.AF. [DOI] [PubMed] [Google Scholar]
- 32.Daugherty A, Manning MW, Cassis LA. Angiotensin II promotes atherosclerotic lesions and aneurysms in apolipoprotein E-deficient mice. J Clin Invest. 2000;105(11):1605–1612. doi: 10.1172/JCI7818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Daugherty A, Cassis L. Chronic angiotensin II infusion promotes atherogenesis in low density lipoprotein receptor -/- mice. Ann N Y Acad Sci. 1999;892:108–118. doi: 10.1111/j.1749-6632.1999.tb07789.x. [DOI] [PubMed] [Google Scholar]
- 34.Hayek T, Pavlotzky E, Hamoud S, Coleman R, Keidar S, Aviram M, Kaplan M. Tissue angiotensin-converting-enzyme (ACE) deficiency leads to a reduction in oxidative stress and in atherosclerosis: studies in ACE-knockout mice type 2. Arteriosclerosis, thrombosis, and vascular biology. 2003;23(11):2090–2096. doi: 10.1161/01.ATV.0000098653.74209.C6. [DOI] [PubMed] [Google Scholar]
- 35.Weiss D, Taylor WR. Deoxycorticosterone acetate salt hypertension in apolipoprotein E-/- mice results in accelerated atherosclerosis: the role of angiotensin II. Hypertension. 2008;51(2):218–224. doi: 10.1161/HYPERTENSIONAHA.107.095885. [DOI] [PubMed] [Google Scholar]
- 36.Petrov V, Fagard R, Lijnen P. Effect of protease inhibitors on angiotensin-converting enzyme activity in human T-lymphocytes. Am J Hypertens. 2000;13(5 Pt 1):535–539. doi: 10.1016/s0895-7061(99)00236-8. [DOI] [PubMed] [Google Scholar]
- 37.Lu H, Rateri DL, Feldman DL, Jr RJ, Fukamizu A, Ishida J, Oesterling EG, Cassis LA, Daugherty A. Renin inhibition reduces hypercholesterolemia-induced atherosclerosis in mice. J Clin Invest. 2008;118(3):984–993. doi: 10.1172/JCI32970. [DOI] [PMC free article] [PubMed] [Google Scholar]