

Abstract
In vitro and in vivo characterization of the cyclomarin/cyclomarazine prenyltransferase CymD revealed its ability to prenylate tryptophan prior to incorporation into both cyclic peptides by the non-ribosomal peptide synthetase CymA. This knowledge was utilized to bioengineer novel derivatives of these marine bacterial natural products by providing synthetic N-alkyl tryptophans to a prenyltransferase deficient mutant of Salinispora arenicola CNS-205.
The anti-inflammatory cyclomarin A1 (Figure 1, 8) and antibacterial cyclomarazine A2 (5) are cyclic hepta- and dipeptides produced by the marine actinobacterium Salinispora arenicola CNS-205.3 Common to both peptides is a N-(1,1-dimethyl-1-allyl)-tryptophan (2) residue, which is further oxidized in 8 and cyclomarin C (9). While prenylated indoles, such as 2, are common structural features in fungal and plant natural products,4 these tryptophan-derived moieties are rare in bacteria. We recently characterized the 47,477-bp cyclomarin/cyclomarazine gene cluster cym, which is dominated by the 23,358-bp cymA gene encoding a heptamodular nonribosomal peptide synthetase (NRPS) responsible for the dual assembly of 5 and 8.2 The discovery of the cym locus led to the in vivo characterization of the prenyltransferase (PTase) CymD, which in turn suggested its role in prenylating a common biosynthetic precursor to both peptides prior to assembly.2 This observation contrasts peptidyl PTase reactions such as in the biosynthesis of the cyanobacterial toxin lyngbyatoxin A5 and various fungal diketopiperazines (DKPs)4a-c in which prenylation occurs post-NRPS assembly of the peptide product, although it has been suggested that prenylation of aromatic amino acid residues precedes the assembly of an unknown Aspergillus fumigatus peptide4d as well as in sirodesmin PL.4e In this study, we established the biological function of CymD as 2 synthase, which provided a general strategy to readily generate unnatural N-alkylated tryptophan analogs of 5 and 8 by a unified mutasynthetic approach.
Figure 1.
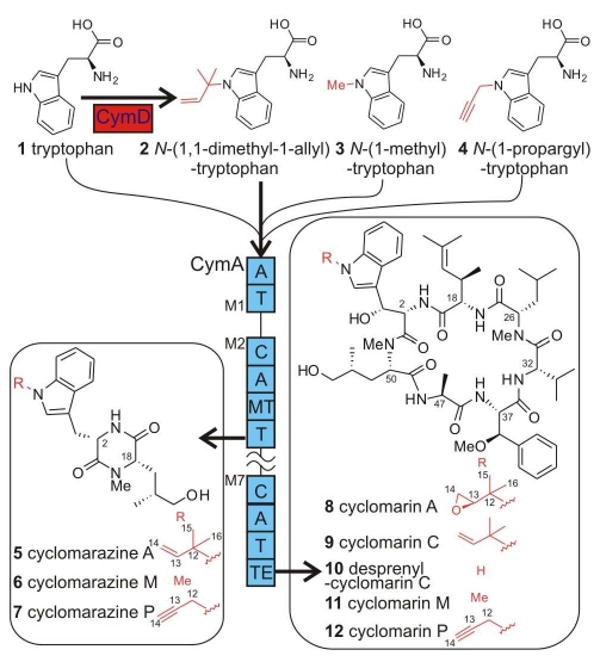
Structures and biosynthesis of natural and unnatural cyclomarin and cyclomarazine analogs. Supplementing cultures of the S. arenicola cymD− mutant with tryptophan analogs yields known and novel cyclomarins and cyclomarazines, thereby establishing 2 as the in vivo substrate for the heptamodular CymA NRPS. Thick arrows denote the pathway operative in wild type S. arenicola CNS-205, while thin lines represent pathways accessible via the cymD-deficient mutant. Abbreviations: A, adenylation domain; C, condensation domain; M1–M7, modules 1–7; MT, methyltransferase; T, thiolation domain; and TE, thioesterase.
Results and Discussion
The targeted disruption of the PTase gene cymD in S. arenicola CNS-205 previously led to a mutant deficient in the known cyclomarin and cyclomarazine chemistry and provided a novel analogue, desprenylcyclomarin C (10).2,6 The >100-fold lower production of 10 in the cymD knockout mutant not only implied that prenylation occurs pre-NRPS assembly, but also suggested that tryptophan is a poor substrate for CymA.2 We thus set out to explore the in vitro function of CymD, which was prepared as a 42 kD octahistidyl-tagged recombinant protein in Escherichia coli. The affinity-purified protein was incubated with l-tryptophan (1) and dimethylallyl pyrophosphate (DMAPP) to yield 2 (Figure 2), which was identical chromatographically and spectroscopically to synthetic 2 prepared by direct prenylation7 of Boc-Trp-OMe (13) followed by deprotection (Scheme 1). Reactions in which DMAPP was replaced by isoprenyl, geranyl, or farnesyl pyrophosphate yielded no detectable products, suggesting a high level of specificity for the isoprene donor. Moreover, addition of Ca2+ or Mg2+ to the reaction mixture did not significantly alter yield, nor did addition of the cation-chelating agent ethylenediaminetetraacetic acid, suggesting that CymD functions cation independently. Alignment of CymD with the fungal indole PTase FgaPT2 (Figure 3) revealed the presence of two conserved lysine residues at positions 146 and 207 consistent with cation-independent fungal PTases8 that belong to an emerging group of divergent prenylating enzymes from bacteria and fungi that share a α/β barrel fold.9
Figure 2.
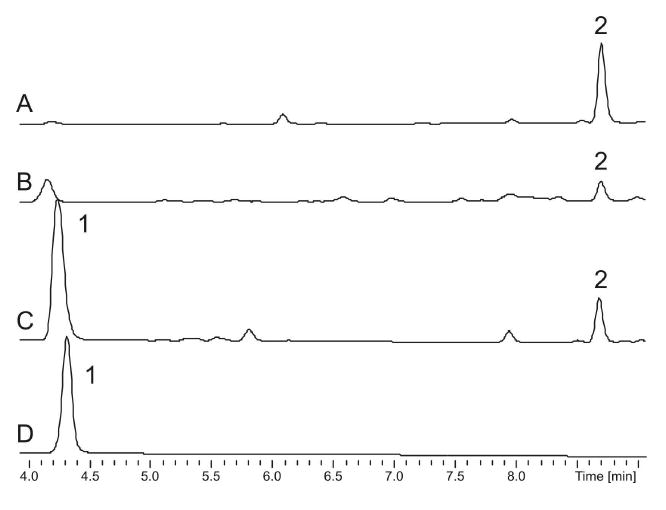
LC-DAD analysis of N-(1,1-dimethyl-1-allyl)-tryptophan (2) at 210 nm. Trace A, synthetic 2; Trace B, natural 2 in the crude organic extract of S. arenicola CNS-205; Trace C, enzymatic 2 from the CymD-catalyzed reaction of DMAPP and tryptophan (1); and Trace D, the CymD boiled control of the enzymatic reaction in C.
Scheme 1.
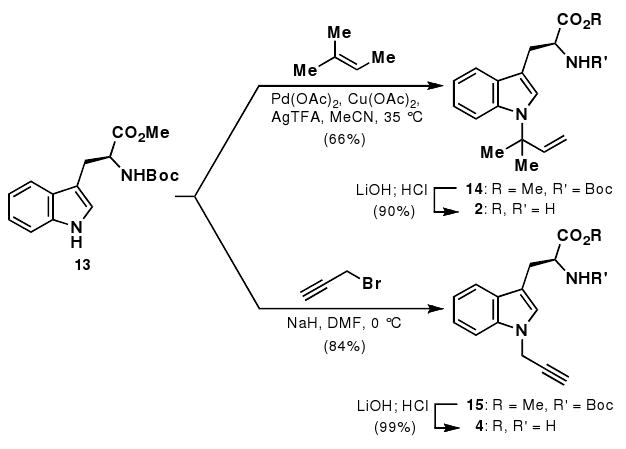
Synthesis of tryptophan substrates.
Figure 3.
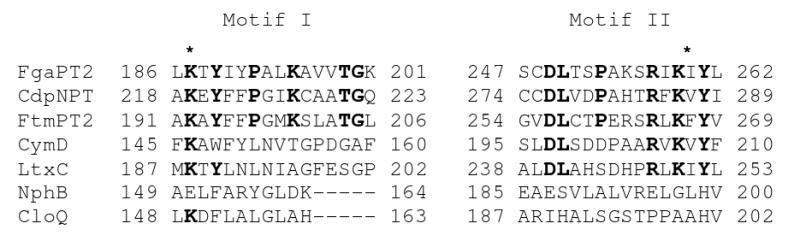
Portion of an alignment of CymD with the active site of FgaPT2 and other prenyltransferases. Fungal indole prenyltransferases (PTases) from Aspergillus fumigatus FgaPT2 (accession no. AAX08549), CdpNPT (ABR14712), and FtmPT2 (AFUA_8G00250) were aligned with bacterial indole PTases CymD from Salinispora arenicola CNS-205 (SARE_4565) and LtxC (Lyngbya majuscula, AAT12285) and bacterial aromatic PTases NphB (Streptomyces sp. CL190, AB187169) and CloQ (Streptomyces roseochromogenes AF329398). Li and coworkers8 have identified the conserved residues in bold, two of which are lysines (*) responsible for the non-metal dependent activity of fungal indole PTases.
Analysis of the S. arenicola CNS-205 fermentation broth revealed that 2 is indeed a natural product produced at 2 mg/L (Figure 2) and that its assembly ceases in the cymD− mutant. The structure of natural 2 was identical in all regards to enzymatic and synthetic 2 (Supporting Information, Figure S3). These data suggested that 2 is preformed and selected by the initiating NRPS module M1 on the heptamodular CymA synthetase to give a common biosynthetic intermediate that ultimately gives rise to natural cyclomarin and cyclomarazine cyclic peptides (Figure 1). We verified this conclusion by the chemical complementation of the cymD− mutant with synthetic 2 that restored the in vivo production of 5 and 8 to wild type levels (Supporting Information, Figures S1 and S2), establishing 2 as the endogenous substrate of the CymA loading didomain M1.
Since prenylation in the cyclomarin/cyclomarazine series occurs on free tryptophan rather than on a peptide precursor as in precedent examples,4a-c,5 it provided us an opportunity to employ a mutasynthesis approach10 to explore whether N-alkyl tryptophan analogues could be simultaneously assimilated into both natural product classes to give rise to novel analogues. Commercially available N-(1-methyl)-tryptophan (3) was first evaluated by administering it to a culture of the cymD− mutant. LC-MS analysis of the resulting organic extract revealed anticipated products that were subsequently isolated and fully characterized by NMR and high-resolution MS to give the novel N-methyl indoles cyclomarin M (Figure 1, 11) and cyclomarazine M (6). NMR analyses (Tables 1 and 2) verified the loss of the respective prenyl groups of 8 and 5 while maintaining the remainder of the peptidic structures. HMBC correlations and chemical shift analyses further established the N-methyl substitution (C-12) on the tryptophan indole ring.
Table 1.
NMR Spectroscopic Data for Cyclomarins M (11) and P (12) in CDC13
| cyclomarin M (11) | cyclomarin P (12) | |||||
|---|---|---|---|---|---|---|
| positiona | δCb | δHc, (J in Hz) | HMBC | δCb | δHc, (J in Hz) | HMBC |
| 1 | 170.75 | 170.46 | ||||
| 2 | 52.90 | 4.57, t (3.9) | 1, 3, 5, 17 | 53.28 | 4.58, t (4.1) | 1, 3, 5, 17 |
| NH-2 | 6.70, d (2.9) | 3, 17 | 6.75, d (1.9) | 2, 3, 17 | ||
| 3 | 67.87 | 5.29, d (3.9) | 1, 4, 5, 6 | 68.34 | 5.28, d (4.4) | 1, 4, 5, 6 |
| OH-3 | 4.13, s | 2, 3 | 4.04d, m | 2, 3, 5 | ||
| 4 | 126.86 | 7.08d, m | 3, 5, 6, 7, 10, 11 | 125.42 | 7.21, s | 2, 3, 5, 10, 11, 12 |
| 5 | 112.33 | 113.52 | ||||
| 6 | 125.32 | 127.93 | ||||
| 7 | 118.82 | 7.50, d (8.0) | 5, 6, 8, 9, 10, 11 | 119.44 | 7.58, d (8.0) | 4, 5, 8, 9, 10, 11 |
| 8 | 119.73 | 7.08d, m | 3, 5, 6, 7, 10, 11 | 120.4 | 7.11d, m | 4, 9, 10, 11 |
| 9 | 122.47 | 7.22, m | 8, 10, 11 | 122.89 | 7.24, m | 6, 7, 11 |
| 10 | 109.89 | 7.28, d (8.3) | 6, 8 | 110.01 | 7.36, d (8.3) | 4, 8, 9 |
| 11 | 137.00 | 136.22 | ||||
| 12 | 32.84 | 3.72, s | 4, 11 | 35.79 | 4.79, d (2.4) | 4, 11, 13, 14 |
| 13 | 77.14 | |||||
| 14 | 74.18 | 2.4, t (2.5) | 12 | |||
| 17 | 172.44 | 172.55 | ||||
| 18 | 58.14 | 4.04, m | 17, 19, 20, 24, 25 | 58.18 | 4.05d, m | 17, 19, 20, 24, 25 |
| NH-18 | 8.00, d (9.4) | 17, 18, 25 | 8.00, d (9.4) | 17, 18, 25 | ||
| 19 | 35.48 | 1.60, s | 18, 20, 21, 24 | 35.55 | 1.61, m | 18, 20, 21, 23 |
| 20 | 124.68 | 4.71d, m | 22, 23, 24 | 124.81 | 4.72, d (9.9) | 19, 22, 23 |
| 21 | 134.56 | 134.55 | ||||
| 22 | 25.71 | 1.70, s | 18, 20, 21, 24 | 25.76 | 1.24, s | 18, 20, 21, 24 |
| 23 | 29.50 | 1.23, s | 18, 20, 21, 22, 24 | 18.86 | 1.72, s | 18, 20, 21, 22, 24 |
| 24 | 18.59 | 0.62, d (6.5) | 18, 19, 20 | 18.51 | 0.62, d (6.5) | 18, 19, 20 |
| 25 | 168.45 | 168.52 | ||||
| 26 | 58.61 | 4.82, m | NMe-26 | 58.63 | 4.85, m | 25, 27, 28, 31, NMe-26 |
| NMe-26 | 29.49 | 2.82, s | 26, 31 | 29.47 | 2.82, s | 26, 31 |
| 27a | 38.84 | 2.28, ddd (13.3, 10.9, 4.4) | 25, 26, 28 | 38.9 | 2.25, m | 25, 26, 28, 29 |
| 27b | 38.84 | 1.02, m | 25, 26, 28 | 38.9 | 1.03, m | 25, 26. 28, 29, 30 |
| 28 | 25.24 | 1.43, m | 25.19 | 1.45, m | 27, 30 | |
| 29 | 23.28 | 0.83, d (6.6) | 27, 28 | 22.83 | 0.85, d (6.6) | 27, 28 |
| 30 | 22.40 | 0.85, d (6.6) | 27, 28 | 23.44 | 0.88, d (6.6) | 27, 28, 29 |
| 31 | 170.4 | 171.39 | ||||
| 32 | 55.3 | 4.34, t (8.8) | 31, 33, 34 | 55.27 | 4.35, t (8.7) | 33, 34, 35, 36 |
| NH-32 | 7.93, d (8.0) | 32, 33, 36 | 7.93, d (8.0) | 32, 33, 36 | ||
| 33 | 30.72 | 2.21, m | 32 | 30.83 | 2.21, m | 32, 34, 35 |
| 34 | 19.29 | 1.04, d (6.7) | 32, 33, 35 | 19.21 | 1.04, d (6.7) | 32, 33, 35 |
| 35 | 19.90 | 0.92, d (6.6) | 32, 33, 34 | 20.52 | 0.92, d (6.7) | 32, 33, 34 |
| 36 | 169.58 | 169.6 | ||||
| 37 | 55.85 | 4.88, m | 36, 38, 39, 46 | 55.89 | 4.88, t (4.9) | 36, 38, 39, 46 |
| NH-37 | 7.14, d (4.7) | 36, 37, 46 | 7.13d, m | 36, 37, 46 | ||
| 38 | 79.81 | 5.04, d (5.3) | 37, 39, 45, 40-44 | 79.92 | 5.05, d (5.3) | 36,37,39,40-44, 45 |
| 39 | 135.08 | 135.15 | 38 | |||
| 40-44 | 127-128 | 7.23-7.25, m | 38, 43 | 127-128 | 7.23-7.25, m | 38, 39 |
| 45 | 57.79 | 3.34, s | 38 | 57.8 | 3.34, s | 38 |
| 46 | 171.68 | 171.79 | ||||
| 47 | 50.58 | 4.86, m | 46, 48 | 50.61 | 4.86, m | 46, 48 |
| NH-47 | 8.19, d (10.3) | 47, 49 | 8.17, d (10.2) | 47, 49 | ||
| 48 | 20.84 | 1.29, d (7.3) | 46, 47 | 20.84 | 1.28, d (7.3) | 46, 47 |
| 49 | 169.7 | 169.13 | ||||
| 50 | 59.30 | 4.72d, m | 51, 52 | 59.28 | 4.68, dd (10.5, 3.0) | 1, 49, 52, NMe-8 |
| NMe-50 | 29.35 | 2.66, s | 1, 50 | 29.33 | 2.64, s | 1,50 |
| 51a | 32.88 | 2.24, m | 49, 52, 53, 54 | 32.79 | 2.16, m | 49, 52, 53, 54 |
| 51b | 32.88 | 0.58, ddd (13.9, 5.7, 3.2) | 49, 52, 53, 54 | 32.79 | 0.41, m | 49, 52, 53, 54 |
| 52 | 33.30 | 1.32, m | 51, 54 | 33.2 | 1.26, m | 50, 51, 53, 54 |
| 53a | 66.09 | 3.23, m | 66.27 | 3.12, m | 51, 54 | |
| 53b | 66.09 | 3.16, m | 66.27 | 3.18, m | 51, 54 | |
| 54 | 17.92 | 0.74, d (6.8) | 51, 52, 53 | 17.68 | 0.65, d | 52, 53 |
Numbering based on Ref. 2.
Assignment by HMQC and HMBC correlations.
600 MHz.
Overlapping signals.
Table 2.
NMR Spectroscopic Data for Cyclomarazines M (6) and P (7) in DMSO-d6
| cyclomarazine M (6) | cyclomarazine P (7) | |||||
|---|---|---|---|---|---|---|
| positiona | δCb | δHc, (J in Hz) | HMBC | δCb | δHc, (J in Hz) | HMBC |
| 1 | 166.19 | 4.10, dd (7.6, 4.3) | 165.52 | |||
| 2 | 55.72 | 8.19, d (2.4) | 1, 3, 5, 17 | 55.62 | 4.12, dd (7.7, 4.3) | 3, 5 |
| NH-2 | 3.19, dd (14.4, 4.9) | 1, 2, 18 | 8.20, d (2.5) | |||
| 3a | 29.67 | 3.00, dd (14.4, 4.5) | 1, 2, 4, 5, 6 | 29.66 | 3.18, dd (14.4, 4.9) | 1, 2, 4, 5, 6 |
| 3b | 29.67 | 7.02, s | 1, 2, 4, 5, 6 | 29.66 | 3.02, dd (14.4, 4.4) | 1, 2, 4, 5, 6 |
| 4 | 128.71 | 3, 5, 6, 7, 11, 12 | 127.18 | 7.11, s | 2, 3, 5, 6, 11, 12 | |
| 5 | 108.37 | 109.28 | ||||
| 6 | 128.60 | 7.50, d (7.9) | 128.36 | |||
| 7 | 118.92 | 6.98, dd (7.5, 7.5) | 5, 6, 8, 11 | 119.06 | 7.53, d (7.9) | 5, 6, 8, 9, 11 |
| 8 | 118.48 | 7.11, dd(7.6, 7.6) | 4, 10 | 118.88 | 7.02, dd(7.3, 7.3) | 6, 7, 10 |
| 9 | 121.04 | 7.34, d (8.2) | 6, 8, 10, 11 | 121.25 | 7.14, dd(7.6,7.6) | 7, 11 |
| 10 | 109.37 | 6, 8, 9 | 109.66 | 7.44, d (8.2) | 8, 6, 9, 11 | |
| 11 | 136.89 | 3.72, s | 135.53 | |||
| 12 | 32.06 | 4, 11 | 34.64 | 5.01, m | 4, 11, 13, 14 | |
| 13 | 79.13 | |||||
| 14 | 75.35 | 3.39, t (2.4) | 12, 13 | |||
| 17 | 167.5 | 3.57, dd (9.0, 3.2) | 166.86 | |||
| 18 | 58.58 | 2.66, s | 1, 17, 19, NMe-18 | 58.57 | 3.57, dd (9.0, 3.1) | 1, 17, 19, 20 |
| NMe-18 | 31.68 | 0.52, ddd (13.1, 9.5, 3.3) | 1, 18 | 31.57 | 2.68, s | 1, 17, 18 |
| 19a | 35.72 | 0.38, ddd (13.7, 9.1, 4.2) | 17, 18, 20, 21, 22 | 35.89 | 0.55d, m | 17, 18, 20, 21, 22 |
| 19b | 35.72 | 1.32, m | 17, 18, 20, 21, 22 | 35.89 | 0.41, ddd (13.7, 9.0, 4.3) | 17, 18, 20, 21, 22 |
| 20 | 32.04 | 2.83, dd (10.3, 5.3) | 19, 21, 22 | 32.17 | 1.34, m | |
| 21a | 65.95 | 2.72, dd (10.1, 7.0) | 19, 20, 22 | 65.91 | 2.81, m | 19, 20 |
| 21b | 65.95 | 4.34, m | 19, 20, 22 | 65.91 | 2.74, m | 19, 20 |
| OH-21 | 0.57, d (6.6) | 4.28, m | ||||
| 22 | 15.92 | 19, 20 | 15.89 | 0.58d, d (6.6) | 17, 19, 20, 21 | |
We next explored incorporating an N-propargyl group to these cyclic peptides in order to prepare analogs appropriate for biological evaluation via semi-synthesis utilizing Click chemistry.11 N-(1-Propargyl)-tryptophan (4) was synthesized by alkylation of Boc-Trp-OMe (13) with propargyl bromide followed by deprotection (Scheme 1). Upon the addition of 4 to the S. arenicola cymD knockout mutant, new cym analogs cyclomarin P (12) and cyclomarazine P (7) carrying the anticipated propargyl side chains (C-12 through C-14) were similarly produced by fermentation. The yields of unnatural 11 and 12 were comparable to that of natural 8 in S. arenicola CNS-205 at 1–3 mg/L with a similar trend observed in the cyclomarazine series. These observations coupled with the inability of tryptophan to restore wild type biosynthetic levels2 suggest that the native CymA loading adenylation (A) domain in module 1 (M1) can accommodate varied N-1 substituted tryptophan substrates for assimilation into the cym hepta- and dipeptides.
Herein we report the characterization of CymD as a bacterial N-(1,1-dimethyl-1-allyl)-tryptophan synthase, which alkylates tryptophan with DMAPP as the prenyl donor in a cation-independent manner prior to NRPS assembly. Although several biochemical studies have been performed on bacterial5 and fungal indole PTases,4 few studies have probed the in vivo timing of the prenyltransfer reaction. In vitro kinetic analysis suggests tryptophan and tyrosine are prenylated by 7-DMATS4d and SirD,4e respectively, prior to incorporation into non-ribosomal peptides, although the final product of the 7-DMATS containing pathway is unknown and in vivo radioisotope incorporation studies contradict the in vitro kinetic analysis of SirD.12 Numerous studies have revealed the promiscuity of bacterial9a and fungal4a prenyltransferases towards related aromatic substrates, making it difficult to solely extend in vitro observations to in vivo biological relevance. N-(1,1-Dimethyl-1-allyl)-tryptophan synthase activity was recently reported for the fungal PTase CdpNPT, which functions to reverse prenylate various tryptophan-containing DKPs at the indole nitrogen yet exhibits a high level of in vitro promiscuity toward indoles such as tryptophan.4b In contrast to CdpNPT's preference to preformed peptides, CymD prenylates free tryptophan, providing both in vitro and in vivo evidence that prenylation occurs prior to NRPS-mediated incorporation into the cyclomarin and cyclomarazine peptides. The preference of CymA module 1 for 2 over tryptophan provided us a unique opportunity to prepare novel N-alkylated cyclomarin and cyclomarazine analogs, which in turn supported the pre-NRPS timing of CymD catalyzed prenyl transfer. This work demonstrates the amenability of cyclomarin/cyclomarazine to diversification via mutasynthesis, providing novel analogues of these di- and heptapeptides and furthering our general understanding of NPRS specificity and amenability to engineering.
Experimental Section
General Experimental Procedures
Chemicals were acquired from Sigma-Aldrich or Fisher Scientific unless noted otherwise. NMR spectroscopic data were obtained on a Bruker 600 MHz spectrometer equipped with a 1.7 mm cryoprobe. LC analysis was performed on a Agilent 1200 series LC or a HP series 1100 LC-MS system utilizing electrospray ionization in positive mode with a linear gradient of 10-90% acetonitrile at 0.7 mL/min over 24 min utilizing a Luna 4.6 × 100 mm, 5 μm C18(2) column (Phenomenex). High-resolution mass spectra were collected by ESI-HR-FTMS at the Chemistry & Biochemistry Mass Spectrometry Facility, University of California San Diego and HR-ESI-TOFMS at the Scripps Research Institute, La Jolla, CA.
Bacterial Strains and Culture Conditions
Wild type S. arenicola CNS-205 and the prenyltransferase deficient mutant S. arenicola cymD- were grown in A1+BFe media (10 g starch, 4 g yeast extract, 2 g peptone, 40 mg of Fe2(SO4)3•4H20, 100 mg KBr, 1 L seawater) at 28 °C and 200 rpm.2 Production cultures were grown for 10 days in 2.8 L fernbach flasks containing 1 L of media unless stated otherwise. Escherichia coli DH5α was used for cloning and expression experiments as described.13 DNA purification and manipulation was performed according to standard procedures.13,14
CymD purification
The cymD gene (GenBank protein accession SARE_4565, genome ascension CP00850) was amplified from genomic S. arenicola CNS-205 DNA via PCR utilizing the forward primer cymDF1 5′-CGTGGTTCGAGCTCTTGACCGAGGAGTTGACGACGGTCCG (SacI site underlined) and the reverse primer cymDR1 5′-GCTCGAATTCAAGCTTTCATTCGGTTCTCCCTCTCG (HindIII site underlined). Ligation into pHIS815 yielded plasmid pHIS8-cymD, which was transformed into E. coli BL21(DE3) (Invitrogen) for expression. Transformants were grown in ZYP-5052 autoinduction media16 containing 50 μg/mL kanamycin at 16 °C for two days. Cells were harvested and lysed by sonication in lysis buffer (50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole, 10% glycerol, 2 mM β-mercaptoethanol) with the addition of 0.5 g/L lysozyme. The lysate was cleared by centrifugation at 20,000 rpm for 50 min, and the resulting supernatant was loaded onto a polypropylene column containing 6 mL Ni-NTA agarose (Qiagen). The column was washed with wash buffer (50 mM NaH2PO4, 300 mM NaCl, 20 mM imidazole, 10% glycerol, 2 mM β-mercaptoethanol), and eluted in elution buffer (50 mM NaH2PO4, 300 mM NaCl, 250 mM imidazole, 10% glycerol, 2 mM β-mercaptoethanol). The resulting eluate was placed in a 10 kD cut off dialysis tube and dialyzed overnight against 2 L of phosphate buffer (50 mM sodium phosphate pH 8.0, 300 mM NaCl, 2 mM DTT). Octahistidyl-tagged CymD was further purified by FPLC (GE Healthcare) utilizing a Superdex 200 26/60 size exclusion column with phosphate buffer as the mobile phase, and fractions containing CymD were pooled and concentrated to a final concentration of 2.34 mg/mL using a Centriprep Ultracel YM-10 (10 kD cutoff, Millipore).
CymD Prenyltransferase Assay
His8-tagged CymD (1 μg, 0.2 μM) was incubated with 1 mM l-tryptophan and 1 mM dimethylallyl pyrophosphate (DMAPP) in 100 μL 50 mM imidazole buffer, pH 6.8, and incubated for 2 h at 37 °C. The reaction was quenched with 10 μL 1.5 M TCA, and centrifuged for 10 min at 13,000 rpm. The supernate was then analyzed by LC or LC-MS. To assay for isoprene pyrophosphate specificity, DMAPP was replaced with 1 mM isopentenyl pyrophosphate, geranyl pyrophosphate, or farnesyl pyrophosphate. Metal dependence was interrogated by including 10 mM Ca2+, 10 mM Mg2+, or no metal added with or without 50 mM EDTA. To characterize the product of of CymD-catalyzed tryptophan conversion, 3.25 mmol l-tryptophan, 3.25 mmol DMAPP, and 380 μg enzyme were incubated in 3.25 ml 50 mM imidazole buffer, pH 6.8, overnight at 37 °C. The reaction was dried in vacuo, redissolved in a minimal volume of MeOH, and was fractionated by preparative HPLC utilizing an Onyx C18 100 × 10 mm column (Phenomenex) in isocratic 40% MeOH at 5 mL/min, with N-(1,1-dimethyl-1-allyl)-tryptophan eluting at 9.5 min, yielding 2.3 mg product after removing the mobile phase in vacuo.
Purification of N-(1,1-dimethyl-1-allyl)-tryptophan from S. arenicola CNS-205
A 4 L production culture of S. arenicola CNS-205 was incubated with XAD-7HP resin for 2 h. The resin and cells were collected by filtration and extracted in acetone, which in turn was evaporated in vacuo. The pH of the remaining aqueous residue was adjusted to ∼14 with 10 N NaOH and washed with EtOAc. The aqueous phase was then adjusted to ∼pH 2 with concentrated HCl and extracted with EtOAc. The EtOAc fraction was dried over MgSO4 and evaporated in vacuo. The resulting residue was fractionated using YMC ODS-A resin (60 mm × 100 mm) with a step gradient of aqueous 20% MeOH followed by 50% MeOH. Fractions containing N-(1,1-dimethyl-1-allyl)-tryptophan (2) were concentrated in vacuo, and the remaining aqueous residue was acidified with glacial acetic acid to pH ∼4. Compound 2 was absorbed onto an equilibrated 1 g SCX cation-exchange SPE cartridge (Phenomenex) from the acidified fractions, which in turn was eluted with 3 M NH4OH. The eluate was desalted using a 1 g C18 SPE cartridge (Thermo), yielding 2.3 mg 2 HRMS (ESI+) m/z 273.1599 [M+H+] (calcd for C16H21N2O2, 273.1597).
Synthesis of N-(1,1-dimethyl-1-allyl)-tryptophan (2)
Literature procedures were utilized for the synthesis of N-(1,1-dimethyl-1-allyl)-Boc-Trp-OMe (14).7 Compound 14 (190.0 mg, 0.492 mmol) was dissolved in THF (5 mL) and MeOH (0.5 mL). LiOH (250 μL, 1 M) was then added and the reaction monitored by TLC. After completion of hydrolysis, the reaction was acidified with 1 M HCl and repeatedly extracted with CH2Cl2. The organic extracts were combined, dried with brine and then further dried over MgSO4. The reaction mixture was concentrated and then deprotected with HCl•dioxane (4 mL, 4 M). After complete deprotection, the reaction mixture was concentrated to afford an off-white product (135.8 mg, 90% over two steps).
N-(1,1-Dimethyl-1-allyl)-tryptophan (2)
mp 135-138 °C; [α]20.0 +21.1 (c 0.5, pyridine); IR (film) vmax 2978, 2930, 1715, 1589, 1515, 1457, 1366, 1315, 1215, 1161, 919 cm-1; 1H NMR (D2O, 600 MHz) δ 7.68 (1 H, d, J = 7.8 Hz), 7.66 (1 H, d, J = 8.4 Hz), 7.48 (1 H, s), 7.21 (t1 H, J = 7.2 Hz), 7.17 (1 H, t, J = 7.8 Hz), 6.13 (1 H, dd, J = 17.4, 10.8 Hz), 5.22 (1 H, d, J = 10.8 Hz), 5.12 (1 H, d, J = 17.4 Hz), 4.28 (1 H, t, J = 6.0 Hz), 3.48 (1 H, dd, J = 15.6, 5.4 Hz), 3.39 (1 H, dd, J = 15.6, 7.2 Hz), 1.73 (H6, s); 13C NMR (D2O, 150 MHz) δ 171.2, 142.4, 134.1, 127.3, 125.1, 120.1, 118.4, 117.4, 113.5, 112.4, 104.0, 58.1, 52.6, 25.9, 24.7; HRMS (ESI+) m/z 273.1600 [M+H+] (calcd for C16H21N2O2, 273.1597)
Synthesis of N-(1-propargyl)-tryptophan
Boc-Trp-OMe (13, 150.0 mg, 0.471 mmol) was dissolved in DMF (3 mL), and NaH (60% dispersion in oil, 24.0 mg) was then added slowly at 0 °C. Propargyl bromide (63 μL, 80% wt solution in toluene) was added dropwise over 5 min. After 1 h at 0 °C, TLC showed complete consumption of starting material and the reaction was quenched with aqueous NH4Cl. The reaction was extracted three times with EtOAc, and the combined EtOAc extracts were then washed with water and brine three times. The crude mixture was dried over MgSO4 and passed through a silica gel plug (1/3 EtOAc/hexanes v/v) to afford a colorless oil (141.8 mg, 84%). The alkylated material (15) was then dissolved in THF (5 mL) and hydrolyzed with LiOH (1.0 mL, 0.5 M). The reaction was complete by TLC after 2 h and was acidified with 1 M HCl. The reaction was then extracted several times with CH2Cl2 and the organic layer was sequentially dried with brine and MgSO4. The crude acid was deprotected with HCl•Dioxane (4 mL, 4 M) and monitored by LC-MS. After complete conversion to product, the reaction was then concentrated to provide a yellow solid (110.1 mg, 99% over two steps).
N-(1-Propargyl)-Boc-Trp-OMe (15)
[α]20.0 +30.3 (c 1.0, CHCl3); IR (film) vmax 3284, 2977, 1742, 1709, 1500, 1467, 1438, 1366, 1250, 1165, 1060, 1015 cm-1; 1H NMR (CDCl3, 600 MHz) δ 7.58 (1 H, d, J = 7.8 Hz), 7.35 (1 H, d, J = 8.4 Hz), 7.26 (1 H, t, J = 6.6 Hz), 7.16 (1 H, t, J = 7.2 Hz), 7.02 (1 H, s), 5.17 (1 H, d, J = 7.8 Hz), 4.78 (2 H, d, J = 2.4 Hz), 4.67 (1 H, br q, J = 7.8 Hz), 3.69 (3 H, s), 3.31 (1 H, dd, J = 15.0, 5.4 Hz), 3.26 (1 H, dd, J = 15.0, 6.0 Hz), 2.41 (1 H, t, J = 2.4 Hz), 1.46 (9 H, s); 13C NMR (CDCl3, 150 MHz) δ 172.7, 155.2, 136.0, 128.6, 125.8, 122.1, 119.7, 119.1, 109.8, 109.4, 79.7, 77.7, 73.6, 54.2, 52.2, 35.6, 28.3, 27.9; HRMS (ESI+) m/z 357.1814 [M+H+] (calcd for C20H25N2O4, 357.1809); TLC Rf 0.59 [EtOAc/Hexanes (3:7)].
N-(1-Propargyl)-tryptophan (4)
mp decomp. > 175 °C; [α]20.0 +44.0 (c 0.5, pyridine); 1H NMR (D2O, 600 MHz) δ 7.70 (1 H, d, J = 7.8 Hz), 7.55 (1 H, d, J = 8.4 Hz), 7.35 (1 H, t, J = 7.2 Hz), 7.32 (1 H, s), 7.23 (1 H, t, J = 7.2 Hz), 4.98 (1 H, d, J = 1.8 Hz), 4.27 (1 H, dd, J = 7.2, 5.4 Hz), 3.48 (1 H, dd, J = 15.0, 4.8 Hz), 3.38 (1 H, dd, J = 15.6, 7.2 Hz), 2.75 (1 H, s); 13C NMR (D2O, 150 MHz) δ 171.5, 135.2, 127.4, 126.8, 121.8, 119.4, 118.1, 109.6, 106.2, 78.0, 73.0, 52.8, 34.6, 25.0; IR (film, cm-1) 3265, 2853, 1731, 1584, 1485, 1464, 1329, 1204, 1181, 1058; HRMS (ESI+) m/z 243.1130, [M+H+] (calcd for C14H15N2O2, 243.1128).
Chemical Complementation of the cymD- Mutant and the Production of Novel Cyclomarin and Cyclomarazine Analogues via Mutasynthesis
Duplicate 50 mL cultures of S. arenicola CNS-205 and the cymD- mutant strain were grown in 250 mL Erlenmeyer flasks containing stainless steel springs. To duplicate mutant cultures, 4 mg tryptophan, N-(1,1-dimethyl-1-allyl)-tryptophan (2), N-(1-methyl)-tryptophan (3), or N-(1-propargyl)-tryptophan (4) were added as 1 mg aliquots daily from day 6 through and including day 9. On the following day 10, the cultures were extracted with EtOAc, and the crude residues analyzed by analytical LC-MS as described above. For the production of analogs, 1 L cultures of the cymD- mutant were administered compounds 3 (1 g) or 4 (50 mg) dissolved in H2O in four equal daily aliquots starting on the 6th day of growth. The cultures were extracted by EtOAc partitioning to yield approximately 130 mg crude organic extracts, which were fractionated utilizing Sephadex LH-20 resin (GE Healthcare, 35 g resin in a 30 mm wide column) with MeOH as the mobile phase. Fractions containing the target compound, as judged on the basis of ESIMS were pooled, dried in vacuo, and subjected to purification via preparative HPLC utilizing a Luna C18(2) 250 × 10 mm column (Phenomenex) under isocratic conditions with a flow rate of 2.5 mL/min and monitored at 210 nm. Methyl indole derivatives cyclomarazine 6 (0.4 mg) eluted at 33 min with 21% MeCN and cyclomarin 11 (2.6 mg) eluted at 36 min with 52% MeCN. Propargyl derivatives cyclomarazine 7 (1.5 mg) eluted at 33 min with 24% MeCN, while cyclomarin 12 (1.0 mg) eluted at 29 min with 56% MeCN.
Cyclomarazine M (6)
NMR data, see Table 2; HRMS (ESI+) m/z 366.1790 [M+Na]+ (calcd for C19H25N3O3Na, 366.1788).
Cyclomarazine P (7)
NMR data see Table 2. HRMS (ESI+) 390.1790 m/z [M+Na]+ (calcd for C21H25N3O3Na 390.1788).
Cyclomarin M (11)
NMR data see Table 1. HRMS (ESI+) 995.5587m/z [M+Na]+ (calcd for C52H76N8O10Na 995.5577).
Cyclomarin P (12)
NMR data see Table 1. HRMS (ESI+) 1019.5582m/z [M+Na]+ (calcd for C54H76N8O10Na 1019.5577).
Supplementary Material
Acknowledgments
The detection of 2 in S. arenicola was originally observed by our UCSD colleagues R. Asolkar and W. Fenical (personal communication). We thank S. Richard and J. P. Noel for assistance with purification of CymD. Financial support was provided by the California Sea Grant Program (R/NMP-98 to B.S.M.) and the NIH (Grants GM085770 to B.S.M. and CA134785 to P.S.B.), and fellowships were provided to A.W.S. (NIH GM067550), C.A.L. (Canadian Institutes of Health Research), and M.R.L. (NIH GM082092).
Footnotes
Dedicated to the late Richard E. Moore of the University of Hawaii at Manoa for his pioneering work on bioactive natural products.
Supporting Information. Additional materials and methods, bioinformatic analysis, and spectrophotometric data. This material is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
- 1.Renner MK, Shen YC, Cheng XC, Jensen PR, Frankmoelle W, Kauffman CA, Fenical W, Lobkovsky E, Clardy J. J Am Chem Soc. 1999;121:11273–11276. [Google Scholar]
- 2.Schultz AW, Oh DC, Carney JR, Williamson RT, Udwary DW, Jensen PR, Gould SJ, Fenical W, Moore BS. J Am Chem Soc. 2008;130:4507–4516. doi: 10.1021/ja711188x. [DOI] [PubMed] [Google Scholar]
- 3.Penn K, Jenkins C, Nett M, Udwary DW, Gontang EA, McGlinchey RP, Foster B, Lapidus A, Podell S, Allen EE, Moore BS, Jensen PR. ISME J. 2009;3:1193–1203. doi: 10.1038/ismej.2009.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Steffan N, Grundmann A, Yin WB, Kremer A, Li SM. Curr Med Chem. 2009;16:218–231. doi: 10.2174/092986709787002772. [DOI] [PubMed] [Google Scholar]; (b) Zou H, Zheng X, Li SM. J Nat Prod. 2009;72:44–52. doi: 10.1021/np800501m. [DOI] [PubMed] [Google Scholar]; (c) Ruan HL, Yin WB, Wu JZ, Li SM. ChemBioChem. 2008;9:1044–1047. doi: 10.1002/cbic.200700723. [DOI] [PubMed] [Google Scholar]; (d) Kremer A, Westrich L, Li SM. Microbiology. 2007;153:3409–3416. doi: 10.1099/mic.0.2007/009019-0. [DOI] [PubMed] [Google Scholar]; (e) Kremer A, Li SM. Microbiology. 2009 doi: 10.1099/mic.0.033886-0. [DOI] [Google Scholar]
- 5.(a) Edwards DJ, Gerwick WH. J Am Chem Soc. 2004;126:11432–11433. doi: 10.1021/ja047876g. [DOI] [PubMed] [Google Scholar]; (b) Read JA, Walsh CT. J Am Chem Soc. 2007;129:15762–15763. doi: 10.1021/ja077374d. [DOI] [PubMed] [Google Scholar]
- 6.Liu WT, Ng J, Meluzzi D, Bandeira N, Gutierrez M, Simmons TL, Schultz AW, Linington RG, Moore BS, Gerwick WH, Pevzner PA, Dorrestein PC. Anal Chem. 2009;81:4200–4209. doi: 10.1021/ac900114t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Luzung MR, Lewis CA, Baran PS. Angew Chem Int Ed. 2009;48:7025–7029. doi: 10.1002/anie.200902761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stec E, Steffan N, Kremer A, Zou H, Zheng X, Li SM. Chembiochem. 2008;9:2055–2058. doi: 10.1002/cbic.200800237. [DOI] [PubMed] [Google Scholar]
- 9.(a) Kuzuyama T, Noel JP, Richard SB. Nature. 2005;435:983–987. doi: 10.1038/nature03668. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Metzger U, Schall C, Zocher G, Unsöld I, Stec E, Li SM, Heide L, Stehle T. Proc Natl Acad Sci USA. 2009;106:14309–14314. doi: 10.1073/pnas.0904897106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kirschning A, Taft F, Knobloch T. Org Biomol Chem. 2007;5:3245–3259. doi: 10.1039/b709549j. [DOI] [PubMed] [Google Scholar]
- 11.Kolb HC, Sharpless KB. Drug Discovery Today. 2003;8:1128–1137. doi: 10.1016/s1359-6446(03)02933-7. [DOI] [PubMed] [Google Scholar]
- 12.Ferezou JP, Quesneau-Thierry A, Servy C, Zissmann E, Barbier M. J Chem Soc Perkin Trans. 1980;1:1739–1746. [Google Scholar]
- 13.Sambrook J, Russell DW. Molecular Cloning: A Laboratory Manual (3-Volume Set) Cold Spring Harbor Laboratory Press; Cold Spring Harbor, New York: 2001. [Google Scholar]
- 14.Kieser T, Bibb M, Buttner M, Chater K, Hopwood D. Practical Streptomyces Genetics. John Innes Foundation; Norwich, United Kingdom: 2000. [Google Scholar]
- 15.Jez JM, Ferrer J, Bowman ME, Dixon RA, Noel JP. Biochemistry. 2000;39:890–902. doi: 10.1021/bi991489f. [DOI] [PubMed] [Google Scholar]
- 16.Studier FW. Protein Expr Purif. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.