

Abstract
In 1992, John Daly et al. reported the isolation and structure determination of epibatidine. Epibatidine’s unique structure and its potent nicotinic agonist activity have had a tremendous impact on nicotine receptor research. This research has led to a better understanding of the nicotinic acetylcholine receptor (nAChR) pharmacophore and to epibatidine analogues with potential as pharmacotherapies for treating various CNS disorders. In this study, we report the synthesis, receptor binding ([3H]epibatidine and [125I]iodoMLA), and in vivo pharmacological properties (mouse tail flick, hot plate, hypothermia, and spontaneous activity) of a series of 3′-(substituted phenyl)epibatidine analogues (5a–m). Results from these studies have added to the understanding of the nAChR pharmacophore and led to nicotinic partial agonists that may have potential for smoking cessation. All the analogues had affinities for the α4β2 nAChR similar to epibatidine (1). 3′-(3-Dimethylaminophenyl)epibatidine (5m) has a nicotinic partial agonist pharmacological profile similar to the smoking cessation drug varenicline. Other analogues are partial agonists with varying degrees of nicotinic functional agonist and antagonist activity. 3′-(3-Aminophenyl)epibatidine (5j) is a more potent functional agonist and antagonist in all tests than varenicline. 3′-(3-Fluorophenyl)epibatidine and 3′-(3-chlorophenyl)epibatidine (5c and 5e) are more potent than varenicline when tested as agonists in four pharmacological tests and antagonists when evaluated against nicotine in the analgesia hot-plate test.
Since it is estimated that four million smoking-related deaths result annually from smoking-related diseases such as lung cancer, chronic obstructive pulmonary disease (COPD), and cardiovascular disease,1 there is great interest in the development of pharmacotherapies for aiding people to stop smoking.2 In addition to nicotine (2) replacement therapy (NRT), the α4β2 nAChR partial agonist varenicline (3), and the antidepressant bupropion (4), which is also a noncompetitive nAChR antagonist, are the first-line treatment drugs for smoking cessation.2–4
Epibatidine (1), a structurally novel nicotinic acetylcholinergic compound, was isolated by Daly et al. from the skin of the Ecuadorian poison frog, Epipedobates tricolor.5 Its unique structure and potent nicotinic acetylcholine receptor (nAChR) activity has had a major impact on nicotinic receptor research. A SciFinder® search on epibatidine reveals 1013 references from 1992 (original report on the isolation by Daly et al.) to September 2009. Even though epibatidine’s acute toxicity limited its therapeutic potential,6–9 it has served as a lead structure to develop pharmacotherapies for treating various CNS disorders including Alzheimer’s and Parkinson’s diseases, pain, schizophrenia, anxiety, depression, Tourette’s syndrome, and smoking cessation.10
During the last several years, we have conducted structure-activity relationship (SAR) studies using epibatidine (1) as our lead structure to help characterize pharmacophores for the nAChR and to identify nAChR agonists, partial agonists, and antagonists as potential pharmacotherapies for treating smokers.11–19 In a preliminary study, we reported that introduction of a phenyl group at the 3′-position on the 2-chloropyridine ring of epibatidine gave 5a, which had high affinity for α4β2 nAChR but was 100–350-times less potent than epibatidine (1) in the mouse tail-flick, hot-plate, hypothermia, and spontaneous activity tests after acute administration.11 The ability of 5a to antagonize nicotine-induced antinociception was not tested in this study. In the present investigation, we report the nicotinic antagonist properties of 5a and compare the nAChR binding and agonist/antagonist pharmacological properties of the 3′-(substituted phenyl)epibatidine analogues 5a–m to those of the nAChR agonists, nicotine (2) and epibatidine (1), and the partial agonist varenicline (3). All analogues (5a–m) had high affinity for the α4β2 nAChR similar to epibatidine (1). Also like epibatidine, they also had weak affinity for the α7 nAChR. Compounds 5a–m showed both agonist and antagonist activity in the mouse acute tail-flick, hot-plate, hypothermia, and spontaneous-activity functional tests and, thus, are partial nAChR agonists.
Results and Discussion
Chemistry
The synthesis of 5b, 5d, 5g, 5i, and 5k is shown in Scheme 1. Palladium acetate-catalyzed coupling of tert-butoxycarbonyl-3′-bromoepibatidine (6)14 with the appropriately substituted phenylboronic acid in dimethoxyethane (DME) in the presence of tris-(o-tolyl)phosphine and sodium carbonate gave the tert-butoxycarbonyl-protected 3′-(substituted phenyl)epibatidine analogues (7). Treatment of 7a–e (X = F, Cl, NO2, tBocNH, and CH3O) with trifluoroacetic acid in methylene chloride removed the protecting tert-butoxycarbonyl group and afforded the desired 3′-(substituted phenyl)epibatidine analogues, 5b, 5d, 5g, 5i, and 5k.
Scheme 1a.

a Reagents: (a) Pd(OAc)2, P(o-tolyl)3, Na2CO3, (X)C6H4B(OH)2, DME; (b) CF3CO2H
Scheme 2 outlines the synthesis of 5j and 5m starting with the previously reported 3′-(3-nitrophenyl)epibatidine (5h).19 Reduction of 5h with iron powder in ethanolic hydrogen chloride gave the desired 5j. In order to prepare 5m, 5h was first converted to the tert-butoxycarbonyl-protected 8 using tert-butoxycarbonyl anhydride catalyzed by dimethylaminopyridine (DMAP) in methylene chloride containing a small amount of triethylamine. Reduction of 8 with nickel borohydride and hydrochloric acid in methanol provided the amino compound 9, which was reductively methylated to the 7-tert-butoxycarbonyl-protected dimethylamino compound 10 using sodium cyanoborohydride and formaldehyde in acetonitrile. Treatment of 10 with trifluoroacetic acid in methylene chloride afforded 5m. Target compounds 5c, 5e, and 5f were all prepared from 5j using various diazotization procedures (Scheme 3). Thus, diazotization of 5j using sodium nitrite in 70% hydrogen fluoride-pyridine yielded 5c and diazotization of 5j using n-butyl nitrite with cuprous chloride or cuprous bromide in acetonitrile afforded 5e and 5f, respectively.
Scheme 2a.

a Reagents: (a) Fe, HCl, C2H5OH; (b) (Boc)2O, DMAP, (C2H5)3N, CH2Cl2; (c) Ni2B, CH3OH, HCl; (d) NaCNBH3, H2CO, CH3CN; (e) CF3CO2H, CH2Cl2
Scheme 3a.

a Reagents: (a) HF pyridine, NaNO2; (b) nC4H9ONO, CuCl, CH3CN, 65 °C; (c) nC4H9ONO, CuBr, CH3,CN, 65 °C
Biological Activity
The nAChR binding affinities and the functional nicotinic pharmacological properties of several 3′-(substituted phenyl)epibatidine analogues were determined. The Ki values for the inhibition of [3H]epibatidine and [125I]iodoMLA binding at the α4β2 and α7 nAChRs, respectively, for compounds 5a–m along with reference compounds (+)-and (−)-epibatidine [(+)-1 and (−)-1], nicotine (2), and varenicline (3), are listed in Table 1. (+)-and (−)-Epibatidine with Ki values of 0.026 and 0.018 nM, respectively, have very similar affinities for the α4β2 nAChR.15 Nicotine and varenicline have Ki values of 1.5 and 0.12 nM, respectively. Since the affinities of the epibatidine isomers are so similar, the (substituted phenyl)epibatidine analogues 5a–m are only compared to the natural epibatidine isomer. The unsubstituted phenyl analogue 5a has a Ki of 0.021 nM at the α4β2 nAChR, which is almost identical to that of epibatidine (Ki = 0.026 nM), and has 71- and 6-times higher affinity at the α4β2 nAChR than nicotine and varenicline, respectively. Substitution of the 3′-phenyl ring of 5a with a 4- or 3-position electron-withdrawing or -releasing substituent had only small effects on binding affinity at the α4β2 nAChR. The Ki values varied from 0.008 and 0.009 nM for the 3-nitrophenyl (5h) and 3-dimethylamino (5m) analogues to 0.034 and 0.039 nM for the 4-aminophenyl (5i) and the 4-chlorophenyl (5d) analogues. In every case the 3′-(3-substituted phenyl) analogue had a slightly lower Ki value than the corresponding 3′-(4-substituted phenyl) analogue (compare 5c, 5e, 5h, 5j, and 5l to the corresponding 5b, 5d, 5g, 5i, and 5k). Similar to epibatidine, all analogues had relatively weak affinity for the α7 nAChR. The α7 nAChR Ki values varied from 30.5 nM for 5e to 1100 nM for 5m, compared to a Ki of 198 nM for epibatidine and 32.5 nM for varenicline. The 4-chlorophenyl analogue 5e with a Ki of 30.5 nM had the highest affinity for the α7 nAChR but was still greater than 2000-fold selective for α4β2 nAChR relative to α7 nAChR.
Table 1.
Comparison of Nicotine, Epibatidine, and Varenicline Radioligand Binding and Antinociception Data to 3′-(Substituted Phenyl)epibatidine Analogues (5a–m)
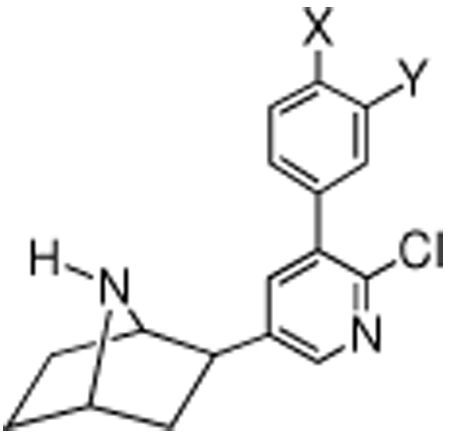 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| compound | X | Y | αβ [3H]epibatidine (Ki, nM) |
α7 [125I]iodoMLA (Ki, nM)b |
ED50 mg/kg tail-flickc |
ED50 mg/kg hot-platec |
ED50 mg/kg hypothermiac |
ED50 mg/kg spontaneous activityc |
AD50 (µg/kg)c |
|
| tail-flick | hot-plate | |||||||||
| (+)-epibatidine [(+)-1]d |
0.026 ± 0.002e | 198 | 0.0061e | 0.004e | 0.004e | 0.001e | ||||
| (−)-epibatidine [(−)-1]d |
0.018 ± 0.001e | 0.0066e | ||||||||
| nicotine (2)e |
1.5 ± 0.3 | 1.3 | 0.65 | 1.0 | 0.5 | |||||
| varenicline (3)f |
0.12 ± 0.02 | 32.5 ± 1.3 | 11% @ 10 | 10% @ 10 | 2.8 | 2.1 | 0.2 | 470 | ||
| 5a | H | H | 0.021 ± 0.005 | 260 ± 5.0 | 0.7g (0.5–1.0) |
1.0g (0.5–2.0) |
0.4g (0.1–0.9) |
0.35g (0.2–0.85) |
280 (80–900) |
30% @ 10,000 |
| 5b | F | H | 0.017 ± 0.003 | 309 ± 13 | 2.48 (1.7–3.5) |
1.27 (0.8–1.9) |
0.33 (0.22–0.67) |
0.33 (0.15–0.7) |
0.5 (0.06–29) |
30% @ 100 |
| 5c | H | F | 0.012 ± 0.001 | 250 ± 44 | 1.0 (0.7–1.5) |
0.43 (0.2–0.94) |
0.19 (0.1–0.7) |
0.07 (0.05–0.10) |
3.9 (5–30) |
86 (20–300) |
| 5d | Cl | H | 0.039 ± 0.005 | 209 ± 50 | 2.6 (2.3–3.3) |
2 (1.4–2.7) |
0.89 (0.5–1.2) |
0.53 (0.3–0.9) |
1 (0.1–27) |
10% @100 |
| 5e | H | Cl | 0.013 ± 0.001 | 30.5 ± 84 | 3.4 (2.6–4.3) |
4.2 (1.5–11.6) |
0.38 (0.2–1) |
0.06 (0.02–0.13) |
2.2 (4–15) |
80 (30–200) |
| 5f | H | Br | 0.016 ± 0.003 | 85 ± 10 | 0.54 (0.44–0.67) |
2.2 (1.2–3.8) |
0.67 (0.5–0.9) |
0.11 (0.06–0.22) |
10 (7–90) |
220 (50–600) |
| 5g | NO2 | H | 0.015 ± 0.001 | 250 ± 25 | 0.46 (0.33–0.64) |
0.38 (0.2–0.5) |
0.23 (0.1–0.6) |
0.24 (0.1–0.5) |
7 (3–10) |
17% @ 50 |
| 5h | H | NO2 | 0.008 ± 0.0003 | 229 ± 43 | 4.6 (3.3–6.5) |
3.2 (2.4–4.5) |
1.4 (1–2.4) |
0.9 (0.7–1.2) |
0.25 (0.02–0.7) |
180 (29–1078) |
| 5i | NH2 | H | 0.034 ± 0.001 | 234 ± 17 | 5 (3.4–7.2) |
4.5 (3.6–5.7) |
1.1 (0.8–1.9) |
0.52 (0.11–2.3) |
6 (5–7) |
0% @100 |
| 5j | H | NH2 | 0.017 ± 0.001 | 256 ± 46 | 0.34 (0.24–0.46) |
0.3 (0.17–0.53) |
0.31 (0.2–0.5) |
0.03 (0.009–0.12) |
0.14 (0.05–0.4) |
26 (5–90) |
| 5k | CH3O | H | 0.022 ± 0.008 | 175 ± 10 | 2.36 (2–3.7) |
1.1 (0.5–2.7) |
0.79 (0.55–1.1) |
0.43 (0.1–1.3) |
10 (1–9) |
10% @ 200 |
| 5l | H | CH3O | 0.019 ± 0.005 | 558 ± 50 | 0.7 (0.3–1.2) |
0.8 (0.3–1.7) |
0.7 (0.5–1.1) |
0.2 (0.18–0.23) |
29 (10–60) |
0% @100 |
| 5m | H | (CH3)2N | 0.009 ± 0.001 | 1100 ± 157 | 0% @ 2 | 0% @ 2 | 3.3 (2.1–5.6) |
2.0 (0.7–5.8) |
20 (10–50) |
560 (100–9200) |
All epibatidine analogues were tested as hydrochloride salts, and all were racemates.
Data represent means ± SE from at least three independent experiments.
Results are provided as ED50 or AD50 values (± confidence limits) or as a percent effect at the individual dose.
Compound (+)-1 is the natural epibatidine hydrochloride, and (−)-1 is the enantiomeric epibatidine hydrochloride.
Data taken from Ref. 15.
Data taken from Ref. 19.
Data taken from Ref. 13.
Natural epibatidine has an ED50 of 0.0061 and 0.004 mg/kg in the tail-flick and hot-plate antinociception tests.15 The unnatural epibatidine isomer has an ED50 of 0.0066 in the tail-flick test.15 Epibatidine also has ED50 values of 0.004 and 0.001 mg/kg in the hypothermia and spontaneous-activity test. The ED50 values for nicotine (2) in the tail-flick, hot-plate, hypothermia, and spontaneous activity tests are 1.3, 0.65, 1.0, and 0.5 mg/kg, respectively. Varenicline (3) is inactive in the tail-flick and hot-plate tests but has ED50 values of 2.8 and 2.1 mg/kg in the hypothermia and spontaneous-activity tests. In addition, varenicline (3) antagonizes the nicotine-induced antinociception in the tail-flick and hot-plate tests with AD50 values of 0.2 and 470 µg/kg, respectively.19
With the exception of the 3-dimethylaminophenyl analogue 5m, which had a profile like varenicline, all compounds showed weak agonists activity compared to epibatidine in the tail-flick, hot-plate, hypothermia, and spontaneous-activity tests. The 3-aminophenyl analogue 5j with ED50 values of 0.34, 0.3, 0.31, and 0.03 mg/kg in the tail-flick, hot-plate, and hypothermia tests was the most potent analogue; however, this potency is 57-, 75-, 76- and 30-times weaker than epibatidine (1) in these four tests. The agonist potencies of these analogues were more similar to nicotine (2) than epibatidine. Similar to varenicline (3), 5m did not have agonist activity in the tail-flick and hot-plate tests but had ED50 values of 3.3 and 2.0 mg/kg in the hypothermia and spontaneous-activity tests, respectively, compared to ED50 values of 2.8 and 2.1 mg/kg for varenicline (3) in these two tests. Most analogues did not exhibit pharmacological selectivity. For the most part they all produced similar potencies in all four tests. The most variation was in the spontaneous-activity test. For example, the 3-aminophenyl analogue 5j was approximately an order of magnitude more potent in this test than in the other three tests.
The high binding affinity of compounds 5a–m for α4β2 nAChRs combined with the relatively weak agonist potency suggested that these analogues might act as nAChR functional antagonists in vivo. Indeed, all analogues were potent antagonists with AD50 values of 0.14–20 µg/kg in the tail-flick test compared to 0.2 µg/kg for the partial agonist varenicline (3). In addition, the 3′-phenyl and 3′-(substituted phenyl) analogues 5a, 5c, 5e, 5h, 5j, and 5m, respectively, were also potent antagonists in the hot-plate test with AD50 values of 26–560 µg/kg compared to 470 µg/kg for varenicline (3). The three most potent analogues were the 3-aminophenyl (5j), 3-chlorophenyl (5e), and 3-fluorophenyl (5c), with AD50 values of 26, 80, and 86 µg/kg, respectively, which is 18-, 5.9-, and 5.5-times more potent as an antagonist than varenicline in the hot-plate test.
In summary, the addition of a 3′-phenyl or electron-withdrawing or -releasing 3′-(3- or 4-substituted phenyl) group to the highly potent nAChR agonist epibatidine (1) provided a series of analogues 5a–m. Like epibatidine these compounds had high affinity for the α4β2 nAChR and weak affinity for the α7 nAChR. In contrast to the high potency of epibatidine (1), these analogues had agonist potency in the tail-flick, hot-plate, hypothermia, and spontaneous-activity tests in mice more like that of nicotine (2). On the other hand, like varenicline, these analogues were potent antagonists in the tail-flick test and to a lesser degree in the hot-plate test. Thus, similar to varenicline, these analogues are functional nAChR partial agonists in vivo. The 3-dimethylaminophenyl analogue 5m has an agonist/antagonist profile most like varenicline. The 3-aminophenyl analogue 5j is a more potent functional agonist and antagonist in all tests than varenicline. The 3-fluorophenyl and 3-chlorophenyl analogues 5c and 5e, respectively, are more potent than varenicline in all four agonist tests and the antagonist hot-plate test. Thus, compounds 5c, 5e, 5j, and 5m represent exciting lead structures for developing a new structural class of nicotinic partial agonists useful in the treatment of nicotine addiction (smokers) and possibly other CNS diseases and disorders.
Experimental Section
General Experimental Procedures
Melting points were determined on a Mel-temp (Laboratory Devices, Inc.) capillary tube apparatus. NMR spectra were recorded on a Bruker Avance 300 using tetramethylsilane as internal standard. Thin-layer chromatography was carried out on Whatman silica gel 60 plates. Visualization was accomplished under UV or in an iodine chamber. Microanalysis was carried out by Atlantic Microlab, Inc. Flash chromatography was carried out using silica gel 60 (230–400 mesh) using various solvent mixtures. CMA is 80% chloroform, 18% methanol, and 2% concentrated ammonium hydroxide.
The [3H]epibatidine was purchased from Perkin Elmer Inc. (Boston, MA). The [125]iodo-MLA was synthesized as previously reported.20
7-tert-Butoxycarbonyl-2-exo-[3-(4-fluorophenyl)-5-pyridinyl]-7-azabicyclo[2.2.1]heptane (7a)
To a resealable reaction tube were added compound 6 (231 mg, 0.6 mmol), 4-fluorophenylboronic acid (168 mg, 1.2 mmol), Pd(OAc)2 (14 mg, 0.06 mmol), tris(o-tolyl)phosphine (37 mg, 0.12 mmol), and Na2CO3 (159 mg, 1.5 mmol) in DME (2 mL) and H2O (0.5 mL). The mixture was purged with argon, sealed, and heated in an 85 °C oil bath overnight. The reaction mixture was cooled, filtered through celite, and diluted with EtOAc. The organic phase was washed with brine, dried (Na2SO4), and concentrated. Flash chromatography of the resulting residue on a silica gel column using EtOAc-hexanes (1:1) yielded 217 mg (90%) of 7a as a yellow oil. 1H NMR (CDCl3) δ 1.40 (9H, s), 1.5–1.9 (5H, m), 2.02 (1H, m), 2.90 (1H, dd, J = 4.5, 8.7 Hz), 4.23 (1H, brs), 4.39 (1H, brs), 7.1–7.2 (2H, m), 7.4–7.5 (2H, m), 7.64 (1H, m), 8.27 (1H, m); 13C NMR (CDCl3) δ 28.6, 29.2, 30.1, 40.8, 45.2, 56.2, 62.2, 80.2, 115.6 (d, JCF = 21.0 Hz), 131.5 (d, JCF = 8.7 Hz), 135.9, 138.5, 140.9, 147.8, 155.2, 163.0 (d, JCF = 247.7 Hz).
3′-(4-Fluorophenyl)epibatidine (5b) Hydrochloride
Compound 7a (217 mg, 0.54 mmol) was dissolved in CH2Cl2 (3 mL). TFA (3 mL) was added dropwise at 0 °C over 30 min. The mixture was stirred at room temperature for 4 h, poured into a cold solution of conc. NH4OH-H2O (1:1), and extracted with CH2Cl2. The organic phase was washed with brine, dried (Na2SO4), and evaporated to dryness. Flash chromatography of the resulting residue on a silica gel column using CH2Cl2-MeOH yielded 136 mg (83%) of 5b. 1H NMR (CDCl3) δ 1.5–1.7 (5H, m), 1.94 (1H, dd, J = 9.0, 12.0 Hz), 2.82 (1H, dd, J = 5.1, 9.0 Hz), 3.62 (1H, brs), 3.81 (1H, brs), 7.1–7.2 (2H, m), 7.4–7.5 (2H, m), 7.76 (1H, d, J = 2.4 Hz), 8.29 (1H, d, J = 2.4 Hz); 13C NMR (CDCl3) δ 30.2, 31.7, 40.6, 44.8, 56.8, 63.1, 115.6 (d, JCF = 21.1 Hz), 131.6 (d, JCF = 8.7 Hz), 135.7, 138.9, 141.8, 147.5, 148.0, 163.0 (d, JCF = 247.8 Hz).
Compound 5b (136 mg, 0.45 mmol) was dissolved in MeOH (4.6 mL) at room temperature. HCl (1 M in ether, 4.6 mL) was added. After stirring for 30 min, the solvent was removed, and the residue was recrystallized from a MeOH-ether mixture to give 5b•HCl as a yellow solid; mp >200 °C (dec); anal. C 60.06%, H 5.16%, N 8.07%, calcd for C17H17Cl2FN2, C 60.19%, H 5.05%, N 8.26%.
7-tert-Butoxycarbonyl-2-exo-[3-(4-chlorophenyl)-5-pyridinyl]-7-azabicyclo[2.2.1]heptane (7b)
To a resealable reaction tube were added compound 6 (233 mg, 0.6 mmol), 4-chlorophenylboronic acid (188 mg, 1.2 mmol), Pd(OAc) (14 mg, 0.06 mmol), tris(o-tolyl)phosphine (37 mg, 0.12 mmol), and Na2CO3 (159 mg, 1.5 mmol) in DME (2 mL) and H2O (0.5 mL). The mixture was purged with argon, sealed, and heated in an 85 °C oil bath overnight. The reaction mixture was cooled, filtered through celite, and diluted with EtOAc. The organic phase was washed with brine, dried (Na2SO4), and concentrated. Flash chromatography of the resulting residue on a silica gel column using EtOAc-hexanes (1:1) yielded 247 mg (98%) of 7b as a yellow oil. 1H NMR (CDCl3) δ 1.40 (9H, s), 1.5–1.9 (5H, m), 2.02 (1H, dd, J = 9.0, 12.3 Hz), 2.92 (1H, dd, J = 4.8, 9.0 Hz), 4.22 (1H, brs), 4.38 (1H, brs), 7.3–7.5 (4H, m), 7.63 (1H, d, J = 2.7 Hz), 8.28 (1H, d, J = 2.4 Hz); 13C NMR (CDCl3) δ 28.6, 29.2, 30.0, 40.8, 45.2, 56.3, 62.2, 80.2, 128.9, 131.1, 131.5, 135.9, 138.4, 141.0, 147.9, 155.2.
3′-(4-Chlorophenyl)epibatidine (5d) Hydrochloride
Compound 7b (247 mg, 0.59 mmol) was dissolved in CH2Cl2 (3 mL). TFA (3 mL) was added dropwise at 0 °C over 30 min. The mixture was stirred at room temperature for 3 h, poured into a cold solution of NH4OH-H2O (1:1), and extracted with CH2Cl2. The organic phase was washed with brine, dried (Na2SO4), and evaporated to dryness. Flash chromatography of the resulting residue on a silica gel column using CH2Cl2-MeOH yielded 147 mg (78%) of 5d as a yellow oil. 1H NMR (CDCl3) δ 1.5–1.7 (5H, m), 1.94 (1H, dd, J = 9.0, 12.0 Hz), 2.82 (1H, dd, J = 5.1, 9.0 Hz), 3.62 (1H, brs), 3.80 (1H, brs), 7.3–7.5 (4H, m), 7.76 (1H, d, J = 2.4 Hz), 8.30 (1H, d, J = 2.4 Hz); 13C NMR (CDCl3) δ 30.4, 31.7, 40.6, 44.8, 56.8, 63.1, 128.6, 128.8, 129.0, 131.1, 131.2, 131.5, 138.9, 141.8, 148.1.
Compound 5d (147 mg, 0.46 mmol) was dissolved in MeOH (4.6 mL) at room temperature. HCl (1 M in ether, 4.6 mL) was added with a syringe pump over 50 min at room temperature. After stirring for 30 min, the solvent was removed. The residue was recrystallized from a MeOH-ether mixture to give 5d•HCl as a yellow solid; mp >200 °C (dec.); anal. C 53.73%, H 4.90%, N 6.81%, calcd for C17H17Cl3N2•1.5 H2O, C 53.35%, H 5.27%, N 7.32%.
7-tert-Butoxycarbonyl-2-exo-[3-(4-nitrophenyl)-5-pyridinyl]-7-azabicyclo[2.2.1]heptane (7c)
To a resealable reaction tube were added compound 6 (233 mg, 0.6 mmol), 4-nitrophenylboronic acid (200 mg, 1.2 mmol), Pd(OAc)2 (14 mg, 0.06 mmol), tris(o-tolyl)phosphine (37 mg, 0.12 mmol), and Na2CO3 (159 mg, 1.5 mmol) in DME (2 mL) and H2O (0.5 mL). The mixture was purged with argon, sealed, and heated in an 85 °C oil bath overnight. The reaction mixture was cooled and diluted with EtOAc. The organic phase was washed with brine, dried (Na2SO4), and concentrated. Flash chromatography of the resulting residue on a silica gel column using EtOAc-hexanes (1:1) yielded 239 mg (93%) of 7c as a yellowish oil. 1H NMR (CDCl3) δ 1.40 (9H, s), 1.5–1.9 (5H, m), 2.06 (1H, m), 2.97 (1H, dd, J = 4.5, 8.7 Hz), 4.24 (1H, brs), 4.40 (1H, brs), 7.6–7.7 (2H, m), 7.70 (1H, d, J = 2.4 Hz), 8.25–8.35 (2H, m), 8.35 (1H, d, J = 2.4 Hz); 13C NMR (CDCl3) δ 28.6, 29.2, 30.0, 40.8, 45.1, 56.3, 62.2, 80.3, 123.8, 124.2, 130.8, 131.1, 138.3, 141.3, 144.4, 149.4, 155.2.
3′-(4-Nitrophenyl)epibatidine (5g) Hydrochloride
Compound 7c (239 mg, 0.56 mmol) was dissolved in CH2Cl2 (3 mL). TFA (3 mL) was added dropwise at 0 °C over 30 min. The mixture was stirred at room temperature for 4 h, poured into a cold solution of NH4OH-H2O (1:1), and extracted with CH2Cl2. The organic phase was washed with brine, dried (Na2SO4), and evaporated to dryness. Flash chromatography of the resulting residue on a silica gel column using CH2Cl2-MeOH yielded 126 mg (69%) of 5g as a yellow oil. 1H NMR (CDCl3) δ 1.5–1.7 (5H, m), 1.97 (1H, dd, J = 9.0, 12.0 Hz), 2.72 (1H, brs), 2.85 (1H, dd, J = 5.1, 9.0 Hz), 3.66 (1H, brs), 3.84 (1H, m), 7.6–7.7 (2H, m), 7.86 (1H, d, J = 2.4 Hz), 7.25–7.35 (2H, m), 8.37 (1H, d, J = 2.4 Hz); 13C NMR (CDCl3) δ 30.4, 31.7, 40.6, 44.7, 56.9, 63.2, 123.9, 130.9, 134.7, 138.8, 142.0, 144.6, 149.1.
Compound 5g (126 mg, 0.38 mmol) was dissolved in MeOH (4 mL) at room temperature. HCl (1 M in ether, 4 mL) was added with a syringe pump over 50 min at room temperature. After stirring for 30 min, the solvent was removed. The residue was recrystallized from MeOH-ether to give 5g•HCl as a yellow solid; mp 168–169 °C; anal. C 54.95%, H 4.78%, N 11.14%, calcd for C17H17Cl2N3O2•0.25 H2O, C 55.07%, H 4.76%, N 11.33%.
7-tert-Butoxycarbonyl-2-exo-[3-(4-tert-butoxylcarbonylaminophenyl)-5-pyridinyl]-7-azabicyclo[2.2.1]heptane (7d)
To a resealable reaction tube were added compound 6 (231 mg, 0.6 mmol), 4-(N-boc-amino)phenylboronic acid (284 mg, 1.2 mmol), Pd(OAc)2 (14 mg, 0.06 mmol), tris(o-tolyl)phosphine (37 mg, 0.12 mmol), and Na2CO3 (159 mg, 1.5 mmol) in DME (2 mL) and H2O (0.5 mL). The mixture was purged with argon, sealed, and heated in an 85 °C oil bath overnight. The reaction mixture was cooled, filtered through celite, and diluted with EtOAc. The organic phase was washed with brine, dried (Na2SO4), and concentrated. Flash chromatography of the resulting residue on a silica gel column using EtOAc-hexanes (1:1) yielded 285 mg (96%) of 7d. 1H NMR (CDCl3) δ 1.40 (9H, s), 1.53 (9H, s), 1.5–1.9 (5H, m), 2.06 (1H, m), 2.90 (1H, dd, J = 4.8, 9.0 Hz), 4.22 (1H, brs), 4.38 (1H, brs), 6.78 (1H, brs), 7.3– 7.5 (4H, m), 7.62 (1H, d, J = 2.4 Hz), 8.24 (1H, d, J = 2.4 Hz); 13C NMR (CDCl3) δ 28.6, 28.7, 29.1, 30.1, 40.7, 45.3, 56.3, 62.2, 80.2, 81.1, 118.4, 130.4, 132.4, 136.4, 138.5, 138.9, 140.8, 147.3, 147.8, 153.0, 155.3.
3′-(4-Aminophenyl)epibatidine (5i) Dihydrochloride
Compound 7d (285 mg, 0.57 mmol) was dissolved in CH2Cl2 (3 mL). TFA (3 mL) was added at 0 °C over 30 min. The mixture was stirred at room temperature for 4 h, poured into a cold solution of conc. NH4OH-H2O (1:1), and extracted with CH2Cl2. The organic phase was washed with brine, dried (Na2SO4), and evaporated to dryness. Flash chromatography of the resulting residue on a silica gel column using CH2Cl2-MeOH yielded 143 mg (84%) of 5i as a yellow oil. 1H NMR (CDCl3) δ 1.5–1.7 (5H, m), 1.91 (1H, dd, J = 9.0, 12.0 Hz), 2.80 (1H, dd, J = 5.1, 9.0 Hz), 3.60 (1H, m), 3.77 (1H, m), 6.6–6.8 (2H, m), 7.2–7.3 (2H, m), 7.70 (1H, d, J = 2.4 Hz), 8.22 (1H, d, J = 2.4 Hz); 13C NMR (CDCl3) δ 30.4, 31.6, 40.6, 45.0, 56.8, 63.1, 114.9, 127.8, 130.8, 136.7, 13838, 141.6, 146.9, 147.0, 147.5.
The free base 5i (143 mg, 0.48 mmol) was dissolved in MeOH (5 mL) at room temperature. HCl (1 M in ether, 5 mL) was added with a syringe pump over 50 min at room temperature. After stirring for 30 min, the solvent was removed, and the residue was recrystallized from MeOH-ether to give 5i•HCl as a yellow solid; mp >265 °C (dec.); anal. C 50.89%, H 5.86%, N 10.14%, calcd for C17H20Cl3N3•1.5 H2O, C 51.08%, H 5.80%, N 10.51%.
7-tert-Butoxycarbonyl-2-exo-[3-(4-methoxyphenyl)-5-pyridinyl]-7-azabicyclo[2.2.1]heptane (7e)
To a resealable reaction tube were added compound 6 (231 mg, 0.6 mmol), 4-methoxyphenylboronic acid (182 mg, 1.2 mmol), Pd(OAc)2 (14 mg, 0.06 mmol), tris(o-tolyl)phosphine (37 mg, 0.12 mmol), and Na2CO3 (159 mg, 1.5 mmol) in DME (2 mL) and H2O (0.5 mL). The mixture was purged with argon, sealed, and heated in an 85 °C oil bath overnight. The reaction mixture was cooled, filtered through celite, and diluted with EtOAc. The organic phase was washed with brine, dried (Na2SO4), and concentrated. Flash chromatography of the resulting residue on a silica gel column using EtOAc-hexanes (1:1) yielded 223 mg (90%) of 7e as a yellow oil. 1H NMR (CDCl3) δ 1.40 (9H, s), 1.5–1.9 (5H, m), 2.01 (1H, dd, J = 9.0, 12.3 Hz), 2.91 (1H, dd, J = 4.8, 8.7 Hz), 3.84 (3H, s), 4.22 (1H, brs), 4.38 (1H, brs), 6.9–7.0 (2H, m), 7.3–7.4 (2H, m), 7.64 (1H, d, J = 2.4 Hz), 8.23 (1H, d, J = 2.4 Hz); 13C NMR (CDCl3) δ 28.6, 29.2, 30.1, 40.7, 45.2, 55.7, 56.3, 62.2, 80.1, 114.1, 130.2, 130.9, 136.5, 138.6, 140.8, 147.2, 155.2, 160.0.
3′-(4-Methoxylphenyl)epibatidine (5k) Dihydrochloride
Compound 7e (223 mg, 0.54 mmol) was dissolved in CH2Cl2 (3 mL). TFA (3 mL) was added dropwise at 0 °C over 30 min. The mixture was stirred at room temperature for 4 h, poured into a cold solution of conc. NH4OH-H2O (1:1), and extracted with CH2Cl2. The organic phase was washed with brine, dried (Na2SO4), and evaporated to dryness. Flash chromatography of the resulting residue on a silica gel column using CH2Cl2-MeOH yielded 128 mg (76%) of 5k as a yellow oil. 1H NMR (CDCl3) δ 1.5–1.7 (5H, m), 1.92 (1H, dd, J = 9.0, 12.3 Hz), 2.80 (1H, dd, J = 4.8, 8.7 Hz), 3.60 (1H, brs), 3.78 (1H, t, J = 3.6 Hz), 3.84 (3H, s), 6.9–7.0 (2H, m), 7.3–7.4 (2H, m), 7.74 (1H, d, J = 2.4 Hz), 8.26 (1H, d, J = 2.4 Hz); 13C NMR (CDCl3) δ 30.1, 31.4, 40.3, 44.6, 55.3, 56.4, 62.8, 113.7, 130.1, 130.6, 136.0, 138.6, 141.4, 147.0, 147.2, 159.530.2, 31.7, 40.6, 44.8, 56.8, 63.1, 115.6 (d, J = 0.855 Hz), 131.6 (d, J = 0.33 Hz), 135.7, 138.9, 141.8, 147.5, 148.0, 161.4, 164.6.
The free base 5k (128 mg, 0.41 mmol) was dissolved in MeOH (4 mL) at room temperature. HCl (1 M in ether, 4 mL) was added with a syringe pump over 20 min at room temperature. After stirring for 30 min, the solvent was removed, and the residue was recrystallized from a MeOH-ether mixture to give 5k•2HCl as a yellow solid; mp >200 °C (dec.); anal. C 53.62%, H 5.63%, N 6.90%, calcd for C18H21Cl3N2O•H2O, C 53.28%, H 5.71%, N 6.90%.
3′-(3-Aminophenyl)epibatidine (5j) Dihydrochloride
Compound 5h (80 mg, 0.241 mmol), ethanol (2 mL), water (0.06 mL), and concentrated HCI (0.01 mL) were stirred at room temperature for 10 min. Iron powder (149.2 mg, 2.66 mmol) was added in small portions. The mixture was heated at 100 °C for 30 min, poured into a cold solution of sodium carbonate (aq. solution), and extracted with ethyl acetate. The organic phase was dried (Na2SO4) and concentrated. The residue was purified by silica gel column chromatography eluting with CMA-EtOAc (1:3) to yield 70 mg (95%) of 5j as a yellow oil. 1H NMR (CDCl3) δ 1.48–1.70 (5H, m), 1.87–1.99 (1H, m), 2.79–2.84 (1H, dd), 3.61 (1H, brs), 3.78 (1H, brs), 6.70–6.74 (2H, m), 6.81 (1H, d, J = 6.0 Hz), 7.20 (1H, d, J = 6.0 Hz), 7.72 (s, 11:1), 8.27 (s, 1H); 13C NMR (CDCl3) δ 30.3, 31.6, 40.6, 44.9, 56.8, 63.1, 115.2, 116.3, 119.9, 129.5, 137.0, 138.9, 139.2, 141.5, 146.7, 147.4, 147.7.
Compound 5j (70 mg, 0.231 mmol) was dissolved in 4 mL of methanol, and 1 M HCI in ether (2 mL) was added. After stirring for 30 min, the solvent was removed. The residue was recrystallized from MeOH-ether (1:3) to yield 5j•HCl as a yellow solid; mp 195 °C (dec.); anal. C 51.08%, H 5.80%, N 10.51%, ca1cd for C17H20Cl3N3•1.5 H20, C 51.14%, H 5.91%, N 9.61%.
7-tert-Butoxycarbonyl-2-exo-[3-(3-nitrophenyl)-5-pyridinyl]-7-azabicyclo[2.2.1]heptane (8)
A solution of compound 5h (0.31 g, 0.94 mmol), tert-butoxycarbonyl anhydride (400 mg, 1.83 mmol), DMAP (10 mg), triethylamine (0.1 mL), and methylene chloride (5 mL) was stirred for 1 h, poured into 100 mL of 1 M K2CO3, and extracted with CH2Cl2. The organic phase was dried (Na2SO4) and concentrated. The residue was purified by flash chromatography on a silica gel column eluting with hexane-EtOAc (3:1) to yield 0.25 g (60%) of 8 as an oil. 1H NMR (CDCl3) δ 1.39 (9H, s), 1.50–1.70 (5H, m), 1.96–2.09 (1H, m), 2.93–2.97 (1H, m,), 4.23 (1H, brs), 4.39 (1H, brs), 7.64–7.70 (2H, m), 7.79–7.80 (1H, m), 8.31–8.35 (1H, m); 13C NMR (CDCl3) δ 27.8, 28.6 (CH3-3), 31.3, 40.8, 45.2, 56.4, 62.3, 80.4, 123.6, 124.7, 129.8, 134.5, 135.9, 138.5, 139.6, 141.4, 147.5, 148.8, 149.1, 155.4.
7-tert-Butoxycarbonyl-2-exo-[3-(3-aminophenyl)-5-pyridinyl]-7-azabicyclo[2.2.1]heptane (9)
To a mixture of compound 8 (80 mg, 0.81 mmol), Ni2B [56.1 mg, prepared from Ni(OAc)2] in MeOH (3.2 mL) was added 1 M HCl (0.8 mL). The reaction mixture was heated at 60 °C for 30 min, poured into 100 mL of a solution of conc. NH4OH-H2O (1:1), and extracted with CH2Cl2, dried (Na2SO4), and concentrated. The residue was purified by flash chromatography eluting with hexane-EtOAc (3:1) to yield 70 mg (93%) of 9 as a yellow oil. 1H NMR (CDC13) δ 1.40 (9H, s), 1.51–1.85 (5H, m), 1.97–2.04 (1H, m), 2.87–2.92 (1H, m), 3.77 (2H, brs), 4.21 (1H, brs), 4.38 (1H, brs), 6.70–6.74 (2H, m), 6.78–6.81 (1H, m), 7.20 (1H, t, J = 6.0 Hz), 7.62 (1H, s), 8.25 (1H, s); 13C NMR (CDC13) δ 28.7 (CH3-3), 29.2, 30.2, 40.7, 45.3, 56.3, 62.3, 80.3, 115.3, 116.3, 120.0, 129.6, 137.1, 138.6, 139.1, 140.7, 146.7, 147.5, 147.8.
7-tert-Butoxycarbonyl-2-exo-[3′-(3-dimethylamino)-5-pyridinyl]-7-azabicyclo[2.2.1]heptane (10)
A mixture of 9 (180 mg, 0.433 mmol), acetonitrile (12 mL), 37% aq. formaldehyde (1.54 mL), and NaCNBH3 (488 mg, 7.76 mmol) was stirred for 3 h at room temperature. Glacial acetic acid (0.642 mL) was added and stirring continued overnight. The reaction mixture was poured into 100 mL of a solution of conc. NH4OH-H2O (1:1), extracted with CH2Cl2, dried (Na2SO4), and concentrated. The residue was purified by flash chromatography on a silica gel column eluting with hexanes-EtOAc (3:l) to yield 179 mg (93%) of 10 as a yellow oil. 1H NMR (CDCl3) δ 1.39 (9H, s), 1.54–1.89 (5H, m), 1.97–2.01 (1H, m), 2.88–2.93 (1H, m), 2.98 (6H, s), 4.22 (1H, brs), 4.36 (1H, brs), 6.73–6.79 (3H, m), 7.29 (1H, d, J = 6.0 Hz), 7.65 (1H, s), 8.26 (1H, s).
3′-(3-Dimethylaminophenyl)epibatidine (5m) Dihydrochloride
Compound 10 (179 mg, 0.403 mmol) in methylene chloride (3 mL) was stirred at 0 °C for 15 min, then trifluroacetic acid (3 mL) was added. After stirring for 30 min, the reaction mixture was poured into 100 mL of a solution of conc. NH4OH-H2O (1:1), extracted with CH2Cl2, dried (Na2SO4), and concentrated. The residue was purified by flash chromatography on a silica gel column eluting with CMA-EtOAc (1:5) to yield 132 mg (95%) of 5m. 1H NMR (CDCl3) δ 1.49–1.69 (5H, m), 1.89–1.96 (1H, m), 2.79–2.84 (1H, m), 2.98 (6H, s), 3.61 (1H, brs), 3.77 (1H, brs), 6.75–6.78 (3H, m), 7.3 (1H, d, J = 6.0 Hz), 7.75 (1H, s), 8.29 (1H, s); 13C NMR (CDCl3) δ 30.5, 31.7, 40.7, 41.0 (2CH3), 45.1, 56.8, 63.1, 112.6, 113.9, 117.9, 129.4, 137.6, 139.0, 141.6, 147.5, 147.6, 150.7. (One tertiary aromatic C was not observed.)
Compound 5m (132 mg, 0.383 mmol) was dissolved in 4 mL of methanol and 1 M HCl in ether (4 mL) was added dropwise. After concentration the residue was recrystallized from a MeOH-ether mixture (1:3) to give 5m•2HCl; mp 69–72 °C; anal. C 54.23%, H 6.49%, N 9.51%, calcd for C19H24Cl3N3•1.25 H2O, C 53.91%, H 6.31%, N 9.93%.
3′-(3-Fluorophenyl)epibatidine (5c) Hydrochloride
To a solution of 5j (150 mg, 0.5 mmol) in 70% HF-pyridine (2.7 mL) at 0 °C was added NaNO2 (266 mg, 3.9 mmol). The reaction mixture was stirred for 30 min at 0 °C and then heated to 100 °C for an additional hour. The reaction mixture was poured into 100 mL of a solution of NH4OH-H2O (1:1), extracted with CH2Cl2, dried (Na2SO4), and concentrated. The residue was purified by flash chromatography on silica gel column chromatography eluting with CMA-EtOAc (1:3) to yield 40 mg (26%) of 5c as an oil. lH NMR (CDCl3) δ 1.53–1.67 (5H, m), 1.90–2.02 (1H, m), 2.79–2.83 (1H, m), 3.61 (1H, brs), 3.80 (1H, brs), 7.08–7.24 (1H, m), 7.38–7.42 (1H, m), 7.78 (1H, s), 8.31 (1H, s, pyridinyl); 13C NMR (CDCl3) δ 30.6, 31.9, 40.8, 44.9, 56.8, 63.2, 115.4 (d, JCF = 20.9 Hz), 116.9 (d, JCF = 22.3 Hz), 125.6 (d, JCF = 2.9 Hz), 130.2 (d, JCF = 8.2 Hz), 135.5, 138.9, 140.2 (d, JCF = 7.7 Hz), 142.1, 147.3, 148.3, 162.8 (d, JCF = 245 Hz).
Compound 5c (40 mg, 0.131 mmol) was dissolved in 4 mL of methanol, and 1 M HCl in ether (4 mL) was added dropwise. The residue obtained on concentration was recrystallized from methanol and ether to give 5c•HCl; mp 101–105 °C; anal. C 57.53%, H 5.43%, N 7.66%, calcd for C17H17Cl2FN2•0.75 H2O, C 57.88%, H 5.29%, N 7.94%.
3′-(3-Chlorophenyl)epibatidine (5e) Hydrochloride
To a mixture of anhydrous CuCl (32.3 mg, 0.24 mmol), butyl nitrite (0.04 mL, 0.3 mmol), and anhydrous acetonitrile (10 mL) warmed to 65 °C was added 5j (60.4 mg, 0.2 mmol) in 2 mL of anhydrous acetonitrile over 5 min. After 20 min, the cooled reaction mixture was poured into 10 mL of 20% HCl aq., stirred 10 min, and poured into 100 mL of a solution of conc. NH4OH-H2O (1:1), extracted with CH2Cl2, dried (Na2SO4), and concentrated. The residue was purified by flash chromatography on a silica gel column eluting with CMA-EtOAc (1:5) to yield 50 mg (78%) of 5e. 1H NMR (CDCl3) δ 1.50–1.72 (5H, m), 1.90–1.97 (1H, m), 2.79–2.83 (1H, m), 3.61 (−lH, brs), 3.79 (1H, brs), 7.34–7.43 (4H, m), 7.77 (1H, s), 8.30 (1H, s); 13C NMR (CDCl3) δ 30.6, 31.8, 40.8, 44.9, 56.8, 63.2, 128.1, 128.6, 129.8, 129.9, 134.5, 135.4, 138.9, 139.9, 142.1, 147.3, 148.4.
Compound 5e (50 mg, 0.383 mmol) was dissolved in 4 mL of methanol, and 1 M HCI in ether (2 mL) was added. The residue obtained on concentration was recrystallized from CH3OH-ether to give 5e•HCl; mp 159 °C (dec.); anal. C 54.78%, H 5.08%, N 7.18%, calcd for C17H17Cl3N2•H2O C 54.64%, H 5.12%, N 7.50%.
3′-(3-Bromophenyl)epibatidine (5f) Hydrochloride
To a mixture of anhydrous CuBr (53.6 mg, 0.24 mmol), butyl nitrite (0.04 mL, 0.3 mmol), and anhydrous acetonitrile (6 mL), warmed to 65 °C, was added 5j (60.4 mg, 0.2 mmol) in 4 mL of anhydrous acetonitrile. After 20 min, the reaction mixture was poured into 10 mL of 20% HCl aq., stirred 10 min, poured into 100 mL of a solution of conc. NH4OH-H2O (1:1), extracted with CH2Cl2, dried (Na2SO4), and concentrated. The residue obtained was purified by flash chromatography on a silica gel column eluting with CMA-EtOAc (1:5) to yield 30 mg (41%) of 5f. 1H NMR (CDC13) δ 1.50–1.73 (5H, m), 1.90–1.99 (1H, m), 2.79–2.83 (1H, m), 3.61 (1H, brs), 3.80 (1H, brs), 7.31–7.41 (4H, m), 7.77 (1H, s), 8.31 (1H, s); 13C NMR (CDCl3) δ 30.6, 31.9, 40.8, 44.9, 56.8, 63.2, 122.6, 128.5, 130.2, 131.6, 132.6, 134.5, 135.3, 138.9, 140.2, 142.1, 148.4.
Compound 5f (60 mg, 0.164 mmol) was dissolved in 4 mL of methanol, and 1 M HCl in ether (3 mL) was added. The residue obtained on concentration was recrystallized from CH3OH-ether to give 5f•HCl; mp 117–120 °C (dec.); anal. C 49.57%, H 4.66%, N, 6.55%, calcd for C17H17BrCl2N2•0.75 H2O, C 49.36%, H 4.51%, N 6.77%.
[3H]Epibatidine Binding Assay
Adult male rat cerebral cortices (Pelfreeze Biological, Rogers, AK) were homogenized in 39 volumes of ice-cold 50 mM Tris buffer (pH 7.4 at 4 °C) containing 120 mM NaCl, 5 mM KCl, 2 mM CaCl2, and 1 mM MgCl2 and sedimented at 37,000 g for 10 min at 4 °C. The supernatant was discarded, the pellet resuspended in the original volume of buffer, and the wash procedure repeated twice more. After the last centrifugation, the pellet was resuspended in 1/10 its original homogenization volume and stored at −80 °C until needed. In a final volume of 0.5 mL, each assay tube contained 3 mg wet weight male rat cerebral cortex homogenate (added last), 0.5 nM [3H]epibatidine (NEN Life Science Products, Wilmington, DE) and one of 10–12 different concentrations of test compound dissolved in buffer (pH 7.4 at room temperature) containing 10% DMSO resulting in a final DMSO concentration of 1%. Total and nonspecific bindings were determined in the presence of vehicle and 300 µM (−)-nicotine, respectively. After a 4-h incubation period at room temperature, the samples were vacuum-filtered over GF/B filter papers presoaked in 0.03% polyethylenimine using a Brandel 48-well harvester and washed with 6 mL of ice-cold buffer. The amount of radioactivity trapped on the filter was determined by standard liquid scintillation techniques in a TriCarb 2200 scintillation counter (Packard Instruments, Meriden, CT) at approximately 50% efficiency. The binding data were fit using the nonlinear regression analysis routines in Prism (Graphpad, San Diego, CA). The Ki values for the test compounds were calculated from their respective IC50 values using the Cheng-Prusoff equation.
[125I]Iodo-MLA Binding Assay
Adult male rat cerebral cortices (Pel-Freez Biologicals, Rogers, AK) were homogenized (polytron) in 39 volumes of ice-cold 50 mM Tris buffer (assay buffer; pH 7.4 at 4 °C) containing 120 mM NaCl, 5 mM KCl, 2 mM CaCl2, and 1 mM MgCl2. The homogenate was centrifuged at 35,000 g for 10 min at 4 °C and the supernatant discarded. The pellet was resuspended in the original volume of buffer and the wash procedure repeated twice more. After the last centrifugation step, the pellet was resuspended in one-tenth the original homogenization volume and stored at −80 °C until needed. Triplicate samples were run in 1.4-mL polypropylene tubes (Matrix Technologies Corporation, Hudson, NH). Briefly, in a final volume of 0.5 mL, each assay sample contained 3 mg wet weight rat cerebral cortex (added last), 40–50 pM [125I]MLA and 50 nM final concentration of test compound dissolved in buffer containing 10% DMSO, giving a final DMSO concentration of 1%. Total and nonspecific binding were determined in the presence of vehicle and 300 µM (−)-nicotine, respectively. After a 2-h incubation period on ice, the samples were vacuum-filtered using a Multimate 96-well harvester (Packard Instruments, Meriden, CT) onto GF/B filters presoaked for at least 30 min in assay buffer containing 0.15% bovine serum albumin. Each well was then washed with approximately 3.0 mL of ice-cold buffer. The filter plates were dried, and 30 µL of Microscint20 (Packard) was added to each well. The amount of radioligand remaining on each filter was determined using a TopCount 12-detector (Packard) microplate scintillation counter at approximately 70% efficiency.
Tail-flick Test
Antinociception was assessed by the tail-flick method of D’Amour and Smith.21 A control response (2–4 sec) was determined for each mouse before treatment, and a test latency was determined after drug administration. In order to minimize tissue damage, a maximum latency of 10 sec was imposed. Antinociceptive response was calculated as percent maximum possible effect (% MPE), where %MPE = [(test-control)/(10-control)] × 100. Groups of eight to twelve animals were used for each dose and for each treatment. The mice were tested 5 min after s.c. injections of epibatidine analogues for the dose-response evaluation. Eight to twelve mice were treated per dose and a minimum of four doses were performed for dose-response curve determination.
Hot-plate Test
Mice were placed into a 10 cm wide glass cylinder on a hot plate (Thermojust Apparatus) maintained at 55.0 °C. Two control latencies at least 10 min apart were determined for each mouse. The normal latency (reaction time) was 8 to 12 sec. Antinociceptive response was calculated as percent maximum possible effect (% MPE), where %MPE = [(test-control)/40-control) × 100]. The reaction time was scored when the animal jumped or licked its paws. Eight mice per dose were injected s.c. with epibatidine analogues and tested 5 min thereafter in order to establish a dose-response curve.
Locomotor Activity
Mice were placed into individual Omnitech photocell activity cages (28 × 16.5 cm) 5 min after s.c. administration of either 0.9% saline or epibatidine analogues. Interruptions of the photocell beams (two banks of eight cells each) were then recorded for the next 10 min. Data were expressed as number of photocell interruptions.
Body Temperature
Rectal temperature was measured by a thermistor probe (inserted 24 mm) and digital thermometer (Yellow Springs Instrument Co., Yellow Springs, OH). Readings were taken just before and 30 min at different times after the s.c. injection of either saline or epibatidine analogues. The difference in rectal temperature before and after treatment was calculated for each mouse. The ambient temperature of the laboratory varied from 21–24 °C from day to day.
Scheme 4.

Acknowledgment
This research was supported by the National Institute on Drug Abuse Grant DA12001.
Footnotes
Dedicated to the late Dr. John W. Daly of NIDDK, NIH, Bethesda, Maryland for his pioneering work on bioactive natural products.
References and Notes
- 1.Ezzati M, Lopez AD. Lancet. 2003;362:847–852. doi: 10.1016/S0140-6736(03)14338-3. [DOI] [PubMed] [Google Scholar]
- 2.Henningfield JE, Fant RV, Buchhalter AR, Stitzer ML. CA Cancer J. Clin. 2005;55:281–299. doi: 10.3322/canjclin.55.5.281. [DOI] [PubMed] [Google Scholar]
- 3.Keating GM, Siddiqui MAA. CNS Drugs. 2006;20:945–960. doi: 10.2165/00023210-200620110-00007. [DOI] [PubMed] [Google Scholar]
- 4.Niaura R, Jones C, Kirkpatrick P. Nat. Rev. Drug Discov. 2006;5:537–538. doi: 10.1038/nrd2088. [DOI] [PubMed] [Google Scholar]
- 5.Spande TF, Garraffo HM, Edwards MW, Yeh HJC, Pannell L, Daly JW. J. Am. Chem. Soc. 1992;114:3475–3478. [Google Scholar]
- 6.Molina PE, Ding YS, Carroll FI, Liang F, Volkow ND, Pappas N, Kuhar M, Abumrad N, Gatley SJ, Fowler JS. Nucl. Med. Biol. 1997;24:743–747. doi: 10.1016/s0969-8051(97)00120-0. [DOI] [PubMed] [Google Scholar]
- 7.Rao TS, Correa LD, Reid RT, Lloyd GK. Neuropharmacology. 1996;35:393–405. doi: 10.1016/0028-3908(96)00013-5. [DOI] [PubMed] [Google Scholar]
- 8.Bonhaus DW, Bley KR, Broka CA, Fontana DJ, Leung E, Lewis R, Shieh A, Wong EH. J. Pharmacol. Exp. Ther. 1995;272:1199–1203. [PubMed] [Google Scholar]
- 9.Sullivan JP, Decker MW, Brioni JD, Donnelly-Roberts D, Anderson DJ, Bannon AW, Kang CH, Adams P, Piattoni-Kaplan M, Buckley MJ, Gopalakrishnan M, Williams M, Arneric SP. J. Pharmacol. Exp. Ther. 1994;271:624–631. [PubMed] [Google Scholar]
- 10.Lloyd GK, Williams M. J. Pharmacol. Exp. Ther. 2000;292:461–467. [PubMed] [Google Scholar]
- 11.Carroll FI. Bioorg. Med. Chem. Lett. 2004;14:1889–1896. doi: 10.1016/j.bmcl.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 12.Carroll FI, Brieaddy LE, Navarro HA, Damaj MI, Martin BR. J. Med. Chem. 2005;48:7491–7495. doi: 10.1021/jm058243v. [DOI] [PubMed] [Google Scholar]
- 13.Carroll FI, Lee JR, Navarro HA, Brieaddy LE, Abraham P, Damaj MI, Martin BR. J. Med. Chem. 2001;44:4039–4041. doi: 10.1021/jm015561v. [DOI] [PubMed] [Google Scholar]
- 14.Carroll FI, Lee JR, Navarro HA, Ma W, Brieaddy LE, Abraham P, Damaj MI, Martin BR. J. Med. Chem. 2002;45:4755–4761. doi: 10.1021/jm0202268. [DOI] [PubMed] [Google Scholar]
- 15.Carroll FI, Liang F, Navarro HA, Brieaddy LE, Abraham P, Damaj MI, Martin BR. J. Med. Chem. 2001;44:2229–2237. doi: 10.1021/jm0100178. [DOI] [PubMed] [Google Scholar]
- 16.Carroll FI, Ma W, Yokota Y, Lee JR, Brieaddy LE, Navarro HA, Damaj MI, Martin BR. J. Med. Chem. 2005;48:1221–1228. doi: 10.1021/jm040160b. [DOI] [PubMed] [Google Scholar]
- 17.Carroll FI, Ware R, Brieaddy LE, Navarro HA, Damaj MI, Martin BR. J. Med. Chem. 2004;47:4588–4594. doi: 10.1021/jm040078g. [DOI] [PubMed] [Google Scholar]
- 18.Abdrakhmanova G, Damaj MI, Carroll FI, Martin BR. Mol. Pharmacol. 2006;69:1945–1952. doi: 10.1124/mol.105.021782. [DOI] [PubMed] [Google Scholar]
- 19.Carroll FI, Yokota Y, Ma W, Lee JR, Brieaddy LE, Burgess JP, Navarro HA, Damaj MI, Martin BR. Bioorg. Med. Chem. 2008;16:746–754. doi: 10.1016/j.bmc.2007.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Navarro HA, Xu H, Zhong D, Abraham P, Carroll FI. Synapse. 2002;44:117–123. doi: 10.1002/syn.10062. [DOI] [PubMed] [Google Scholar]
- 21.D'Amour FE, Smith DL. J. Pharmacol. Exp. Ther. 1941;72:74–79. [Google Scholar]