

Summary
The Y-family translesion DNA polymerases enable cells to tolerate many forms of DNA damage, yet these enzymes have the potential to create genetic mutations at high rates. Although this polymerase family was defined less than a decade ago, more than 90 structures have already been determined so far. These structures show that the individual family members bypass damage and replicate DNA with either error-free or mutagenic outcomes, depending on the polymerase, the lesion and the sequence context. Here, these structures are reviewed and implications for polymerase function are discussed.
Keywords: error-prone DNA polymerase, translesion synthesis, lesion bypass, mutagenic replication
1. Introduction
Our understanding of how cells tolerate DNA damage has grown explosively in the past decade, sparked by the discovery of polymerase (pol) η as the protein that is non-functional in individuals with the variant form of xeroderma pigmentosum [1, 2], a disease that causes extreme sensitivity to UV light and a predisposition for the development of skin cancer at an early age. Pol η accurately performs translesion DNA synthesis across cyclobutane pyrimidine dimers (CPDs), which are DNA lesions induced by UV light [3, 4]. When pol η function is absent, other polymerases take over the replication of CPDs, but they copy the damaged DNA much less accurately [5, 6], leading to an accumulation of mutations that eventually cause cells to become cancerous. A number of proteins evolutionarily related to pol η were quickly identified, some of which either had been known for years to be involved in the DNA damage response [7] or had been shown previously to possess nucleotidyl transferase activity [8]. In 2001, these enzymes were designated the Y-family polymerases [9], and the first structures of family members were published later that same year [10-13].
Phylogenetic trees of the Y family divide the polymerases into six branches: the DinB enzymes, found in bacteria, archaea and eukaryotes; the Rad30A (pol η), Rad30B (pol ι) and Rev1 proteins, found only in eukaryotes; and two UmuC branches, found in gram-positive and gram-negative bacteria [9]. Each branch displays a different specificity as to the types of lesions that are replicated efficiently and the types of mistakes made during replication. None of these polymerases contains a proofreading exonuclease domain. The Y-family polymerases display a wide range of error rates, but tend to be 100- to 1000-fold less accurate than polymerases from other families [14]. The DinB polymerases are generally the most accurate, making about 1 mistake per 103 to 104 bases copied. At the other extreme are the pol ι enzymes, which preferentially (mis)incorporate deoxyguanosine (dG) rather than deoxyadenosine (dA) opposite a template thymidine (dT) [15]. The Rev1 proteins specifically incorporate deoxycytidine (dC) [8], and are technically terminal nucleotidyl transferases rather than polymerases, because they ignore the basepairing information that is present in the templating strand of DNA. Frequently, more than one specialized polymerase is required to fully bypass a lesion [15-17]: one inserts a nucleotide opposite the damaged template DNA and a second extends DNA synthesis until a replicative polymerase is able to resume high fidelity DNA synthesis.
More than 90 Y-family DNA polymerase structures have been published to date (Supplementary Table S1). Nearly two-thirds of the structures are of the archaeal DNA polymerase IV (Dpo4) from Sulfolobus solfataricus, which has become the archetype for the family, due to the relative ease with which it forms crystals that diffract to high resolution (beyond 2 Å in some cases) [13]. The remaining structures solved are of: Dbh from Sulfolobus acidocaldarius [10, 12]; pol η from Saccharomyces cerevisiae [11]; pol ι and pol κ, from humans [18, 19]; and Rev1 from both Saccharomyces cerevisiae and humans [20, 21]. The C-terminal domain of DinB (pol IV) from E. coli has also been crystallized [22], leaving the UmuC enzymes as the only branches of the Y-family polymerase tree for which crystal structures have not yet been determined.
The aim of this review is to provide a relatively concise yet comprehensive summary of the wide range of Y-family polymerase structures that have been determined. The major focus is on the structural features that give each branch of the polymerase family its particular specificity on damaged and undamaged template DNA. The lesions are grouped according to the type of structural problem that they pose for replication, rather than on their chemical similarity. A final topic is the interactions between the Y-family polymerases and sliding-clamp processivity factors.
2. Structural overview: conserved and distinctive features
2.1. Two-domain polymerase structure
The Y-family polymerases contain, at a minimum, two domains tethered together by a relatively unstructured linker (Fig. 1A). The N-terminal domain contains all of the residues required for catalysis (Fig. 1B), while the C-terminal domain contributes substantially to DNA binding and is required for full polymerase activity [10, 11]. The conserved sequences that define the polymerase family are all located within the N-terminal catalytic core domain. Even though the C-terminal domain does not contain primary sequence motifs that are conserved throughout the Y-family, this domain has a conserved tertiary structure, a four-stranded β sheet supported on one side by two long α helices. This domain is commonly referred to as the little finger (LF) in the archaeal [13] and bacterial enzymes [22] and as the polymerase-associated domain (PAD) in the eukaryotic proteins [11]. This review will use the combined acronym, LF/PAD, to emphasize the underlying structural relationship of the domain among all the Y-family polymerases.
Fig. 1. Structural overview of Dpo4, a model Y-family polymerase.

(A) Ribbons diagram of a ternary complex of Dpo4 (PDB code 2AGQ [37]) and a diagram showing the linear order of the polymerase domains. The palm (magenta), fingers (blue), and thumb (green) comprise the N-terminal catalytic core domain that is linked to the C-terminal LF/PAD domain (orange). The B/PIP (β-clamp / PCNA interaction peptide) sequence at the very C-terminus of the protein is disordered. The loop located between strands β2 and β3 of the fingers domain is identified (β2-3 loop). The primer and template DNAs are white, and the incoming nucleotide (dNTP) is shown in ball and stick representation. (B) Stereo view of the Dpo4 polymerase active site showing a ribbons representation of the palm domain sequences that contain motifs A and C that are found in all polymerases belonging to the “classical” polymerase superfamily. Key residues binding incoming dNTP and divalent metals A and B are shown. Coloring is by atom: carbon (white), oxygen (red), nitrogen (blue), phosphate (yellow), and calcium (green). In this and subsequent figures: template bases are numbered with position zero as the nucleotide in the nascent basepair binding pocket, pairing with dNTP; bases to the 5′ (downstream) side are given sequential positive numbers; bases to the 3′ (upstream) side are give sequential negative numbers; hydrogen bonds are shown as dotted lines.
The catalytic core is composed of three subdomains, the fingers, palm and thumb, named based on the resemblance to an open right hand. Because the palm has the same topology as found in most other polymerases families (the A- and B-family polymerases, the reverse transcriptases, and the viral RNA-directed RNA polymerases), the Y-family polymerases clearly belong to the classical polymerase superfamily [10, 11]. The other structural features (fingers, thumb and LF/PAD) are unique to the Y-family polymerases.
2.2. General features of substrate binding and catalysis
The Y-family polymerases use the same two-metal-ion catalytic mechanism [23] as is used by both the classical and β-nucleotidyl transferase polymerase superfamilies (Fig. 1B). One of the two metal ions (metal B) is coordinated by the catalytic aspartates (D7 and D105 in Dpo4), by the α, β and γ phosphates of the incoming nucleotide, and by a backbone oxygen atom (F8 in Dpo4). The second metal ion (metal A) is coordinated by the two catalytic aspartates and, in some structures, by an adjacent glutamate (E106 in Dpo4). In the vast majority of the structures, the divalent metal ions are Ca2+, which is present in most of the crystallization conditions that have been used. In cells, however, Mg2+ is expected to be the divalent cation at both the metal A and B positions. In addition to participating in coordination of metal B, the phosphates of the incoming nucleotide are bound by conserved residues in the fingers (Y48 and R51 in Dpo4) and the palm (K159 in Dpo4).
As is true for other DNA polymerases, the ribose ring of the incoming nucleotide is positioned by stacking on top of a “steric gate” residue (Y12 in Dpo4; Y or F in other Y-family polymerases) and by the 3′ OH's formation of a hydrogen bond with the backbone nitrogen of the same residue (Fig. 1B). The steric gate residue selects against the incorporation of ribonucleotides by the polymerase [24]; steric conflicts between the residue and the 2′ OH of an rNTP would prevent the nucleotide from being aligned for catalysis. The base of the incoming nucleotide stacks between the previously formed basepair and the fingers, forming hydrogen bonds with the templating base (Fig. 1B).
In all of the Y-family polymerases, the DNA duplex is held between the two domains of the polymerase, with the phosphoribose backbones of both the primer and template strands being contacted across the minor groove by the thumb and across the major groove by the LF/PAD (Fig. 1A and Fig. 2). The 5′ single stranded template DNA is guided into the polymerase active site through contacts with the fingers and/or LF/PAD.
Fig. 2. Archaeal and eukaryotic Y-family polymerases.

Crystal structures of ternary complexes of (A) Dpo4 (PDB code 2AGQ [37]), (B) Dbh (PDB code 3BQ1 [34]), (C) human pol kappa (PDB code 3HED [53]), (D) yeast Rev1 (PDB code 2AQ4 [20]), (E) yeast pol eta (PDB code 2R8J [35]), and (F) human pol iota (PDB code 3GV8 [38]) are shown with the conserved protein domains shown in surface representation. The N-clasp of pol kappa, the N-digit of Rev1, and the insert between the fingers and palm of pol eta are shown in ribbons representation, colored yellow. The polymerase palms were aligned and are shown to scale from the same orientation.
Compared to the higher-fidelity DNA polymerases, the Y-family polymerases impose relatively few constraints on the nascent basepair. The fingers are much smaller than in most other polymerases, resulting in virtually no contact with the major groove side of the nascent basepair, and the palm subdomain similarly has very little contact with the minor groove. The major constraint on the geometry of the nascent basepair (other than stacking between the fingers and the terminal basepair) seems to be on the width of the duplex [25]; the incoming nucleotide is fixed by the active site, and the templating nucleotide is constrained by contacts between the DNA backbone and residues from the fingers and/or the LF/PAD.
2.3. Structural variations among the Y-family polymerases
Some of the largest differences among the Y-family enzymes occur in regions of the protein that are outside of the two-domain structure of the minimal polymerase. The eukaryotic enzymes have long N- and/or C-terminal extensions that, for the most part, are poorly characterized in terms of structure. Both pol κ and Rev1 contain N-terminal sequences that form integral parts of the polymerase: the N-clasp [26], and the N-digit [20], respectively (Fig. 2C and 2D).
The N-clasp of pol κ crosses over the DNA, linking the fingers and thumb subdomains (Fig. 2C) [26]. The N-clasp also contacts the LF/PAD, so that the DNA duplex is completely encircled by protein. Additionally, a relatively unstructured sequence at the N-terminus of pol κ reaches over the fingers toward the nascent basepair, partially closing off the very spacious active site (Fig. 2C). The N-digit of Rev1 is a long α-helix that fits between the palm and LF/PAD (Fig. 2D) [20], forming a critical part of the nascent basepair binding pocket (see section 3.4).
To the C-terminal side of the LF/PAD domain, all of the Y-family enzymes except Rev1 contain a short β-clamp or PCNA interaction peptide (B/PIP) that is required for interaction with the sliding clamp processivity factors in bacteria, eukaryotes, and archaea. Rev1 also interacts with the sliding clamp, but through a BRCA1 C-terminal (BRCT) domain rather than a B/PIP [27]. In Dpo4, Dbh and E. coli DinB, the B/PIP is located immediately following the LF/PAD (Fig. 1A). The B/PIP is disordered [12, 13], unless it is bound to the β-clamp or PCNA (see section 6.1).
The C-terminal extensions of the eukaryotic proteins contain: ubiquitin-binding motifs (UBM) and ubiquitin-binding zinc finger (UBZ) domains [28]; sites for interaction of polymerases κ, η and ι with Rev1 [29, 30]; and nuclear localization signals. Rev1 is unique among the Y-family polymerases in containing a BRCT domain, part of its N-terminal extension [27]. The only structures available for these C-terminal extensions are of the BRCT domain of Rev1 (PDB code 2EBW [31]) and the UBZ domain of pol η (PDB code 2I5O [32]), both of which have been determined by NMR spectroscopy as isolated domains. A recent review illustrates the locations of these motifs in the various Y-family polymerases, and discusses their functions in more detail [33].
Within the minimal Y-family polymerase structure, one of the most significant variations is in the size of the loop between strands β2 and β3 (β2-3 loop) in the fingers domain (Fig. 1A), since the loop is located where it can substantially alter the protein-DNA contacts as the template DNA enters the active site (Fig. 1A). In Dpo4, the β2-3 loop contacts the LF/PAD, helping to form a channel through which the single-stranded DNA passes (Fig. 2A) [13]. In Dbh, the loop makes some contacts with the template DNA, but is very poorly ordered and does not contact the LF/PAD (Fig. 2B) [34]. In pol κ, the loop is much shorter, and does not contact the LF/PAD; consequently, a large, unobstructed gap is left between the polymerase and LF/PAD domains in this region (Fig. 2C), even though the N-clasp forms contacts between these domains on the other side of the DNA [26]. The β2-3 loop in Rev1 is even longer; together with the N-digit and the LF/PAD, it forms a tunnel through which the template DNA must pass before gaining access to the active site (Fig. 2D) [20]. In pol η [11] and ι [18] (Fig. 2E-F), there is a tight turn instead of a loop between β2 and β3. In the case of pol η (see section 4.1), a second base is able to fit into the active site area on the 5' side of the templating base [35]. Pol η also contains an insertion between the fingers and palm (Fig. 2E), providing contacts between the polymerase domain and the LF/PAD (Fig. 2E) [11].
Most of the other structural variations found in the Y-family polymerases are insertions that occur either between domains or in surface loops within a domain. Most of these additional sequences are located away from substrate-binding sites and are less likely to affect substrate binding and catalysis.
2.4. Conformational flexibility
A notable aspect of the Y-family polymerases is the lack of flexibility of the fingers subdomain, since in other DNA polymerase families movement of the fingers is required for assembling the active site around the incoming nucleotide. No large-scale movement of the fingers has been observed for any of the Y-family polymerases, although smaller rearrangements of tri-phosphate-binding residues have been observed in Dbh [34] and Dpo4 [36]. These rearrangements correlate with movement of an α-helix in the fingers subdomain upon binding of the incoming nucleotide. The functional significance of these movements has not been determined, but could correspond to a rate-limiting conformational change. The thumb subdomain of the Y-family polymerases frequently moves upon primer-template duplex binding, as is often observed in other polymerase families.
The catalytic core and LF/PAD domains of the Y-family polymerases, in contrast, have an enormous potential for adopting different positions relative to one another. The polypeptide sequence that links the thumb of the catalytic core to the LF/PAD, about 10 amino acids long, is sensitive to cleavage by proteases in Dbh [10] and Dpo4 [13], and is partially disordered in apoenzyme structures of pol κ [19]. The LF/PAD of pol κ adopts at least two positions, in the absence of ligands, and must be able to move a distance >50 Å to reach the location observed in ternary complexes [19, 26].
The LF/PAD may not be as mobile in all of the Y-family polymerases, however. Although the linker is sensitive to proteolysis in Dbh, the LF/PAD of Dbh shows relatively little movement in the apo, binary and ternary structures that have been determined, with only a 15° rotation and 1 Å translation needed to position the LF/PAD upon binding DNA [34]. Pol η shows even less domain motion: structures of the apo [11] and ternary [35] complexes superimpose with root-mean-squared deviations of less than 1 Å over the 387 residues contained in the catalytic core and LF/PAD domains.
In Dbh, the LF/PAD movement appears to be restricted by a short segment of the linker that forms a β strand and interacts with both the palm and the LF/PAD [10, 12]. In the eukaryotic polymerases, the N-clasp, N-digit, and other sequences have interactions with both the catalytic core and the LF/PAD, such that movements could easily be restricted. In this regard, it is worth noting that the apo-enzyme structure of pol κ [19] does not contain the N-clasp helices that contact the fingers and LF/PAD.
3. Replication of undamaged DNA
3.1. Mutagenic and non-mutagenic basepairing
Highly accurate DNA replication (~1 mistake in 105 bases replicated, in the absence of exonuclease proofreading [14]) is, in large part, a consequence of the polymerase's formation of a tight steric fit around the nascent basepair, such that only correct Watson-Crick pairing will allow the substrates to be accurately positioned at the active site, for efficient catalysis. It was apparent, even from the first apo-enzyme structures [10-12, 14], that the Y-family polymerases would not be able to constrain the nascent basepair to the same extent as seen in the higher-fidelity DNA polymerases. The first ternary complexes of Dpo4 showed that, despite the minimal constraints, Watson-Crick basepairs form readily at the active site and are preferred over at least some types of mispairs [13], a finding that is perhaps not surprising given the relatively high accuracy of the DinB subfamily of polymerases. An example of a dT/dATP Watson-Crick pair at the active site of Dpo4 [37] is shown in Fig. 3A.
Fig. 3. Replication of undamaged DNA.

Three different ways in which a template dT can be copied: (A) Dpo4 with a standard Watson-Crick dT/dATP nascent basepair at the active site (PDB code 2AGQ [37]); (B) human pol iota with a Hoogsteen dT/ddADP nascent basepair PDB code 3GV5 [38]); and (C) human pol iota with a non-standard dT/dGTP nascent basepair (PDB code 3GV8 [38]). Polymerase skipping of a template base (green) to form a single-base deletion: (D) Dbh extending DNA synthesis on a template containing an extrahelical base at position −3 (PDB code 3BQ1 [34]; (E) Dpo4 type II complex (PDB code 1JXL [13]). (F) Protein-templated dCTP incorporation by human Rev1 (PDB code 3GQC [21]). View in A-C and F is looking towards the fingers, along the axis of the DNA duplex; view in (D) and (E) is looking down on to the active site from above, as in Fig. 1 and 2. Template bases are numbered as in Fig. 1. Dotted lines indicate hydrogen bonds. The protein surface is colored white, except in (E) where atoms within 4 Å of the bulged base are colored yellow. Steric gate residues (Y12 in Dpo4, Y39 in pol iota, F367 in Rev1) are shown as a reference point.
What is incredibly surprising, in contrast, is the observation that pol ι replicates template dA bases accurately, but through Hoogsteen pairing rather than Watson-Crick pairing [18]. In this case, the templating dA is found in an anti-conformation opposite an incoming dTTP, with two hydrogen bonds forming between the bases. A more recent structural study shows that the same type of Hoogsteen pair forms between a template dT and an incoming ddADP (Fig. 3B). However, the incoming nucleotide is not optimally positioned at the active site: the nitrogen of the steric gate residue forms a water-mediated hydrogen bond to the α phosphate, rather than a direct bond to the 3′ OH of the nucleotide [38]. The same study provided a structural explanation as to why pol ι preferentially pairs dGTP with a template dT: the incoming nucleotide is stabilized both by a direct hydrogen bond to the backbone of the steric gate and by an additional hydrogen bond between Q59 of the fingers and N2 of dGTP (Fig. 3C). In this case, both bases are in anti conformation, but the template base is twisted so that a bifurcated hydrogen bond forms between the bases. Pol ι's preference for Hoogsteen pairs appears to arise from the enzyme's imposition of a narrow groove width on the nascent basepair [25].
Other types of non-Watson-Crick base pairs have been observed in structures of Dpo4, including a G/3'dT reverse wobble (1S97, [39]) and a T/dTMP reverse pair (2RDJ, [36]). In each of these cases, however, significant rearrangements would have to occur at the primer terminus for synthesis to continue, since the 3′ end of the primer points away from the α-phosphate of the incoming nucleotide, a conformation that is incompatible with catalysis.
The Y-family polymerases generally seem to be more dependent than other polymerase familes on hydrogen-bonding interactions, as evidenced by their lower-efficiency utilization of base analogs that form pairs sterically similar to standard basepairs, but lacking the ability to form hydrogen bonds [40, 41].
3.2. Deletions and insertions
The DinB and pol η Y-family polymerases make single-base deletion errors at high rates (reviewed in [14]). For the bacterial and archaeal DinB polymerases, these mistakes occur especially frequently in template sequences containing runs of identical pyrimidines that are flanked on the 5′ side by a guanosine [42-44]. Structures of Dbh extending DNA synthesis after a single base deletion [34] show the skipped template base in an extrahelical conformation, stabilized by interactions with the LF/PAD (Fig. 3D). Biochemical data indicate that deletions of this type occur through a template-slippage mechanism, with an extrahelical base as an intermediate in the reaction [45]. These deletions arise most frequently when the skipped template base is located three bases upstream, 3′ of the templating base [34], consistent with the base at position −3 being stabilized in an extrahelical conformation.
One of the first structures of Dpo4 suggested that deletions are formed by a dNTP-stabilized misalignment mechanism [13]. In this “type II” structure, containing a non-repetitive sequence, the incoming nucleotide skips over the next available (non-complementary) templating base, and instead pairs correctly with next 5′ base downstream (Fig. 3E). The skipped template base is stacked between the bases on either side, opposite a gap between the bases of the incoming nucleotide and primer terminus.
The process of creating or extending insertion mutations by Y-family polymerases has not been observed on undamaged DNA. Nevertheless, one structure, in which Dpo4 is copying an abasic site, shows that the penultimate nucleotide in the primer can adopt an extrahelical conformation in the minor groove of the DNA duplex, which could result in a single-nucleotide insertion that would cause a +1 frameshift mutation [46]. A similar structure is observed in one complex of Dpo4 replicating an O6-benzyl-dG lesion [56].
3.3. Protein-templated nucleotide incorporation
A remarkable type of DNA synthesis catalyzed by the Y-family polymerases is seen in structures of the Rev1p terminal transferase, in which an arginine (R324 in yeast, R327 in human) acts as the template to specify incorporation of dCTP (Fig. 3F) [20, 21]. The base that would normally template nucleotide incorporation (position 0) is swung out of the DNA duplex toward the major groove, and is stablilized in that position by stacking and hydrogen bonding interactions with residues of a pocket in the LF/PAD. The templating arginine is located in the N-digit, and it forms hydrogen bonds with both the incoming dCTP and the phosphate of the nucleotide at position 0. Two additional residues in the N-digit (L325 and L328 in yeast; L358 and I360 in human) stack against the primer-terminal basepair, isolating the incoming base from contact with the primer-template DNA. The terminal transferase activity is non-mutagenic when dG is in position 0, as was the case for both the yeast and human Rev1 structures that have been published [20, 21].
4. Translesion synthesis
4.1. Cross-linked bases
Cross-linked DNA bases, resulting from exposure to UV light, ionizing radiation, and some types of chemotherapy, are some of the most challenging lesions for high fidelity DNA polymerases to replicate. The challenge arises because each template base is unstacked from its 5′ neighbor as it enters the enzyme's active site [47]; no such unstacking can occur when two bases are covalently linked together.
Crystal structures available for pol η depict three stages of the replication of a 1,2-dGpG cisplatin (Pt-GG) chemotherapy adduct (Fig. 4; [35]). At the first stage (Fig. 4A), the lesion is interacting with the residues in the fingers and dCTP is correctly positioned at the active site, but the 3′ crosslinked dG has not entered the active site far enough to form a basepair. The two bases of the lesion are oriented almost perpendicular to one another, being forced together on the major-groove side by the covalent bonds to the platinum. In this pre-entry structure, M74 fits between the cross-linked bases, interacting with both through Van der Waals contacts. R73 stacks against the base of the incoming nucleotide and forms a single hydrogen bond with the α phosphate. At the next stage (Fig. 4B), the 3′dG moves into the nascent basepair binding pocket, forming Watson-Crick hydrogen bonds with dCTP. The complex is stabilized by an additional hydrogen bond that forms between R73 and the triphosphate, and by M74 moving along with the lesion, maintaining the same interactions between the bases as in the pre-entry structure. Additionally, Q55 hydrogen bonds to N3 of the templating 3′ base of the cross-linked Pt-GG. These first two stages are visualized separately in the same crystal form, suggesting that only a minimal energy difference exists between the two conformations. In the final stage (Fig. 4C), the 3′ dG is paired with the primer-terminal dC, and the 5′ dG forms one hydrogen bond with an incoming dATP and a second to the backbone carbonyl of R73. M74 has moved back up into its pre-entry position, and Q55 does not contact the nascent basepair. A single hydrogen bond could also be formed with an incoming dCTP, but not with other bases, explaining why dATP and dCTP are the two nucleotides that are preferentially incorporated opposite the 5′ base of the Pt-GG lesion [35].
Fig. 4. Replication of cross-linked bases by pol eta.
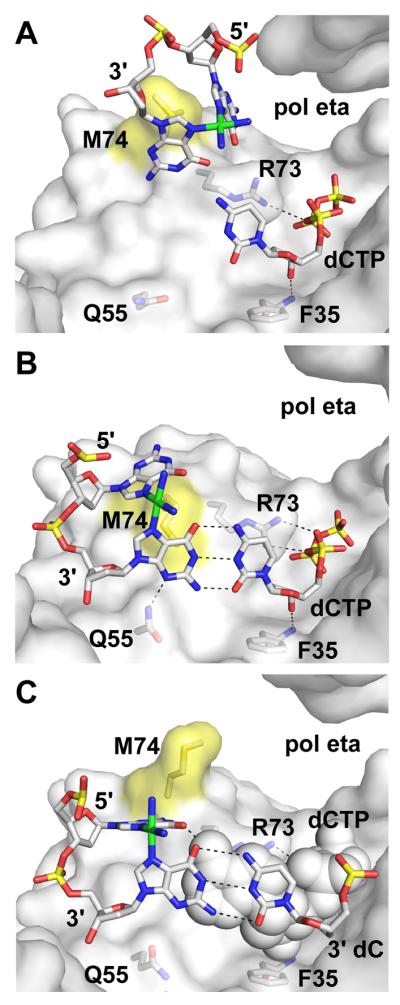
Three stages in the replication of cis-PtdG template lesion by yeast pol eta: (A) pre-insertion (PDB code 2R8J, protein chain B [35]), (B) insertion of dCTP opposite the 3′ cross-linked dG (PDB code 2R8J, protein chain A [35]), and (C) insertion of dATP opposite the 5′ cross-linked dG (PDB code 2R8K, protein chain A [35]). View and coloring are as in Fig. 3A, except that the protein surface contributed by M74 is colored yellow and the platinum atom of the lesion is colored green. The steric gate residue in pol eta is F35. The incoming nucleotide in (C) is shown in sphere representation.
Similar structures show Dpo4 in the process of accurately copying a cis-syn thymidine dimer (TT) [48]. The 3′ dT is copied via Watson-Crick pairing with a ddATP, while the 5′ dT forms a Hoogsteen pair with an incoming ddATP. Dpo4 is not as efficient as pol η in replicating CPD lesions [48, 49], however, and comparison with the pol η Pt-GG structures suggests that the shorter β2-3 loop in the pol η fingers allows the crosslinked lesion to fit more easily into the active site.
4.2. 8-oxo-dG
Although an 8-oxo-dG lesion differs from an undamaged base by the addition of just a single atom, it is a highly mutagenic lesion in cells. When the base is in an anti conformation, required for correct Watson-Crick pairing with dCTP, the additional oxygen closely approaches the backbone phosphates. The steric conflict is relieved when the 8-oxo-dG rotates into a syn conformation, where it is able to form a Hoogsteen pair with dATP.
The archaeal DinB polymerase Dpo4 replicates 8-oxo-dG lesions not only accurately, preferring to incorporate dCTP opposite the lesion, but also efficiently, incorporating this nucleotide up to 10-fold more efficiently than an undamaged dG [50, 51]. Dpo4 accomplishes this nonmutagenic replication by an arginine (R332 in the LF/PAD) forming either a direct or a water-mediated hydrogen bond to the extra oxygen atom of the damaged base (Fig. 5A) [50, 51]. This contact constrains the base in an anti conformation, where it can pair correctly with an incoming dCTP. The hydrogen bond between R332 and 8-oxo-dG is maintained as DNA synthesis continues, and the lesion translocates to the -1 position, where it pairs with the primer terminal dC [51]. Interestingly, an R332E mutation in Dpo4 is able to form a similar water-mediated contact, and the polymerase is still able to replicate the lesion accurately [52].
Fig. 5. Correct and mutagenic replication of 8-oxo-dG.

(A) Dpo4 with an incoming dCTP forming a correct Watson-Crick pair with the template lesion (PDB code 2ASD [50]). 8-oxo-dG is stabilized in the anti conformation by a water-mediated hydrogen bond with R332. (B) Human pol kappa with an incoming dATP forming a Hoogsteen pair with the 8-oxo-dG lesion in a syn conformation (PDB code 3HED [53]). View and coloring are as in Fig. 3A. Arrows point at the oxygen added at position 8 of dG.
Even though pol κ is also a DinB polymerase, it prefers to insert dATP opposite the 8-oxo-dG lesion [17]. A co-crystal structure (Fig. 5B) shows that the lesion is in syn conformation, stacking against M135 in the fingers and Hoogsteen pairing with dATP [53]. O8 of the lesion is solvent-exposed in the minor groove, making no contacts with the enzyme. L508 in the LF/PAD replaces the arginine present in the equivalent position in Dpo4. These observations all help explain the preference of pol κ to bypass this lesion in a mutagenic manner. These structures of Dpo4 and pol κ emphasize how varied replication specificities can be even within a single class of the Y-family polymerases.
4.3. Minor-groove adducts
Base adducts that would protrude into the minor groove in a standard Watson-Crick pair pose another difficult replication problem for high fidelity polymerases, which generally constrain that side of the DNA duplex extensively, through hydrogen bonds and Van der Waals contacts [54]. Bulky adducts at the N2 position of dG fall into this category, and either block DNA synthesis by the more accurate polymerases, or significantly decrease the efficiency and fidelity of DNA synthesis Some minor-groove adducts can be replicated relatively efficiently and accurately by Y-family polymerases: one polymerase may insert a nucleotide insertion opposite the lesion, and a second one may extend DNA synthesis (as occurs for certain other adducts as well). The DinB polymerases are able to replicate N2-adducted deoxyguanosine bases more efficiently than can many of the other Y-family polymerases.
Two structures showing Dpo4 extending DNA synthesis from an N2-benzo[a]pyrene-dG (N2-BPdG) lesion show how translesion synthesis can be accomplished, with either a base-substitution (Fig. 6A) or single-base deletion mutation arising as a consequence [55]. In both crystal structures, the adduct and the linked base are extrahelical, bulged out on the minor-groove side of the DNA. The damaged nucleotide is located between the LF/PAD and the palm, and is stabilized by Van der Waals interactions and hydrogen bonds. In the structure containing a base-substitution, the extrahelical lesion in the −1 position leaves a space opposite the 3′ terminal dA. In the structure with a single base deletion, the bases on either side of the damaged base (at position −2) stack together; there are no unpaired bases in the primer, thus the primer is one base shorter than the template DNA to which it is paired.
Fig. 6. Extension of DNA synthesis beyond lesions with bulky minor- or major-groove adducts.

Dpo4 replicating (A) an N2-BP-dG lesion (PDB code 2IA6 [55]), (B) an N2-napthyldG lesion (PDB code 2W8K [56]), (C) an N6-BP-dA lesion (PDB code 1S0M [61]), and (D) an O6-benzyl-dG lesion (PDB code 2JEF [59]). View and coloring are as in Fig. 3A, with the carbon atoms in each of the adducts colored green. In this and subsequent figures, nascent basepairs are shown in sphere representation.
In contrast to the above structures, a structure showing Dpo4 with an N2-naphthyl-dG lesion at the −1 template position reveals that the adduct is accommodated via the damaged base's rotation into a syn conformation (Fig. 6B) [56]. This rotation puts the napthyl group into the major groove above the nascent basepair and allows the primer terminal dC to form a bifurcated hydrogen bond with the “Hoogsteen edge” of the adducted dG.
Similarly, in a structure of pol ι with a smaller N2-ethyl-dG lesion at position 0, the base is found in a syn conformation, forming a Hoogsteen pair with an incoming dCTP, and the adduct is in the major groove above the DNA backbone [57]. In a final related structure, of Dpo4 with N2-dimethyl-dG template lesion at position 0, the damaged base shifts toward the minor groove, but remains in an anti conformation and forms a wobble pair with an incoming dTTP [58].
All of the structures containing N2-adducts of dG have companion structures that depict how the same lesion can alternatively be bound in a way that would disrupt continued DNA synthesis [55-58]. Such disruption occurs when the base is in an anti conformation, and the adduct stacks within the DNA duplex. Substrate positioning at the active site becomes disrupted, often with the primer-terminal base disordered or folded back into the minor groove of the DNA, arising due to the stacking of the lesion against the penultimate base of the primer.
4.4. Major-groove adducts
In a standard Watson-Crick basepair, adducts that are covalently attached to either the N6 position of dA or the O6 position of dG would protrude into the major groove. Such lesions strongly inhibit DNA synthesis by high-fidelity polymerases, even though steric constraints to the major groove are much less extensive than those to the minor groove. As for the minor-groove adducts, the structures of the major-groove adducts discussed below typically have companion structures in which the lesion is stacked with neighbor bases [59-61], thus demonstrating that the two types of adducts can inhibit replication in similar ways. Currently, the structures with major-groove DNA adducts are all of Dpo4, with the lesions in anti conformation at the −1 position, opposite the primer terminal base.
The first such structure shows Dpo4 with N6-benzo[a]pyrene-dA (N6-BP-dA) (Fig. 6C) [61]. The adduct is located in the major groove; it is largely solvent-exposed, but is also stabilized by two hydrogen bonds (direct and water-mediated) to the basepairs at positions −2 and −3 of the upstream duplex. The dA to which the adduct is attached is forced to shift toward the major groove and consequently does not form hydrogen bonds with the primer-terminal dT (Fig. 6C). A non-polar additive was included in the crystallization solution to stabilize the solvent-exposed conformation. The more energetically favorable conformation, found in aqueous solution, shows the bulky lesion stacking against the primer-terminal base pair, where it prevents further DNA synthesis by occupying the space where an incoming nucleotide would bind.
Structures of Dpo4 with either a benzyl (Fig. 6D) [59] or a methyl [60] adduct at the O6 position of dG also show the adduct on the major-groove side of the duplex. O6-benzyl-dG is shifted so that it forms a wobble pair with the primer-terminal dC (Fig. 6D), as does the O6-methyl-dG lesion. For both of these lesions, alternate conformations exist in which the adduct stacks within the DNA duplex, causing the primer-terminal dC to be extrahelical and poorly ordered.
4.5. Adducts that prevent Watson-Crick basepairing
Base modifications that involve a covalent linkage to N1 of purine bases sterically prevent the formation of Watson-Crick basepairs by such lesions. All of the adducts of this type that have been studied in complexes with Y-family polymerases form cyclic attachments to the N1 and to either the N6 of dA or the N2 of dG.
The structure of pol ι complexed with 1, N6-etheno-dA (Fig. 7A) shows the damaged base rotated into the syn conformation where it forms a Hoogsteen-type pair with an incoming dCTP in the nascent-basepair binding pocket [62]. Additionally, Q59 in the fingers subdomain forms a water-mediated hydrogen bond with the O2 of dCTP.
Fig. 7. Adducts preventing Watson-Crick pairing.

(A) Pol iota with a 1,N2-etheno-dA template lesion in the anti conformation pairing with an incoming dCTP (PDB code 2DPI [62]). (B) Rev1 bypassing a 1,N2-propano-dG lesion by protein-templated incorporation of dCTP (PDB code 3BJY [63]). (C) Dpo4 attempting to extend DNA synthesis beyond a 1,N2-propanodG lesion at the −1 position (PDB code 2R8G [64]). The primer terminal dC is disordered in this structure.
In the crystal structure of Rev1 with 1, N2-propano-dG (also called 1, N2-γ-HOP-dG) at position 0 (Fig. 7B) [63], the adducted base is bulged out of the DNA duplex, and is stabilized in the same way as is an undamaged dG in this position (Fig. 3F). An incoming dCTP is templated by R324 in the N-digit; the result is non-mutagenic DNA synthesis. In contrast, the same lesion at position −1 is stacked within the DNA duplex in a co-crystal structure with Dpo4 (Fig. 7C) [64]. The latter structure shows a “type II” complex, in which no base is situated opposite the lesion, and the primer terminus is paired with the template base at position −2. Nucleotide incorporation from this conformation would result in a single-base deletion.
4.6. Abasic sites
Abasic sites pose similar problems for all polymerases: in addition to containing no information to specify which nucleotide should be incorporated, the lesion cannot fully occupy the space available in the nascent basepair binding pocket, thus reducing the constraints on the position of the incoming nucleotide.
Translesion synthesis of abasic sites often leads to deletion mutations, and two structures of Dpo4 [46] show how this can occur. In a structure with an abasic site at the −1 position (Fig. 8A), the ribose group is bulged out of the DNA duplex on the minor-groove side, in the space between the LF/PAD and palm, thereby allowing the primer terminal base to stack against the nascent basepair. Contact between R247, in the LF/PAD domain, and the template backbone phosphate adjacent to the abasic site help stabilize this conformation (Fig. 8A). Similar arrangements occur with the lesion located at the −2 position. Additional structures of Dpo4 show that the abasic site sometimes remains in the B-form helical structure of the DNA, opposite a base in the primer strand; in this latter situation, a deletion mutation would not occur.
Fig. 8. Replication of a non-instructional abasic sites.

(A) Dpo4 with the lesion bulged out of the DNA duplex at the −1 position (PDB code 1S0N [46]) to form a single-base deletion. (B) Pol iota positioning an incoming dGTP for incorporation opposite an abasic site (PDB code 3G6X [65]).
Structures of pol ι with an abasic site opposite either a dGTP (Fig. 8B), dATP, or dTTP incoming nucleotide show that any one of these three nucleotides can stack appropriately on the steric gate residue and hydrogen bond to the backbone nitrogen [65]. Additionally, the incoming dGTP forms a direct hydrogen bond between N2 of the base and the side-chain oxygen of Q59 (Fig. 8B), just as is formed when dGTP is paired with template dT (Fig. 3C).
5. Functional implications Y-family polymerase structures
5.1. DNA translocation
Two series of structures shed light on how the Y-family polymerases translocate along the DNA during a nucleotide incorporation cycle. Pre-insertion binary, insertion ternary, and post-insertion binary complexes of Dpo4 replicating an 8-oxo-dG lesion portray together stepwise movements: the LF/PAD contacts with the DNA shift by one basepair upon nucleotide binding, to form the ternary complex, but the thumb contacts shift only after nucleotide incorporation [50]. In both of the binary complexes (pre- and post-insertion), the primer terminus occupies the site where the next incoming nucleotide will bind (see Fig. 11 in [50]).
A comparable series of complexes, showing Dbh extending DNA synthesis with a bulged template nucleotide at position −3, tells a different story [34]. In the pre-insertion binary complex, the templating base and the primer terminus are already positioned so that space is available for the incoming nucleotide to bind and form the ternary complex. In the post-insertion binary complex, the DNA is located in nearly the same position on the polymerase. Thus, in the situation depicted in this series of structures, translocation is not associated with nucleotide binding, or incorporation, or pyrophosphate release (see Fig. 1 in [34]).
Like the Dbh complexes above, two pre-insertion binary complexes of pol ι [66] depict a configuration in which space is already available for an incoming nucleotide, indicating that translocation can occur prior to nucleotide binding. The pre-insertion binary complex of Dbh may be stabilized by interactions between the LF/PAD domain and the bulged base (Fig. 3E), but it is not apparent what specific interactions stabilize the pol ι pre-insertion complex. Interestingly, however, binding of an incoming nucleotide suffices to induce the dA or the dG templating base to switch from the anti conformation in the pre-insertion binary complex to the syn conformation in the ternary complex [66].
The above structures, in their variety, indicate that no single translocation mechanism is common to all of the Y-family polymerases. Mechanistic variability could arise from structural differences both among the polymerases and among the templates. Additionally, the Y-family polymerases appear not to couple DNA translocation to a conformational change in the enzyme itself, in contrast to the A- and B-family polymerases. For the two latter polymerase families, nucleotide binding and pyrophosphate release trigger conformation changes in the fingers, and those changes are coupled to DNA translocation [47, 67-70].
5.2. Difficulty in positioning the substrates for catalysis
One notable characteristic of the Y-family polymerases becomes apparent when all of the available structures (Supplementary Table 1) are reviewed: they have great difficulty in positioning the substrates precisely at the active site for catalysis. Variability, both in divalent metal binding and triphosphate conformation, has been carefully analyzed [37]; however, the variability in substrate binding extends also to the locations of the primer terminus and the incoming nucleotide, relative to the active-site residues. Two straightforward measures that can provide an indication of how substrates are positioned at the active site are the distance between the primer terminus and the α-phosphate of the incoming nucleotide, and the distance between the ribose of the incoming nucleotide and the steric gate residue.
In the vast majority of Y-family polymerase structures (including those solved in my lab), some type of structural change needs to occur before catalysis can proceed. Such a change can be relatively small, such as binding of a divalent cation in the metal A position, or adoption by the 3′ terminal ribose of a C3′ endo conformation, or it can be somewhat larger, such as movement of the incoming nucleotide and primer terminus toward the metal-binding catalytic residues at the active site. Some of the solved complexes are obviously inactive; for instance, structures in which the primer terminus is not located at the active site, but is instead disordered or folded back into the minor groove away from the active site. The conformations observed in all of the structures are obviously thermodynamically stable; further experiments, however, will be required to determine which of these conformations are directly on the kinetic pathway to catalysis.
For some of the structures, the magnitude of change needed is not readily apparent. An example is the “type II” structures in which an unpaired base occupies the −1 template position, and a gap exists between the bases of the incoming nucleotide and primer terminus. The rearrangement required in this case could simply be movement of the 3' OH and α-phosphate toward each other. Some of the “type II” structures show signs of this occurring, but often at the expense of mispositioning the triphosphate of the incoming nucleotide. Alternatively, the unpaired base may need to swing out of the DNA duplex into an extrahelical conformation, thereby allowing the reactive groups to more readily approach one another.
The difficulty in positioning the substrates arises from the spacious but rigid nature of the Y-family polymerase active site [37]; it is likely to account for many of the distinctive activities displayed by the various enzymes in the family. Small variations in contacts between the polymerase and substrate, such as the presence or absence of a single residue that can stabilize 8-oxo-dG in an anti conformation, can determine whether or not translesion DNA synthesis is mutagenic. As DNA synthesis continues and the polymerase translocates along a damaged (or undamaged) template, the specific contacts between the protein and the DNA will change, influencing the ultimate outcome of replication by the Y-family polymerase.
6. Switching of replicative and translesion polymerases
6.1. Y-family polymerase interactions with sliding clamps
An understanding, in structural terms, of how the Y-family polymerases interact with sliding-clamp processivity factors, while replicating DNA, is still at an early stage. Structures that have been determined are: from E. coli, the LF/PAD of pol IV in complex with the β-clamp [22]; from S. solfataricus, full-length Dpo4 in complex with PCNA subunits 1 and 2 [71]; and, from humans, B/PIP sequences from pol κ, pol η and pol ι bound to PCNA [72].
In each of the above-listed structures (Fig. 9), the B/PIP binds to a largely hydrophobic area on the surface of the sliding clamp, overlapping the binding sites that have been observed of B/PIP sequences present in other proteins. In the bacterial homodimeric clamp (Fig. 9A) [22] and the eukaryotic homotrimeric clamp (Fig. 9C) [72], each subunit is bound to a B/PIP sequence. In the archaeal clamp (Fig. 9B) [71], only subunit 2 is bound to the polymerase, while subunit 1 is empty (note that subunit 3 was not included in the crystallization experiment).
Fig. 9. Y-family polymerase interactions with sliding clamp processivity factors.

(A) The LF/PAD of E. coli DinB in complex with the beta-clamp homodimer (PDB code 1UNN [22]). (B) Full-length Dpo4 in complex with subunits 1 and 2 of the PCNA heterotrimer (PDB code 3FDS [71]). (C) The B/PIP sequence of human pol kappa bound to homotrimeric PCNA (PDB code 2ZVL [72]). Individual subunits of the sliding clamps are shown in surface representation colored light green, light blue or pink, with the residues within 4 Å of the B/PIP sequences colored yellow. The polymerases are colored as in Fig. 1A. Residues within the B/PIP are shown in stick representation colored yellow.
In the bacterial and archaeal structures, the polymerase forms additional contacts with the sliding clamp. These additional contacts constrain the polymerase so that it is unable to contact the DNA that is encircled by the clamp; by this mechanism, a Y-family polymerase may be held away from the primer-template junction until translesion synthesis is necessary.
6.2. Structural models for polymerase switching
Several, rather similar models have been proposed to explain how replicative and translesion polymerases switch places at sites of DNA damage; the models are derived from crystal structures of bacterial C-family replicative polymerase catalytic subunits (pol IIIα) [73-76] together with structures of E. coli β-clamp bound to DNA [77], and to the LF/PAD of pol IV [22]. In these models, the β-clamp encircles the upstream DNA duplex, thereby positioning the pol IIIα B/PIP sequence adjacent to one of the two clamp subunits; the second clamp subunit binds pol IV.
The modeling, as well as an analysis of the structure of Dpo4 with PCNA subunits 1 and 2, suggests that a series of movements, involving conformational changes in both polymerases, must occur to allow a switch between replicative and translesion synthesis [71, 73, 75, 76]. Movement of the primer-template junction between polymerase active sites may also be facilitated by a change in the tilt of the DNA, with respect to the sliding clamp [77]. Some additional considerations become apparent from a three dimensional model. First, the pol IIIα enzymes from gram-negative bacteria contain a second B/PIP sequence that could potentially bind the second clamp subunit during replication [74]; such a binding contact would need to be disrupted prior to a polymerase switch. Second, the proofreading exonuclease (a domain of the polymerase in gram-positive bacteria, but a second subunit in gram-negative bacteria) may restrict a translesion polymerase from binding to the clamp during active replicative synthesis [73-75]. Third, the primer terminus is likely to pass near the exonuclease active site as it moves between polymerase active sites [75].
Some structural aspects of polymerase switching will differ from the bacterial process, for both eukaryotes and the archaea, in which B-family rather than C-family polymerases are responsible for replicative DNA synthesis. In the C-family polymerases, the conserved B/PIP is located at the distal end of a separate structural domain (called the β-binding [73], duplex-binding [75], or extended fingers domain [74]), while in the B-family polymerases, the B/PIP is located adjacent to the thumb domain [78-80]. The exonuclease is also situated differently between the B- and C-polymerase families: near the thumb in the C-family, but adjacent to the fingers in the B-family [78]. These structural differences suggest different pathways for the DNA, as it moves between the replicative and translesion polymerase active sites; differences in pathway could lead to differences in the balance between polymerization and proofreading.
Switching to the eukaryotic Y-family polymerases is also distinguished from the bacterial and archaeal processes by the requirement for mono-ubiquitination of PCNA [81]. The structural role of ubiquitin has not yet been elucidated, but is expected to involve direct contacts between the ubiquitin molecule covalently linked to PCNA and the UBZ or UBM motifs located in the eukaryotic Y-family polymerases [28].
7. Concluding remarks
The remarkably wide variety of Y-family polymerase structures that have been determined makes difficult any detailed generalization how these enzymes interact with different substrates. The very accessible active sites of these enzymes allow ready access for damaged DNA that is to be replicated, but this the same accessibility hinders us in predicting precisely how various lesions interact with each enzyme. An additional complication is that the structural differences that confer specificity to each polymerase can be subtle. While analysis of further structures will almost certainly reveal additional ways in which the Y-family polymerases can replicate DNA, many of the advances to come in the next few years should also help to elucidate how the Y-family polymerases interact with other replication and repair proteins, and how such interactions are regulated.
Supplementary Material
Acknowledgements
I thank Joachim Jaeger and members of the Pata and Jaeger groups for many stimulating discussions about polymerase structure and function, the anonymous referees for their helpful comments, and Adriana Verschoor for expert editing of the manuscript. J.D.P is funded by NIH grant GM080573.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Masutani C, Kusumoto R, Yamada A, Dohmae N, Yokoi M, Yuasa M, Araki M, Iwai S, Takio K, Hanaoka F. The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase eta. Nature. 1999;399:700–704. doi: 10.1038/21447. [DOI] [PubMed] [Google Scholar]
- 2.Johnson RE, Kondratick CM, Prakash S, Prakash L. hRAD30 mutations in the variant form of xeroderma pigmentosum. Science. 1999;285:263–265. doi: 10.1126/science.285.5425.263. [DOI] [PubMed] [Google Scholar]
- 3.Masutani C, Araki M, Yamada A, Kusumoto R, Nogimori T, Maekawa T, Iwai S, Hanaoka F. Xeroderma pigmentosum variant (XP-V) correcting protein from HeLa cells has a thymine dimer bypass DNA polymerase activity. EMBO J. 1999;18:3491–3501. doi: 10.1093/emboj/18.12.3491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johnson RE, Prakash S, Prakash L. Efficient bypass of a thymine-thymine dimer by yeast DNA polymerase, Poleta. Science. 1999;283:1001–1004. doi: 10.1126/science.283.5404.1001. [DOI] [PubMed] [Google Scholar]
- 5.Ziv O, Geacintov N, Nakajima S, Yasui A, Livneh Z. DNA polymerase zeta cooperates with polymerases kappa and iota in translesion DNA synthesis across pyrimidine photodimers in cells from XPV patients. Proc Natl Acad Sci U S A. 2009;106:11552–11557. doi: 10.1073/pnas.0812548106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Y, Woodgate R, McManus TP, Mead S, McCormick JJ, Maher VM. Evidence that in xeroderma pigmentosum variant cells, which lack DNA polymerase eta, DNA polymerase iota causes the very high frequency and unique spectrum of UV-induced mutations. Cancer Res. 2007;67:3018–3026. doi: 10.1158/0008-5472.CAN-06-3073. [DOI] [PubMed] [Google Scholar]
- 7.Kenyon CJ, Walker GC. DNA-damaging agents stimulate gene expression at specific loci in Escherichia coli. Proc Natl Acad Sci U S A. 1980;77:2819–2823. doi: 10.1073/pnas.77.5.2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nelson JR, Lawrence CW, Hinkle DC. Deoxycytidyl transferase activity of yeast REV1 protein. Nature. 1996;382:729–731. doi: 10.1038/382729a0. [DOI] [PubMed] [Google Scholar]
- 9.Ohmori H, Friedberg EC, Fuchs RP, Goodman MF, Hanaoka F, Hinkle D, Kunkel TA, Lawrence CW, Livneh Z, Nohmi T, Prakash L, Prakash S, Todo T, Walker GC, Wang Z, Woodgate R. The Y-family of DNA polymerases. Mol Cell. 2001;8:7–8. doi: 10.1016/s1097-2765(01)00278-7. [DOI] [PubMed] [Google Scholar]
- 10.Zhou BL, Pata JD, Steitz TA. Crystal structure of a DinB lesion bypass DNA polymerase catalytic fragment reveals a classic polymerase catalytic domain. Mol Cell. 2001;8:427–437. doi: 10.1016/s1097-2765(01)00310-0. [DOI] [PubMed] [Google Scholar]
- 11.Trincao J, Johnson RE, Escalante CR, Prakash S, Prakash L, Aggarwal AK. Structure of the catalytic core of S. cerevisiae DNA polymerase eta: implications for translesion DNA synthesis. Mol Cell. 2001;8:417–426. doi: 10.1016/s1097-2765(01)00306-9. [DOI] [PubMed] [Google Scholar]
- 12.Silvian LF, Toth EA, Pham P, Goodman MF, Ellenberger T. Crystal structure of a DinB family error-prone DNA polymerase from Sulfolobus solfataricus. Nat Struct Biol. 2001;8:984–989. doi: 10.1038/nsb1101-984. [DOI] [PubMed] [Google Scholar]
- 13.Ling H, Boudsocq F, Woodgate R, Yang W. Crystal structure of a Y-family DNA polymerase in action: a mechanism for error-prone and lesion-bypass replication. Cell. 2001;107:91–102. doi: 10.1016/s0092-8674(01)00515-3. [DOI] [PubMed] [Google Scholar]
- 14.McCulloch SD, Kunkel TA. The fidelity of DNA synthesis by eukaryotic replicative and translesion synthesis polymerases. Cell Res. 2008;18:148–161. doi: 10.1038/cr.2008.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Johnson RE, Washington MT, Haracska L, Prakash S, Prakash L. Eukaryotic polymerases iota and zeta act sequentially to bypass DNA lesions. Nature. 2000;406:1015–1019. doi: 10.1038/35023030. [DOI] [PubMed] [Google Scholar]
- 16.Johnson RE, Haracska L, Prakash S, Prakash L. Role of DNA polymerase zeta in the bypass of a (6-4) TT photoproduct. Mol Cell Biol. 2001;21:3558–3563. doi: 10.1128/MCB.21.10.3558-3563.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haracska L, Prakash L, Prakash S. Role of human DNA polymerase kappa as an extender in translesion synthesis. Proc Natl Acad Sci U S A. 2002;99:16000–16005. doi: 10.1073/pnas.252524999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nair D, Johnson R, Prakash S, Prakash L, Aggarwal A. Replication by human DNA polymerase-iota occurs by Hoogsteen base-pairing. Nature. 2004;430:377–380. doi: 10.1038/nature02692. [DOI] [PubMed] [Google Scholar]
- 19.Uljon SN, Johnson R, Edwards TA, Prakash S, Prakash L, Aggarwal A. Crystal structure of the catalytic core of human DNA polymerase kappa. Structure. 2004;12:1395–1404. doi: 10.1016/j.str.2004.05.011. [DOI] [PubMed] [Google Scholar]
- 20.Nair D, Johnson R, Prakash L, Prakash S, Aggarwal A. Rev1 employs a novel mechanism of DNA synthesis using a protein template. Science. 2005;309:2219–2222. doi: 10.1126/science.1116336. [DOI] [PubMed] [Google Scholar]
- 21.Swan MK, Johnson RE, Prakash L, Prakash S, Aggarwal AK. Structure of the human Rev1-DNA-dNTP ternary complex. J Mol Biol. 2009;390:699–709. doi: 10.1016/j.jmb.2009.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bunting KA, Roe SM, Pearl LH. Structural basis for recruitment of translesion DNA polymerase Pol IV/DinB to the beta-clamp. EMBO J. 2003;22:5883–5892. doi: 10.1093/emboj/cdg568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Steitz TA. DNA- and RNA-dependent DNA polymerases. Current Opinion in Structural Biology. 1993;3:31–38. [Google Scholar]
- 24.DeLucia AM, Grindley ND, Joyce CM. An error-prone family Y DNA polymerase (DinB homolog from Sulfolobus solfataricus) uses a ‘steric gate’ residue for discrimination against ribonucleotides. Nucleic Acids Res. 2003;31:4129–4137. doi: 10.1093/nar/gkg417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nair D, Johnson R, Prakash L, Prakash S, Aggarwal A. Human DNA polymerase iota incorporates dCTP opposite template G via a G.C + Hoogsteen base pair. Structure. 2005;13:1569–1577. doi: 10.1016/j.str.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 26.Lone S, Townson SA, Uljon SN, Johnson R, Brahma A, Nair D, Prakash S, Prakash L, Aggarwal A. Human DNA polymerase kappa encircles DNA: implications for mismatch extension and lesion bypass. Mol Cell. 2007;25:601–614. doi: 10.1016/j.molcel.2007.01.018. [DOI] [PubMed] [Google Scholar]
- 27.Guo C, Sonoda E, Tang TS, Parker JL, Bielen AB, Takeda S, Ulrich HD, Friedberg EC. REV1 protein interacts with PCNA: significance of the REV1 BRCT domain in vitro and in vivo. Mol Cell. 2006;23:265–271. doi: 10.1016/j.molcel.2006.05.038. [DOI] [PubMed] [Google Scholar]
- 28.Bienko M, Green CM, Crosetto N, Rudolf F, Zapart G, Coull B, Kannouche P, Wider G, Peter M, Lehmann AR, Hofmann K, Dikic I. Ubiquitin-binding domains in Y-family polymerases regulate translesion synthesis. Science. 2005;310:1821–1824. doi: 10.1126/science.1120615. [DOI] [PubMed] [Google Scholar]
- 29.Guo C, Fischhaber PL, Luk-Paszyc MJ, Masuda Y, Zhou J, Kamiya K, Kisker C, Friedberg EC. Mouse Rev1 protein interacts with multiple DNA polymerases involved in translesion DNA synthesis. EMBO J. 2003;22:6621–6630. doi: 10.1093/emboj/cdg626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ohashi E, Murakumo Y, Kanjo N, Akagi J, Masutani C, Hanaoka F, Ohmori H. Interaction of hREV1 with three human Y-family DNA polymerases. Genes Cells. 2004;9:523–531. doi: 10.1111/j.1356-9597.2004.00747.x. [DOI] [PubMed] [Google Scholar]
- 31.Nagashiima T, Hayashi F, Yokoyama S. Solution structure of the BRCT domain from human DNA repair protein Rev1. RIKEN Structural Genomics / Proteomics Initiative To be published. 2007 [Google Scholar]
- 32.Bomar MG, Pai MT, Tzeng SR, Li SS, Zhou P. Structure of the ubiquitin-binding zinc finger domain of human DNA Y-polymerase eta. EMBO Rep. 2007;8:247–251. doi: 10.1038/sj.embor.7400901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang W, Woodgate R. What a difference a decade makes: insights into translesion DNA synthesis. Proc Natl Acad Sci U S A. 2007;104:15591–15598. doi: 10.1073/pnas.0704219104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wilson RC, Pata JD. Structural insights into the generation of single-base deletions by the Y family DNA polymerase dbh. Mol Cell. 2008;29:767–779. doi: 10.1016/j.molcel.2008.01.014. [DOI] [PubMed] [Google Scholar]
- 35.Alt A, Lammens K, Chiocchini C, Lammens A, Pieck JC, Kuch D, Hopfner KP, Carell T. Bypass of DNA lesions generated during anticancer treatment with cisplatin by DNA polymerase eta. Science. 2007;318:967–970. doi: 10.1126/science.1148242. [DOI] [PubMed] [Google Scholar]
- 36.Wong JH, Fiala KA, Suo Z, Ling H. Snapshots of a Y-family DNA polymerase in replication: substrate-induced conformational transitions and implications for fidelity of Dpo4. J Mol Biol. 2008;379:317–330. doi: 10.1016/j.jmb.2008.03.038. [DOI] [PubMed] [Google Scholar]
- 37.Vaisman A, Ling H, Woodgate R, Yang W. Fidelity of Dpo4: effect of metal ions, nucleotide selection and pyrophosphorolysis. EMBO J. 2005;24:2957–2967. doi: 10.1038/sj.emboj.7600786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kirouac KN, Ling H. Structural basis of error-prone replication and stalling at a thymine base by human DNA polymerase iota. EMBO J. 2009;28:1644–1654. doi: 10.1038/emboj.2009.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Trincao J, Johnson R, Wolfle W, Escalante C, Prakash S, Prakash L, Aggarwal A. Dpo4 is hindered in extending a G.T mismatch by a reverse wobble. Nat Struct Mol Biol. 2004;11:457–462. doi: 10.1038/nsmb755. [DOI] [PubMed] [Google Scholar]
- 40.Irimia A, Eoff RL, Pallan PS, Guengerich FP, Egli M. Structure and activity of Y-class DNA polymerase DPO4 from Sulfolobus solfataricus with templates containing the hydrophobic thymine analog 2,4-difluorotoluene. J Biol Chem. 2007;282:36421–36433. doi: 10.1074/jbc.M707267200. [DOI] [PubMed] [Google Scholar]
- 41.Washington MT, Johnson RE, Prakash L, Prakash S. The mechanism of nucleotide incorporation by human DNA polymerase eta differs from that of the yeast enzyme. Mol Cell Biol. 2003;23:8316–8322. doi: 10.1128/MCB.23.22.8316-8322.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wagner J, Nohmi T. Escherichia coli DNA polymerase IV mutator activity: genetic requirements and mutational specificity. J Bacteriol. 2000;182:4587–4595. doi: 10.1128/jb.182.16.4587-4595.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Potapova O, Grindley ND, Joyce CM. The mutational specificity of the Dbh lesion bypass polymerase and its implications. J Biol Chem. 2002;277:28157–28166. doi: 10.1074/jbc.M202607200. [DOI] [PubMed] [Google Scholar]
- 44.Boudsocq F, Kokoska RJ, Plosky BS, Vaisman A, Ling H, Kunkel TA, Yang W, Woodgate R. Investigating the role of the little finger domain of Y-family DNA polymerases in low fidelity synthesis and translesion replication. J Biol Chem. 2004;279:32932–32940. doi: 10.1074/jbc.M405249200. [DOI] [PubMed] [Google Scholar]
- 45.DeLucia AM, Grindley ND, Joyce CM. Conformational changes during normal and error-prone incorporation of nucleotides by a Y-family DNA polymerase detected by 2-aminopurine fluorescence. Biochemistry. 2007;46:10790–10803. doi: 10.1021/bi7006756. [DOI] [PubMed] [Google Scholar]
- 46.Ling H, Boudsocq F, Woodgate R, Yang W. Snapshots of replication through an abasic lesion; structural basis for base substitutions and frameshifts. Mol Cell. 2004;13:751–762. doi: 10.1016/s1097-2765(04)00101-7. [DOI] [PubMed] [Google Scholar]
- 47.Doublie S, Tabor S, Long AM, Richardson CC, Ellenberger T. Crystal structure of a bacteriophage T7 DNA replication complex at 2.2 A resolution. Nature. 1998;391:251–258. doi: 10.1038/34593. [DOI] [PubMed] [Google Scholar]
- 48.Ling H, Boudsocq F, Plosky BS, Woodgate R, Yang W. Replication of a cis-syn thymine dimer at atomic resolution. Nature. 2003;424:1083–1087. doi: 10.1038/nature01919. [DOI] [PubMed] [Google Scholar]
- 49.Washington MT, Johnson RE, Prakash S, Prakash L. Accuracy of thymine-thymine dimer bypass by Saccharomyces cerevisiae DNA polymerase eta. Proc Natl Acad Sci U S A. 2000;97:3094–3099. doi: 10.1073/pnas.050491997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rechkoblit O, Malinina L, Cheng Y, Kuryavyi V, Broyde S, Geacintov NE, Patel DJ. Stepwise translocation of Dpo4 polymerase during error-free bypass of an oxoG lesion. PLoS Biol. 2006;41:e11. doi: 10.1371/journal.pbio.0040011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zang H, Irimia A, Choi JY, Angel KC, Loukachevitch LV, Egli M, Guengerich FP. Efficient and high fidelity incorporation of dCTP opposite 7,8-dihydro-8-oxodeoxyguanosine by Sulfolobus solfataricus DNA polymerase Dpo4. J Biol Chem. 2006;281:2358–2372. doi: 10.1074/jbc.M510889200. [DOI] [PubMed] [Google Scholar]
- 52.Eoff RL, Irimia A, Angel KC, Egli M, Guengerich FP. Hydrogen bonding of 7,8-dihydro-8-oxodeoxyguanosine with a charged residue in the little finger domain determines miscoding events in Sulfolobus solfataricus DNA polymerase Dpo4. J Biol Chem. 2007;282:19831–19843. doi: 10.1074/jbc.M702290200. [DOI] [PubMed] [Google Scholar]
- 53.Vasquez-Del Carpio R, Silverstein TD, Lone S, Swan MK, Choudhury JR, Johnson R, Prakash S, Prakash L, Aggarwal A. Structure of human DNA polymerase kappa inserting dATP opposite an 8-OxoG DNA lesion. PLoS ONE. 2009;4:e5766. doi: 10.1371/journal.pone.0005766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Doublie S, Ellenberger T. The mechanism of action of T7 DNA polymerase. Curr Opin Struct Biol. 1998;8:704–712. doi: 10.1016/s0959-440x(98)80089-4. [DOI] [PubMed] [Google Scholar]
- 55.Bauer J, Xing G, Yagi H, Sayer JM, Jerina DM, Ling H. A structural gap in Dpo4 supports mutagenic bypass of a major benzo[a]pyrene dG adduct in DNA through template misalignment. Proc Natl Acad Sci USA. 2007;104:14905–14910. doi: 10.1073/pnas.0700717104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang H, Eoff RL, Kozekov ID, Rizzo CJ, Egli M, Guengerich FP. Versatility of Y-family Sulfolobus solfataricus DNA polymerase Dpo4 in translesion synthesis past bulky N2-alkylguanine adducts. J Biol Chem. 2009;284:3563–3576. doi: 10.1074/jbc.M807778200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pence MG, Blans P, Zink CN, Hollis T, Fishbein JC, Perrino FW. Lesion bypass of N2-ethylguanine by human DNA polymerase iota. J Biol Chem. 2009;284:1732–1740. doi: 10.1074/jbc.M807296200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang H, Eoff RL, Kozekov ID, Rizzo CJ, Egli M, Guengerich FP. Structure-function relationships in miscoding by Sulfolobus solfataricus DNA polymerase Dpo4: guanine N2,N2-dimethyl substitution produces inactive and miscoding polymerase complexes. J Biol Chem. 2009;284:17687–17699. doi: 10.1074/jbc.M109014274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Eoff RL, Angel KC, Egli M, Guengerich FP. Molecular basis of selectivity of nucleoside triphosphate incorporation opposite O6-benzylguanine by sulfolobus solfataricus DNA polymerase Dpo4: steady-state and pre-steady-state kinetics and x-ray crystallography of correct and incorrect pairing. J Biol Chem. 2007;282:13573–13584. doi: 10.1074/jbc.M700656200. [DOI] [PubMed] [Google Scholar]
- 60.Eoff RL, Irimia A, Egli M, Guengerich FP. Sulfolobus solfataricus DNA polymerase Dpo4 is partially inhibited by “wobble” pairing between O6-methylguanine and cytosine, but accurate bypass is preferred. J Biol Chem. 2007;282:1456–1467. doi: 10.1074/jbc.M609661200. [DOI] [PubMed] [Google Scholar]
- 61.Ling H, Sayer JM, Plosky BS, Yagi H, Boudsocq F, Woodgate R, Jerina DM, Yang W. Crystal structure of a benzo[a]pyrene diol epoxide adduct in a ternary complex with a DNA polymerase. Proc Natl Acad Sci USA. 2004;101:2265–2269. doi: 10.1073/pnas.0308332100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nair D, Johnson R, Prakash L, Prakash S, Aggarwal A. Hoogsteen base pair formation promotes synthesis opposite the 1,N6-ethenodeoxyadenosine lesion by human DNA polymerase iota. Nat Struct Mol Biol. 2006;13:619–625. doi: 10.1038/nsmb1118. [DOI] [PubMed] [Google Scholar]
- 63.Nair D, Johnson R, Prakash L, Prakash S, Aggarwal A. Protein-template-directed synthesis across an acrolein-derived DNA adduct by yeast Rev1 DNA polymerase. Structure. 2008;16:239–245. doi: 10.1016/j.str.2007.12.009. [DOI] [PubMed] [Google Scholar]
- 64.Wang Y, Musser SK, Saleh S, Marnett LJ, Egli M, Stone MP. Insertion of dNTPs opposite the 1,N2-propanodeoxyguanosine adduct by Sulfolobus solfataricus P2 DNA polymerase IV. Biochemistry. 2008;47:7322–7334. doi: 10.1021/bi800152j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nair D, Johnson R, Prakash L, Prakash S, Aggarwal A. DNA synthesis across an abasic lesion by human DNA polymerase iota. Structure. 2009;17:530–537. doi: 10.1016/j.str.2009.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nair D, Johnson R, Prakash L, Prakash S, Aggarwal A. An incoming nucleotide imposes an anti to syn conformational change on the templating purine in the human DNA polymerase-iota active site. Structure. 2006;14:749–755. doi: 10.1016/j.str.2006.01.010. [DOI] [PubMed] [Google Scholar]
- 67.Berman AJ, Kamtekar S, Goodman JL, Lazaro JM, de Vega M, Blanco L, Salas M, Steitz TA. Structures of phi29 DNA polymerase complexed with substrate: the mechanism of translocation in B-family polymerases. EMBO J. 2007;26:3494–3505. doi: 10.1038/sj.emboj.7601780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Johnson SJ, Taylor JS, Beese LS. Processive DNA synthesis observed in a polymerase crystal suggests a mechanism for the prevention of frameshift mutations. Proc Natl Acad Sci U S A. 2003;100:3895–3900. doi: 10.1073/pnas.0630532100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li Y, Korolev S, Waksman G. Crystal structures of open and closed forms of binary and ternary complexes of the large fragment of Thermus aquaticus DNA polymerase I: structural basis for nucleotide incorporation. EMBO J. 1998;17:7514–7525. doi: 10.1093/emboj/17.24.7514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yin YW, Steitz TA. The structural mechanism of translocation and helicase activity in T7 RNA polymerase. Cell. 2004;116:393–404. doi: 10.1016/s0092-8674(04)00120-5. [DOI] [PubMed] [Google Scholar]
- 71.Xing G, Kirouac K, Shin YJ, Bell SD, Ling H. Structural insight into recruitment of translesion DNA polymerase Dpo4 to sliding clamp PCNA. Mol Microbiol. 2008 doi: 10.1111/j.1365-2958.2008.06553.x. [DOI] [PubMed] [Google Scholar]
- 72.Hishiki A, Hashimoto H, Hanafusa T, Kamei K, Ohashi E, Shimizu T, Ohmori H, Sato M. Structural basis for novel interactions between human translesion synthesis polymerases and proliferating cell nuclear antigen. J Biol Chem. 2009;284:10552–10560. doi: 10.1074/jbc.M809745200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bailey S, Wing RA, Steitz TA. The structure of T. aquaticus DNA polymerase III is distinct from eukaryotic replicative DNA polymerases. Cell. 2006;126:893–904. doi: 10.1016/j.cell.2006.07.027. [DOI] [PubMed] [Google Scholar]
- 74.Lamers MH, Georgescu RE, Lee SG, O'Donnell M, Kuriyan J. Crystal structure of the catalytic alpha subunit of E. coli replicative DNA polymerase III. Cell. 2006;126:881–892. doi: 10.1016/j.cell.2006.07.028. [DOI] [PubMed] [Google Scholar]
- 75.Evans RJ, Davies DR, Bullard JM, Christensen J, Green LS, Guiles JW, Pata JD, Ribble WK, Janjic N, Jarvis TC. Structure of PolC reveals unique DNA binding and fidelity determinants. Proc Natl Acad Sci U S A. 2008;105:20695–20700. doi: 10.1073/pnas.0809989106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wing RA, Bailey S, Steitz TA. Insights into the replisome from the structure of a ternary complex of the DNA polymerase III alpha-subunit. J Mol Biol. 2008;382:859–869. doi: 10.1016/j.jmb.2008.07.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Georgescu RE, Kim SS, Yurieva O, Kuriyan J, Kong XP, O'Donnell M. Structure of a sliding clamp on DNA. Cell. 2008;132:43–54. doi: 10.1016/j.cell.2007.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wang J, Sattar AK, Wang CC, Karam JD, Konigsberg WH, Steitz TA. Crystal structure of a pol alpha family replication DNA polymerase from bacteriophage RB69. Cell. 1997;89:1087–1099. doi: 10.1016/s0092-8674(00)80296-2. [DOI] [PubMed] [Google Scholar]
- 79.Shamoo Y, Steitz TA. Building a replisome from interacting pieces: sliding clamp complexed to a peptide from DNA polymerase and a polymerase editing complex. Cell. 1999;99:155–166. doi: 10.1016/s0092-8674(00)81647-5. [DOI] [PubMed] [Google Scholar]
- 80.Franklin MC, Wang J, Steitz TA. Structure of the replicating complex of a pol alpha family DNA polymerase. Cell. 2001;105:657–667. doi: 10.1016/s0092-8674(01)00367-1. [DOI] [PubMed] [Google Scholar]
- 81.Zhuang Z, Johnson RE, Haracska L, Prakash L, Prakash S, Benkovic SJ. Regulation of polymerase exchange between Poleta and Poldelta by monoubiquitination of PCNA and the movement of DNA polymerase holoenzyme. Proc Natl Acad Sci U S A. 2008;105:5361–5366. doi: 10.1073/pnas.0801310105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zang H, Goodenough AK, Choi JY, Irimia A, Loukachevitch LV, Kozekov ID, Angel KC, Rizzo CJ, Egli M, Guengerich FP. DNA adduct bypass polymerization by Sulfolobus solfataricus DNA polymerase Dpo4: analysis and crystal structures of multiple base pair substitution and frameshift products with the adduct 1,N2-ethenoguanine. J Biol Chem. 2005;280:29750–29764. doi: 10.1074/jbc.M504756200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.