

Summary
Mitochondrial DNA polymerase gamma (Pol γ) is the sole polymerase responsible for replication of the mitochondrial genome. The study of human Pol γ is of key importance to clinically relevant issues such as nucleoside analog toxicity and mitochondrial disorders such as progressive external ophthalmoplegia. The development of a recombinant form of the human Pol γ holoenzyme provided an essential tool in understanding the mechanism of these clinically relevant phenomena using kinetic methodologies. This review will provide a brief history on the discovery and characterization of human mitochondrial DNA polymerase γ, focusing on kinetic analyses of the polymerase and mechanistic data illustrating structure-function relationships to explain drug toxicity and mitochondrial disease.
Keywords: DNA polymerase gamma, mitochondrial genome, nucleoside analog toxicity, progressive external ophthalmoplegia, pre-steady-state kinetics
1. Introduction
The presence of mitochondria in eukaryotic cells represents an important step in evolution from prokaryotes, as it provided increased energy output by utilizing oxidative phosphorylation. The origins of mitochondria are proposed to have occurred after envelopment of a prokaryotic cell that adapted to serve as a “powerhouse” organelle specializing in ATP production, a hypothesis referred to as the endosymbiotic theory [1]. The mitochondrial dual membrane, replication analogous to binary fission within the eukaryotic cell, and circular mitochondrial genome akin to a prokaryotic plasmid are several points of evidence that provide rationale for this theory. Replication of mitochondrial DNA (mtDNA) is performed by a nuclear encoded protein, mitochondrial DNA polymerase γ (Pol γ), the sole replicative polymerase of the mitochondrial genome. This review will focus on the structure and function of Pol γ, as well as its clinical relevance in fields wide ranging from toxicity of antiviral nucleoside analogs to genetic mitochondrial disorders.
The presence of a mitochondrial DNA polymerase was first suggested in the late 1960's, with the discovery of a polymerase in mitochondrial fractions that exhibited distinct characteristics from known mammalian polymerases [2, 3]. Several years later, a novel human polymerase was identified in HeLa cells that could utilize DNA/RNA primer-templates, [4] eventually found to be the mitochondrial polymerase [5]. Pol γ was characterized by its ability to utilize synthetic DNA/RNA primer-templates for dNTP incorporation [4, 6, 7], sensitivity to dideoxynucleotides [8-10], inhibition by N-ethylmalemide (NEM) [7] and resistance to aphidicolin [11]. Comparison of the amino acid sequence demonstrated high sequence homology with family A polymerases including T7 DNA polymerase and DNA Pol I [12, 13]. In addition to polymerization, Pol γ also catalyzes additional enzymatic activities attributed to DNA repair. A 3′-5′ exonuclease proofreading activity was identified originally in Pol γ purified from chick embryos [14] and further characterized in Drosophila [15]. Pol γ also demonstrated the ability to perform base excision repair through its intrinsic 5′-deoxyribonucleic phosphate lyase activity [16, 17].
Replication of the mitochondrial genome has been extensively studied and debated. This 16.5 kb circular DNA consists of a heavy chain and light chain and contains 13 genes which encode proteins essential for oxidative phosphorylation, as well as the 22 tRNAs and 2 rRNAs responsible for intramitochondrial protein synthesis. Similar to nuclear DNA, the mitochondrial genome exists in complex with proteins responsible for transcription and DNA maintenance in structures called nucleoids (recently reviewed in [18]). Two models have been suggested for replication of the mitochondrial genome, the strand-displacement theory and the strand-coupled theory (Figure 1). The strand displacement theory suggests that replication is one directional and continuous, not requiring the processing of Okazaki fragments on the displaced strand [19, 20]. Copying of the mitochondrial genome begins at the origin of heavy chain replication in the non-coding D-loop region of the mitochondrial genome, displacing the light chain until progressing 2/3 of the way around the circular DNA. The synthesis of the light chain then begins after the formation of a stem-loop structure of the displaced heavy chain, which signifies the origin of light chain replication [20]. The strand-coupled model conversely suggests that this process occurs simultaneously and bi-directionally from multiple sites of initiation in a zone near the origin of heavy chain replication [21, 22]. Interestingly, it has also been shown that there is a high prevalence of ribonucleotides present in the lagging strand during mtDNA replication [23]. This has lead to an alternative view of the strand-displacement theory, named RITOLS (RNA incorporated throughout the lagging strand) replication, in which large patches of RNA act to protect the displaced strand during one directional replication [24, 25]. Subsequent maturation steps are then required to leave behind short RNA templates that can be used as primers to complete replication of the lagging strand, which may explain the lag between synthesis of the heavy and light chains of the mitochondrial genome [24, 25].
Figure 1. Proposed models of mitochondrial genome replication.
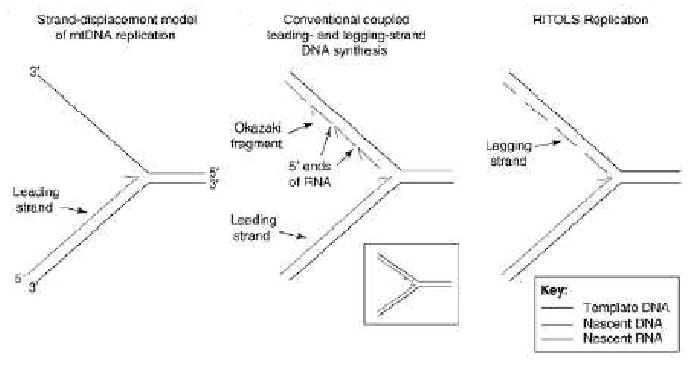
Schematic representation of strand displacement, strand-coupled and RITOLS replication of the mitochondrial genome. (Reprinted from Trends in Biochemical Sciences, Vol 34 / issue 7, Ian J. Holt, Mitochondrial DNA replication and repair: all a flap, Pages 358-365, Copyright 2009, with permission from Elsevier)
2. Kinetic characterization and analysis of human mitochondrial DNA Pol γ
2.1 Initial Characterization and Cloning of the Catalytic Subunit
While human Pol γ had been successfully purified from HeLa cells [26], the low yield of protein made extensive biochemical characterization difficult. Therefore, cloning and expression of a recombinant version was of the utmost importance for extensive biochemical analyses of Pol γ. The 140 kDa catalytic or large subunit of human Pol γ was expressed and purified successfully utilizing a baculoviral expression system to produce a truncated (mature) version of the protein lacking the mitochondrial targeting sequence [27, 28]. Initial kinetic analyses utilizing pre-steady-state kinetics demonstrated that the catalytic subunit alone was somewhat inefficient, with relatively weak binding affinity to DNA (39 nM) and slow maximum rate of polymerization (3.5 s-1). Determination of processivity yielded a value of ∼ 100 nucleotides [27], similar to results establishing that Pol γ incorporated ∼ 50 nucleotides prior to dissociation from the primer-template [28]. Therefore, it was apparent that the catalytic subunit alone was insufficient for processive replication of the mitochondrial genome, as this process would take several hours to complete and require multiple binding events. As previous reports demonstrated the co-purification of a second smaller molecular weight protein with human [26] and Drosophila Pol γ [29], it was hypothesized that this smaller protein could function as an processivity factor acting in analogous fashion to the interaction between T7 DNA polymerase and thioredoxin [30].
2.2 Characterization of Pol γ Holoenzyme: Role of the Accessory Subunit
Later publications demonstrated that this in fact was the case, in experiments showing that recombinant Xenopus laevis [31], mouse [32] and human polymerase γ accessory subunits [32-34] could significantly enhance processivity of the catalytic subunit of human Pol γ. Processivity was found to increase from 50 to hundreds of nucleotides for the catalytic subunit alone [27, 28] to up to thousands of nucleotides after association with stoichiometric amounts of the accessory subunit [33, 34]. Kinetic analysis demonstrated that the accessory and catalytic subunit bound with a Kd of 35 nM and that the enhancement of processivity provided by this interaction was not linked to a significant decrease in the dissociation rate of the holoenzyme from primer/template [34]. Although koff is largely not affected (from 0.03 s-1 to 0.02 s-1), the accessory subunit provided a 3.5-fold increase in DNA binding affinity. The formation of the holoenzyme also results in ∼ 5-fold enhancement of kpol (8.7 ± 1.1 s-1 to 45 ± 1.0 s-1) and ∼ 6-fold decreased Kd (4.7 ± 2.0 μM to 0.78 ± 0.065 μM) for dATP incorporation compared to the catalytic subunit alone. This established that the increase in kpol is the largest determinant of the increase in processivity, defined as kpol/koff [34]. Furthermore, this indicates that the interaction between subunits is enhancing the rate of the hypothesized rate limiting conformational change in the enzyme that limits processive dNTP incorporation. Preliminary experiments also indicated that the functional complex of human mitochondrial DNA polymerase was a heterodimer of p140 and p55 accessory subunits [33, 34], similar to what had been established for Drosophila Pol γ [35]. Binding of the accessory subunit to the catalytic subunit was also found to provide tolerance to physiological salt concentrations as well as protection from NEM inhibition [33].
Interestingly, the accessory subunit demonstrated high sequence homology with glycyl-tRNA synthetases [31, 36], enzymes responsible for attachment of glycine to its cognate tRNA (reviewed in [37]). This indicated that the accessory subunit may also play a role in primer recognition [31, 36], as the formation of DNA secondary structure is important in the initiation of light chain synthesis [20]. This similarity was confirmed by the crystal structure of the murine Pol γ accessory subunit, illustrating that it was a homodimer [38]. Experiments by Yakubovskaya et al. demonstrated that the human Pol γ holoenzyme was in fact a heterotrimer, of one catalytic subunit bound to the homodimeric accessory subunit [39]. Utilization of mutants of the accessory subunit that prevented dimerization illustrated that a monomer alone bound to the catalytic subunit with similar binding affinity as the dimeric accessory subunit. This indicates that the interactions of the subunits of the heterotrimeric holoenzyme predominantly occurs between one half of the accessory subunit complex and the catalytic subunit [39]. Elucidation of the cryo-EM structure of Pol γ provided further rationale for these findings, demonstrating that one subunit of the accessory subunit complex contacts the catalytic subunit through interaction with the spacer region of the polymerase between the exonuclease and polymerase domains, whereas the other subunit provides little surface interaction [40]. In addition to kinetic experiments demonstrating no significant decrease in the rate of primer-template release upon accessory subunit binding [34], this provides evidence that the accessory subunit complex is likely not acting as a processivity clamp that encircles the primer-template as had previously been suggested [41].
Recently, a crystal structure of the Pol γ heterotrimeric holoenzyme [42] was solved by Lee, et al., providing additional insight into the mechanism of processivity enhancement by the accessory subunit (Figure 2). As had been previously predicted [39, 40], the majority of the interaction between subunits occurs between one monomer of the dimeric accessory subunit and the catalytic subunit. Several unique features were evident in the crystal structure. One such feature is an intrinsic processivity subdomain in the spacer region, thought to impart additional processivity of the catalytic subunit relative to other family A polymerases, due to additional interactions with the primer-template [42]. The accessory interacting determinant (AID) subdomain of the spacer region, nearly 50 Å from the polymerase domain, provides rationale for the mechanism of DNA binding enhancement (Figure 2). This subdomain forms extensive hydrophobic interactions between the L-helix of the catalytic subunit with the proximal accessory subunit. Modeling of the holoenzyme bound to primer-template suggests that this interaction exposes a positively charged tract of residues predicted to interact with the negatively charged sugar phosphate backbone of the primer-template resulting in enhanced DNA binding affinity [42].
Figure 2. Crystal structure of Pol γ holoenzyme.
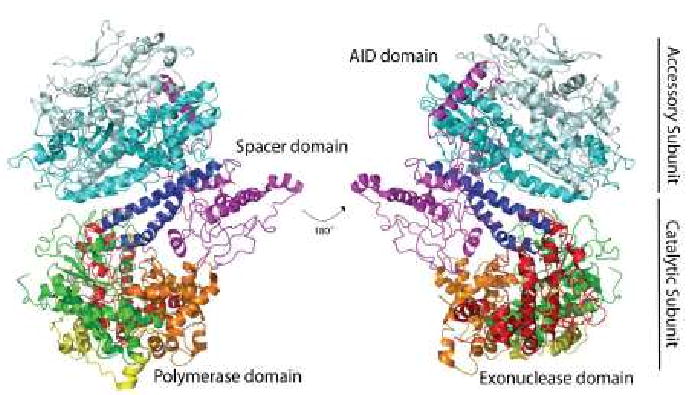
Image depicts the crystal structure of the heterotrimeric Pol γ holoenzyme [42]. Fingers (orange), palm (green) and thumb (blue) subdomains of the canonical right hand organization of the polymerase domain are shown. Additional domains highlighted include the mitochondrial localization sequence (yellow), exonuclease (red) and spacer subdomain (purple). The proximal (cyan) and distal (light cyan) monomers of the dimeric accessory subunit are shown.
These findings however, did not address the mechanism of accessory subunit enhancement of kpol, which provides the largest improvement of processivity [34]. A follow-up study by Lee, et al. delved further into the contribution of each accessory subunit monomer (distal and proximal) to processivity enhancement [43]. By comparing pre-steady state incorporation kinetics of holoenyzmes formed with dimerization deficient accessory subunit mutants, it was established that the proximal monomer alone tightens the binding affinity between Pol γ and the primer-template. This is not surprising, as the crystal structure demonstrates that the AID subdomain predominantly interacts with the proximal monomer (Figure 2) [42]. However, rate enhancement is only achieved in the presence of the dimeric accessory subunit, indicating a role for the distal monomer of the accessory subunit complex. The authors speculate that perhaps this effect could be mediated through proper positioning of the primer-template that is only accomplished when the catalytic subunit is bound to the dimeric accessory subunit [43].
The accessory subunit has also demonstrated the ability to bind nucleic acids, an uncommon trait of processivity factors [38, 44]. Specifically it had been demonstrated that the accessory subunit preferentially bound dsDNA and that tight binding required interaction from both subunits of the dimer [44]. Deletion of regions important for the DNA binding properties of the accessory subunit did not affect its ability to enhance Pol γ polymerization [38]. This indicated a potential role of DNA binding in a function not directly related to enhancement of processivity. A recent report has illustrated that the nucleic acid binding of the accessory subunit may play a role in mitochondrial DNA maintenance, specifically having a role in organization of mtDNA in nucleoids [45]. The authors showed that altering expression levels of the accessory subunit using RNAi modulated the number of genomes per nucleoid in cultured cells. The Pol γ accessory subunit was found to preferentially bind plasmids containing a D-loop region significantly more tightly than dsDNA, indicating that it is likely a physiologically significant interaction with the mitochondrial genome that may mitigate these effects. [45].
2.3 Fidelity and Exonuclease Activity of Pol γ
Fidelity of a polymerase is determined by the ability of the polymerase to actively discriminate against the incorporation of improper base pairs at the active site of the enzyme. If an error does occur, polymerases having a 3′-5′ exonuclease activity are able to correct these errors providing an increase in replication fidelity. In the case of Pol γ, mispairs are highly discriminated against through impairing both rate of incorporation and the binding affinity of mismatched bases [46, 47]. If a misincorporation does occur, formation of proper base pair incorporation after a mismatch is unfavorable, as the rate and binding affinity decrease by 80- and 440-fold respectively, causing the polymerase to stall [46]. This allows for shuttling of the primer-template to the exonuclease active site, where the mispair can be excised [48]. This has been shown to occur without dissociation of the primer-template [49], allowing processive DNA synthesis to continue immediately following exonuclease proofreading. Alternatively, dissociation of the mismatched primer-template may also occur, as koff is increased compared to properly base paired primer-templates. The exonuclease rate also increases significantly when presented with a buried mismatch, indicating an ability to repair buried mispairs as well [49]. The kinetic scheme for dNTP incorporation by Pol γ as determined by pre-steady-state kinetics is shown in Figure 3. A later study provided a full kinetic analysis of all possible mispairs and provided a fidelity for Pol γ of 1 error in every 440,000 nucleotides incorporated [47]. The fidelity of the polymerase is increased by 4- 200-fold due to the exonuclease activity [49], which would imply an overall fidelity of 1 error in 1.8 million to 36 million bases incorporated [47].
Figure 3. Kinetic Scheme for Pol γ Polymerization.
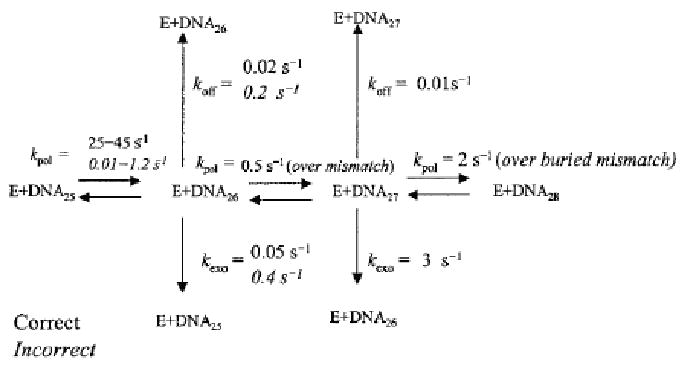
Schematic demonstrating rate constants for dNTP incorporation and removal by Pol γ as determined by pre-steady state kinetic analysis. Figure adapted from [49].
2.4 Reverse Transcription by Pol γ
As mentioned previously, one of the earliest characteristics of Pol γ was the ability to utilize synthetic DNA/RNA primer-templates. Initially, the biological significance of this was not understood until it was demonstrated that ribonucleotides are present in detectable quantities throughout the mitochondrial genome [23]. Our lab demonstrated that Pol γ can in fact perform reverse transcription with similar efficiency as HIV-1 RT, with comparable kpol values [50]. Furthermore, Pol γ can perform exonuclease cleavage of mismatched dNMPs opposite a RNA template [50]. The reverse transcriptase activity of Pol γ was later found to be not processive, incorporating one dNTP and dissociating from the primer/template [51]. This indicates that the physiological significance of this phenomenon may be linked to single ribonucelotides present throughout the template strand. The weaker interaction between Pol γ and DNA/RNA primer-templates also serves to explain the increased steady-state rate of incorporation versus DNA/DNA primer-templates, as the steady-state rate is governed by the rate of primer-template release [51].
3. Importance of Pol γ in Nucleoside Analog Therapy
3.1 Nucleoside Analogs and Molecular Basis for Potent ddNTP Inhibition
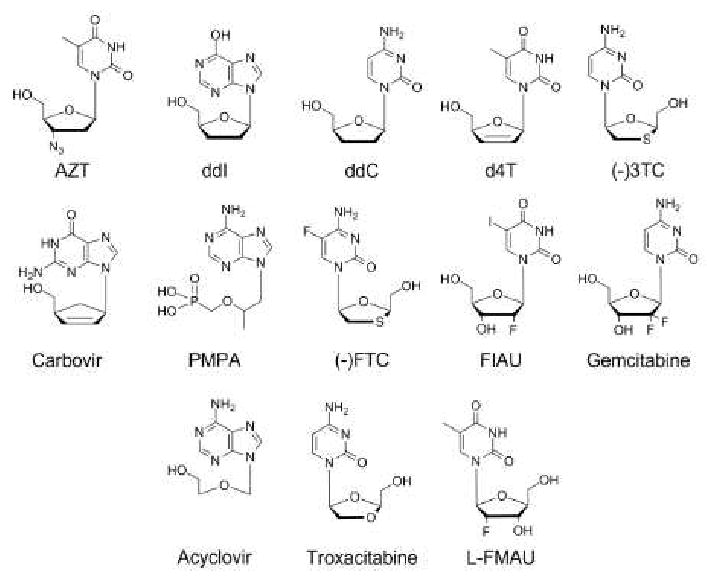
Nucleoside analogs have a long history of clinical use in the treatment of cancer and viral infection. Nucleoside RT inhibitors (NRTIs) represent the first FDA approved class of therapeutics for treatment of HIV infection (Figure 4). As substrate analogs, NRTIs are competitive inhibitors administered as prodrugs that must be transported into the cell and phosphorylated to the metabolically active triphosphate in order to exert their therapeutic effect (reviewed in [52]). All currently approved NRTIs lack a 3′-hydroxyl group required to perform nucleophilic attack on an incoming dNTP, therefore AZT and other NRTIs elicit their antiviral effect by halting primer extension [53]. These NRTIs can also serve as substrates for inhibition of host cell polymerases, leading to host cell toxicity.
Figure 4. Structures of selected clinically relevant nucleoside analogs.
The sensitivity of Pol γ to dideoxynucleoside therapies might have been predicted, as one of the initial defining characteristics of Pol γ was susceptibility to dideoxynucleotide inhibition [8-10]. Early experiments linked inhibition of Pol γ to side effects of NRTIs, illustrating high susceptibility to inhibition by the triphosphate form of NRTI, ddC [54] and that treating cells with ddC provoked a reversible decrease in mtDNA levels [55]. Later clinical findings also suggested mitochondrial toxicity in response to treatment with various NRTIs (reviewed in [56]). The most common side effects of AZT treatment include bone marrow suppression [57] and myopathies [58] with concomitant decreases in mtDNA [59], and cardiomyopathy [60]. ddC and d4T treatment are associated with peripheral neuropathy [61, 62]. More rare and severe toxicities to NRTI treatment include lactic acidosis and pancreatitis [56]. The similarities of these side effects to genetic mitochondrial disorders lead to the hypothesis that Pol γ inhibition was the primary source of NRTI mitochondrial toxicity [63-65].
The molecular basis of this sensitivity was explained by experiments demonstrating that one single amino acid acts as a “gatekeeper” for ddNTP susceptibility in family A polymerases [66, 67]. DNA Pol I, which demonstrates high selectivity against ddNTP incorporation, has a phenylalanine residue on the highly conserved O-helix, opposite the 3′-OH of the bound dNTP. Other family A polymerases that efficiently incorporate ddNTPs, such as T7 DNA polymerase, have a tyrosine at the analogous position on the O-helix. Mutation of this residue to tyrosine in Pol I was demonstrated to decrease ddNTP discrimination thousands of fold [66, 67]. It was hypothesized that the phenolic hydroxyl group of the tyrosine residue could substitute for the missing 3′-OH of the bound ddNTP, allowing efficient incorporation. Molecular modeling of the Pol γ active site illustrated that the analogous residue, tyrosine 951 was responsible for dideoxynucleotide sensitivity [28, 68] (Figure 5), which ultimately results in susceptibility to NRTI inhibition and subsequent mitochondrial toxicity. It is important to recognize toxicity is a multifactorial phenomenon which encompasses metabolic activation by cellular kinases and transport into the mitochondria in addition to inhibition of Pol γ.
Figure 5. Molecular basis for Pol γ sensitivity to ddNTP inhibition.
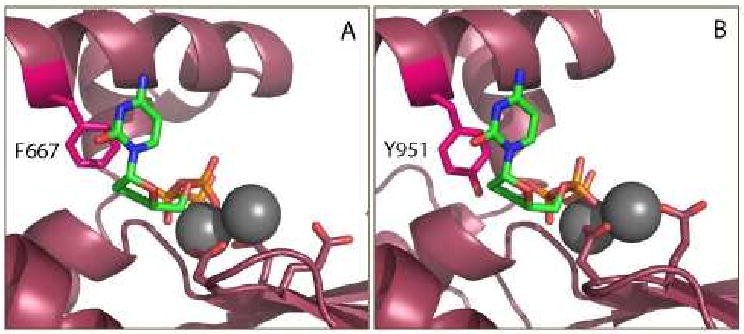
A) Pol I is resistant to dideoxynucleotide inhibition, which has been linked to the presence of a phenyalanine at position 667 on the O-helix, shown in fuschia. Image created from the ternary complex of the catalytic subunit ternary complex of Klentaq [120]. B) Molecular model of the Pol γ active site [112] demonstrates the phenolic hydroxyl group of Y951 mimicking the 3′-OH of a bound dNTP, allowing efficient incorporation of ddNTPs. ddCTP is shown bound to the Pol I and Pol γ active sites in green, with coordinated Mg2+ ions (gray), and catalytic residues shown in stick form.
3.2 Toxicity of FIAU Marks A Call for Understanding NRTI Toxicity
The failure of hepatitis B inhibitor FIAU illustrated the importance of understanding the mechanism of mitochondrial toxicity attributed to nucleoside analog therapy (Figure 4). Tragically, 5 patients in a phase II clinical trial died and 5 others required liver transplants after exhibiting hallmark symptoms of mitochondrial dysfunction. These side effects included liver failure, lactic acidosis, skeletal- and cardiomyopathies, neuropathy, and pancreatitis [69, 70]. The effect of FIAU on hepatic cell lines was examined after the clinical trial ended, illustrating induced lactic acid production and enlarged mitochondria with lipid droplet formation [71, 72]. Although FIAUTP was demonstrated to be a substrate and potent competitive inhibitor of Pol γ [72-74], the presence of FIAUMP in mtDNA [71, 75] suggested that it serving as a substrate for Pol γ, but not acting as a chain terminator. Studies also demonstrated that FIAU treatment was not associated with a decrease in mtDNA in response to FIAU treatment [71, 76], indicating a unique mechanism of toxicity compared to dideoxynucleotides. After the failure of FIAU, it became clear that considerations had to be made to ensure that new nucleoside analogs were potent against virus, but also did not inhibit the mitochondrial polymerase.
3.3 Mechanistic Studies of Pol γ Inhibition by Antiviral Nucleoside Analogs
Whereas the goal of designing antiviral nucleoside analogs is to maximize incorporation efficiency by the target polymerase, it became clear that the potency of these compounds must be weighed against their inhibition of the mitochondrial polymerase. Studies by our lab and others have illustrated that inhibition of Pol γ is correlated to the clinical toxicity of nucleoside analogs [77-79]. Inhibition of Pol γ can be assayed by determining the kinetic parameters for incorporation of the NRTI, as well as the exonuclease removal of the NRTI from the 3′-termini of chain-terminated primers. Pre-steady-state kinetic studies from our laboratory demonstrated the mechanism of mitochondrial toxicity of cytidine analogs with varying degrees of mitochondrial toxicity in vivo [77]. The high toxicity of ddC was explained by kinetic data indicating that ddCTP was incorporated only 3-fold less efficiently than the natural substrate by Pol γ with high binding affinity (41 nM versus 1.1 μM for dCTP), while the rate of exonuclease removal was nearly undetectable (< 0.0001 s-1). In contrast unnatural L-enantiomer (-)3TC, which demonstrates low clinical toxicity, is incorporated ∼ 2900-fold less efficiently than the natural substrate, caused by a 350-fold drop in incorporation rate and an 8-fold decrease in binding affinity. The rate of exonuclease removal of (-)3TC however, was comparable to the removal rate of the natural substrate [77]. A comprehensive study of FDA approved NRTIs at the time provided further mechanistic information combining incorporation and removal data, yielding a hierarchy of Pol γ inhibition that mirrored mitochondrial toxicity in the clinic as follows (ddC > ddA > d4T ≫ (-)3TC > PMPApp > AZT > CBV) [79, 80]. Pre-steady-state kinetic analysis of the most recently approved NRTI, (-) FTC also demonstrated a further decrease in Pol γ incorporation efficiency compared to (-)3TC [81]. Mechanistic information regarding the severe toxicity associated with FIAU also was examined, which is unique with respect to currently approved anti-HIV nucleoside analogs as it is not a chain terminator. Incorporation of the next correct nucleotide after FIAUMP incorporation was several fold faster than the exonuclease rate, indicating FIAU gets internalized into mtDNA [79]. This internalization allows FIAU to act as a mutagen, which explains its delayed and severe toxicity that is not reversible upon cessation of treatment.
These studies have shown that several design aspects of newer generation NRTIs have dramatically decreased Pol γ inhibition, including the use of unnatural L-nucleosides ((-)3TC and (-)FTC), acyclic nucleosides (tenofovir) and carbocyclic nucleosides (abacavir). The use of L-nucleosides such as (-)3TC and (-)FTC allows for potent inhibition of RT while serving as poor substrates for the mitochondrial polymerase imparting low mitochondrial toxicity [77, 81-83]. Fluorination of the C-5 position of cytosine base in (-)FTC results in further decreased incorporation by Pol γ compared to (-)3TC [81]. Acyclic nucleoside analog PMPA and carbocyclic analog carbovir, the active metabolites of tenofovir and abacavir respectively, present among the highest therapeutic indicies by also minimizing incorporation by Pol γ [79]. Utilization of pre-steady-state kinetics in the analysis of nucleoside analog therapeutics represents a powerful tool in understanding and predicting mitochondrial toxicity. Recent publications have utilized a similar approach to examine inhibition of Pol γ by anti-cancer agent gemcitabine [84] and herpes simplex virus drug acyclovir (Figure 4) [85].
Although use of L-nucleosides typically provides an improved toxicity profile compared to their D-enantiomers, it should be noted there are exceptions to this generalization. One notable example is highly toxic L-nucleoside analog troxacitabine (L-dioxolane cytosine)[86, 87], which is in phase II clinical trials as an anti-cancer agent. Furthermore, phase III clinical trials of clevudine (L-FMAU) for the treatment of HBV infection were recently halted due to unexpected mitochondrial toxicity [88-90], which was undocumented in preclinical studies (Figure 4) [91, 92]. Biochemical analysis showed L-FMAUTP was not a substrate of Pol γ or other host polymerases [93], suggesting the possibility of an alternative mechanism of mitochondrial toxicity. Further study is required to understand the mechanism of clevudine toxicity.
4. Predisposition to NRTI Toxicity
4.1 Identification and characterization of R964C mutation
Although significant strides have been made in characterizing the toxicity of NRTIs, one question that remains unclear was why certain individuals tolerate treatment with NRTIs, whereas others encounter issues with mitochondrial toxicity. Clearly, this is a multi-factorial problem, but one possible answer could be found in a genetically inherited predisposition to NRTI toxicity caused by a mutation or polymorphism in the Pol γ coding sequence. In 2007, Yamanaka, et al. described the identification and initial characterization of the first such proposed mutation of Pol γ linked to stavudine (d4T) mediated mitochondrial toxicity [94]. In this report, an HIV infected individual undergoing stavudine treatment, presenting to the clinic with lactic acidosis, was found to harbor a homozygous R964C mutation in Pol γ. Steady-state inhibition assays using the exonuclease proficient catalytic subunit alone suggested little change in Ki values for inhibition by ddTTP, AZTTP, or d4TTP between mutant and wild type Pol γ. However, a decrease in specific enzyme activity, as well as steady-state dTTP incorporation rate, was noted [94]. This preliminary kinetic data, coupled with data from experiments demonstrating a significant decrease in mtDNA levels in hetero- and homozygous R964C cultured lymphoblast cell lines in response to d4T treatment, lead the authors to speculate that the mutation impaired the normal enzymatic activity of the polymerase. Further challenge with d4T treatment then was hypothesized to overcome a threshold for mitochondrial toxicity, leading to lactic acidosis [94].
To address this question at a molecular level, we recently performed a pre-steady-state kinetic analysis to assess the functional impact of this mutation and to determine its effect on natural dNTP and d4TTP binding and incorporation. [95]. Our experiments determined that the R964C mutation imparted a 25% decrease in overall dTTP incorporation efficiency relative to wild type. This was caused by a decrease in the maximum rate of polymerization, with no significant change in nucleotide binding affinity. The mutation also imparted a 2-fold increased binding affinity for d4TTP by the R964C mutant versus wild type with similar incorporation rates, resulting in a 2-fold increased incorporation efficiency. Therefore, the mutant enzyme was 3-fold less able to discriminate between the natural substrate and d4TTP, implying a mechanism of increased stavudine-induced mitochondrial toxicity [95]. Identification of a Pol γ mutant that demonstrates greater susceptibility to NRTI inhibition represents an important step in utilizing pharmacogenomics to prevent antiviral drug toxicity.
4.2 Future Possibilities in Pharmacogenomics for NRTI therapy
This goal of pharmacogenomics in NRTI therapy is limited by the identification of additional mutations attributable to the side effects and the prevalence of such mutations. A recent report predicted a link between the E1143G polymorphism, one of seventeen documented polymorphisms of Pol γ, and an increased prevalence of lipodystrophy in response to d4T treatment [96]. E1143G is found combination with the W748S mutation in patients with ataxia-neuropathy syndrome and progressive external ophthalmoplegia patients (described in greater detail in the following section) [97, 98]. Biochemical analyses of the single mutants and the double mutant were performed illustrating the effect of each mutation on Pol γ activity. The E1143G polymorphism demonstrated increased steady-state catalytic activity, DNA binding affinity and thermostability relative to the wild-type enzyme, with no apparent change in fidelity [99]. This improvement in polymerase activity served to counteract the decrease in DNA binding and catalytic activity of the W748S mutant, as the double mutant exhibited increased DNA binding affinity and polymerase activity compared to the W748 mutant alone, albeit with decreased thermostability [99]. Therefore, it is currently unclear how the E1143G mutation alone may be affecting d4T mediated toxicity. Future experiments will focus on understanding this phenomenon, as well as serve as a call for more genetic analysis of patients demonstrating lactic acidosis and mitochondrial toxicity in response to NRTI treatment.
5. Mutation of the Mitochondrial DNA Polymerase and Genetic Disorders
5.1 Linking Pol γ Mutation to Genetic Disease
Mutation of Pol γ has been linked to several different genetic mitochondrial disorders. These include progressive external ophthalmoplegia (PEO), Alpers syndrome, parkinsonism, male infertility, SANDO (sensory ataxic neuropathy, dysarthria, and ophthalmoparesis) among others (reviewed by Chan and Copeland [100]). As the majority of mutations to Pol γ are associated with PEO, this review will focus on the molecular mechanisms of several mutations of Pol γ linked to this disease. PEO is a rare autosomal disorder, characterized by ophthalmoplegia, general muscle weakness and wasting, peripheral neuropathy, and ataxia [101-104]. A number of genetic loci have been linked to PEO, including ANT1, an adenine nucleotide translocator [105]; Twinkle, the mitochondrial helicase [106]; and mitochondrial DNA polymerase γ catalytic (POLG1) [107] and its accessory subunit (POLG2) [108]. All of these genes encode proteins with critical functions for stability of the mitochondrial genome. Age of onset varies from between the ages of 18-40 depending on the severity of the disease and initially presents with progressive weakening of the eye muscles leading to ophthalmoparesis. Muscle biopsies from PEO patients demonstrate hallmarks of mitochondrial dysfunction, including electron dense “parking lot inclusions” in the mitochondria and concentric cristae. These effects have been linked to depletion of mtDNA, as well as deletions and mutations in the mitochondrial genome [109].
5.2 Molecular Mechanisms of Mitochondrial Toxicity Linked to Pol γ Mutation
The most prominently studied and identified mutations involved in these disorders are linked to Pol γ, specifically adPEO. Mutations span the entire coding region of the mitochondrial DNA polymerase (http://tools.niehs.nih.gov/polg/), indicating the possibility of several mechanisms of inhibiting the natural function of the polymerase. Effects of autosomal dominant mutations near the polymerase active site of Pol γ have been the most studied, specifically focusing on the Y955C mutation, the first mutation identified linking PEO to mutation of the mitochondrial polymerase [107]. Subsequently Y955C was illustrated to be one of the most severe mutations with rapid onset of disease symptoms. Y955 is an invariant member of the highly conserved O-helix of family A polymerases (Figure 6). The analogous residue in T7 DNA polymerase, Y530 hydrogen bonds with steric gate residue E408 to prevent incorporation of ribonucleotides. Along with residue Y526 (analogous to Pol γ Y951), these residues form a hydrophobic pocket for binding of the incoming dNTP [110]. The effect of this mutation on polymerase function was first characterized in 2002 [111], demonstrating severe impairment of nucleotide binding affinity, resulting in compromised polymerization efficiency. Furthermore, it was demonstrated that the mutation decreased the fidelity of the polymerase rendering a mutator phenotype that may contribute to the disease severity [111].
Figure 6. Molecular Model of Pol γ Active Site.
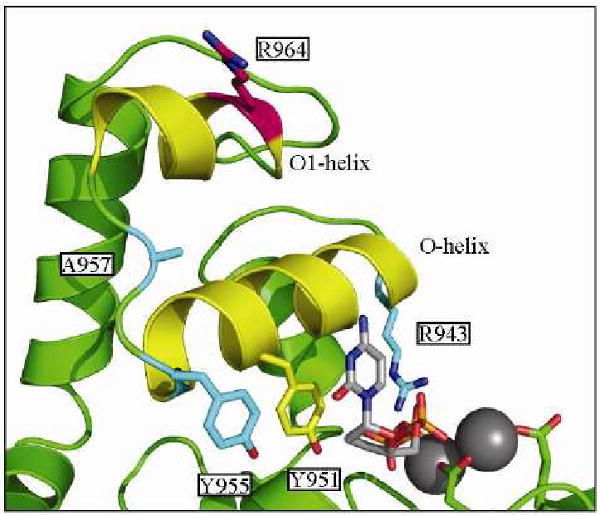
Several residues mutated in PEO and discussed in the text are shown in cyan. NRTI susceptibility is linked to mutation of R964, shown in hot pink. ddCTP is shown bound to the active site in grey. Molecular model was created using the ternary structure of T7 polymerase and was previously described [112].
This study was followed by a more comprehensive report that examined several mutations associated with adPEO localized to the active site of the enzyme, G923D, R943H, Y955C, and A957S (Figure 6) [112]. The authors of this study demonstrated structure-activity relationships between these mutants using biochemical assays and molecular modeling to correlate their results with disease phenotype [112]. As predicted by the severity of the phenotype, Y955C imparted the largest decrease in steady-state polymerization efficiency (3800-fold relative to wild type), as well as severe decreases in processivity and fidelity. R943 is another highly conserved residue on the O-helix analogous to R518 that hydrogen bonds to the γ-phosphate of the incoming dNTP in the T7 DNA polymerase structure [110]. Mutation to histidine then demonstrates a 475-fold impairment of steady-state incorporation and decreased processivity. This is predicted to be caused by improper orientation of the triphosphate tail of the bound dNTP, impairing catalysis. Mutation of G923 and A957 to D and S respectively were found to decrease steady-state polymerization approximately 5-fold with minimal effects on processivity and fidelity, in line with their mild phenotype in heterozygous patients. Further analysis of these mutants forms of Pol γ were analyzed utilizing a yeast model system, which has commonly been used as a powerful tool in the understanding of mitochondrial disorders (reviewed by Shadel [113]). Stuart et al. determined the viability of yeast as a model system for understanding the cellular mechanisms of PEO caused by mutation of Pol γ, yielding results that parallel the severity of the clinical phenotype of several mutations [114].
The A467T mutation is the most common Pol γ mutation and is associated with Alper's syndrome, and arPEO. Initially thought to reside in the spacer region, A467 is part of the conserved γ1 segment conserved amongst mtDNA polymerases across species. Deletion or mutation of several residues of this conserved segment in Pol γ from Drosophila demonstrated several catalytic defects, including decreased polymerase activity and a weakened interaction between the catalytic and accessory subunits [115]. Initial experiments demonstrated that the mutation impaired polymerization catalyzed by the catalytic subunit, with only 4% of steady-state activity [116]. The authors also demonstrated that several biochemical features provided by binding of the accessory subunit were lost, namely the increase in processivity and protection from NEM inactivation associated with formation of the holoenzyme. Impairment of the binding of catalytic and accessory subunits was further verified using immunopreciptation experiments, demonstrating a weak interaction between the subunits [116]. Although the decrease in activity caused by the A467T was also seen in a later publication, it was also demonstrated that this effect could be ameliorated by addition of the accessory subunit [117]. However, a majority of evidence for the impairment of this interaction is further supported by the cryo-EM and crystal structures [40, 42] of the polymerase gamma holoenzyme suggesting that only one monomer of the accessory subunit dimer interacts with the p140 subunit in the region of A467. Recently, the crystal structure indicated that A467 actually is part of the thumb subdomain which is known to interact with the primer-template and is very near the interface of the catalytic subunit and the accessory subunit [42]. Mutation to threonine could impair enzymatic function by disrupting hydrophobic interactions in this area, altering the orientation of the thumb subdomain, impairing DNA binding and decreasing the binding affinity of the catalytic and accessory subunits. Furthermore, the association of arPEO with mutations to the accessory subunit and experiments denoting impaired interaction between p140 and p55 provides further rationale that impaired interaction between these subunits can lead to mitochondrial dysfunction [108].
6. Conclusions
Research of the mitochondrial polymerase γ has provided tremendous insight into understanding of mitochondrial replication, nucleoside analog toxicity, and mitochondrial disorders. What remains unclear however is exactly how impairment of the mitochondrial polymerase by mutation or nucleoside analogs can yield such, in some cases, drastically different phenotypes. Accumulation of mutations in mtDNA has also been associated with aging as transgenic mice expressing an exonuclease deficient form of Pol γ develop a progeroid phenotype [118]. However, mutations in the exonuclease domain demonstrated to increase the prevalence of mtDNA mutations and deletions are linked to mitochondrial disorder such as PEO. In some cases, the exact same mutation of Pol γ is linked to multiple diseases with different phenotypes. Side effects of NRTIs also vary greatly, although inhibition of Pol γ is considered the main cause for NRTI toxicity. Only recently has a report surfaced demonstrating the appearance of PEO-like symptoms in individuals undergoing NRTI treatment for HIV infection [119]. One patient noted symptoms of ptosis that decreased upon ceasing NRTI treatment, although underlying genetic disease cannot be ruled out as no genetic analysis was performed to assess mutation in the Pol γ gene, or other genetic loci linked to PEO [119].
Although homology modeling allows for accurate predictions of active site residues of Pol γ [112], the mechanism of accessory subunit enhancement of the rate limiting conformational change has yet to be explained by current structural data, calling for a structure with primer-template and dNTP bound. Moreover, comprehensive structure-function studies are essential for delineating mechanisms of mitochondrial disease, as well as the design of safer, less toxic nucleoside analogs. Many mutations of Pol γ associated with mitochondrial disease have yet to be characterized biochemically. We believe that using a pre-steady-state kinetic approach to examine mutant forms of Pol γ will allow more precise assessment of nucleotide incorporation efficiency and may yield important mechanistic data that would contribute to understanding mitochondrial disease.
Acknowledgments
This work was supported by NIH GM49551 to KSA
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lang BF, Gray MW, Burger G. Mitochondrial genome evolution and the origin of eukaryotes. Annu Rev Genet. 1999;33:351–397. doi: 10.1146/annurev.genet.33.1.351. [DOI] [PubMed] [Google Scholar]
- 2.Kalf GF, Ch'ih JJ. Purification and properties of deoxyribonucleic acid polymerase from rat liver mitochondria. J Biol Chem. 1968;243:4904–4916. [PubMed] [Google Scholar]
- 3.Meyer RR, Simpson MV. DNA biosynthesis in mitochondria: partial purification of a distinct DNA polymerase from isolated rat liver mitochondria. Proc Natl Acad Sci U S A. 1968;61:130–137. doi: 10.1073/pnas.61.1.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fridlender B, Fry M, Bolden A, Weissbach A. A new synthetic RNA-dependent DNA polymerase from human tissue culture cells (HeLa-fibroblast-synthetic oligonucleotides-template-purified enzymes) Proc Natl Acad Sci U S A. 1972;69:452–455. doi: 10.1073/pnas.69.2.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bolden A, Noy GP, Weissbach A. DNA polymerase of mitochondria is a gamma-polymerase. J Biol Chem. 1977;252:3351–3356. [PubMed] [Google Scholar]
- 6.Fry M, Weissbach A. The utilization of synthetic ribonucleic acid templates by a new deoxyribonucleic acid polymerase from cultured murine cells. J Biol Chem. 1973;248:2678–2683. [PubMed] [Google Scholar]
- 7.Knopf KW, Yamada M, Weissbach A. HeLa cell DNA polymerase gamma: further purification and properties of the enzyme. Biochemistry. 1976;15:4540–4548. doi: 10.1021/bi00665a032. [DOI] [PubMed] [Google Scholar]
- 8.Edenberg HJ, Anderson S, DePamphilis ML. Involvement of DNA polymerase alpha in simian virus 40 DNA replication. J Biol Chem. 1978;253:3273–3280. [PubMed] [Google Scholar]
- 9.Zimmermann W, Chen SM, Bolden A, Weissbach A. Mitochondrial DNA replication does not involve DNA polymerase alpha. J Biol Chem. 1980;255:11847–11852. [PubMed] [Google Scholar]
- 10.van der Vliet PC, Kwant MM. Role of DNA polymerase gamma in adenovirus DNA replication. Mechanism of inhibition by 2′,3′-dideoxynucleoside 5′-triphosphates. Biochemistry. 1981;20:2628–2632. doi: 10.1021/bi00512a041. [DOI] [PubMed] [Google Scholar]
- 11.Geuskens M, Hardt N, Pedrali-Noy G, Spadari S. An autoradiographic demonstration of nuclear DNA replication by DNA polymerase alpha and of mitochondrial DNA synthesis by DNA polymerase gamma. Nucleic Acids Res. 1981;9:1599–1613. doi: 10.1093/nar/9.7.1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Braithwaite DK, Ito J. Compilation, alignment, and phylogenetic relationships of DNA polymerases. Nucleic Acids Res. 1993;21:787–802. doi: 10.1093/nar/21.4.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Delarue M, Poch O, Tordo N, Moras D, Argos P. An attempt to unify the structure of polymerases. Protein Eng. 1990;3:461–467. doi: 10.1093/protein/3.6.461. [DOI] [PubMed] [Google Scholar]
- 14.Kunkel TA, Soni A. Exonucleolytic proofreading enhances the fidelity of DNA synthesis by chick embryo DNA polymerase-gamma. J Biol Chem. 1988;263:4450–4459. [PubMed] [Google Scholar]
- 15.Olson MW, Kaguni LS. 3′-->5′ exonuclease in Drosophila mitochondrial DNA polymerase. Substrate specificity and functional coordination of nucleotide polymerization and mispair hydrolysis. J Biol Chem. 1992;267:23136–23142. [PubMed] [Google Scholar]
- 16.Longley MJ, Prasad R, Srivastava DK, Wilson SH, Copeland WC. Identification of 5′-deoxyribose phosphate lyase activity in human DNA polymerase gamma and its role in mitochondrial base excision repair in vitro. Proc Natl Acad Sci U S A. 1998;95:12244–12248. doi: 10.1073/pnas.95.21.12244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pinz KG, Bogenhagen DF. Characterization of a catalytically slow AP lyase activity in DNA polymerase gamma and other family A DNA polymerases. J Biol Chem. 2000;275:12509–12514. doi: 10.1074/jbc.275.17.12509. [DOI] [PubMed] [Google Scholar]
- 18.Holt IJ, He J, Mao CC, Boyd-Kirkup JD, Martinsson P, Sembongi H, Reyes A, Spelbrink JN. Mammalian mitochondrial nucleoids: organizing an independently minded genome. Mitochondrion. 2007;7:311–321. doi: 10.1016/j.mito.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 19.Clayton DA. Replication of animal mitochondrial DNA. Cell. 1982;28:693–705. doi: 10.1016/0092-8674(82)90049-6. [DOI] [PubMed] [Google Scholar]
- 20.Shadel GS, Clayton DA. Mitochondrial DNA maintenance in vertebrates. Annu Rev Biochem. 1997;66:409–435. doi: 10.1146/annurev.biochem.66.1.409. [DOI] [PubMed] [Google Scholar]
- 21.Holt IJ, Lorimer HE, Jacobs HT. Coupled leading- and lagging-strand synthesis of mammalian mitochondrial DNA. Cell. 2000;100:515–524. doi: 10.1016/s0092-8674(00)80688-1. [DOI] [PubMed] [Google Scholar]
- 22.Bowmaker M, Yang MY, Yasukawa T, Reyes A, Jacobs HT, Huberman JA, Holt IJ. Mammalian mitochondrial DNA replicates bidirectionally from an initiation zone. J Biol Chem. 2003;278:50961–50969. doi: 10.1074/jbc.M308028200. [DOI] [PubMed] [Google Scholar]
- 23.Yang MY, Bowmaker M, Reyes A, Vergani L, Angeli P, Gringeri E, Jacobs HT, Holt IJ. Biased incorporation of ribonucleotides on the mitochondrial L-strand accounts for apparent strand-asymmetric DNA replication. Cell. 2002;111:495–505. doi: 10.1016/s0092-8674(02)01075-9. [DOI] [PubMed] [Google Scholar]
- 24.Yasukawa T, Reyes A, Cluett TJ, Yang MY, Bowmaker M, Jacobs HT, Holt IJ. Replication of vertebrate mitochondrial DNA entails transient ribonucleotide incorporation throughout the lagging strand. EMBO J. 2006;25:5358–5371. doi: 10.1038/sj.emboj.7601392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Holt IJ. Mitochondrial DNA replication and repair: all a flap. Trends Biochem Sci. 2009;34:358–365. doi: 10.1016/j.tibs.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 26.Gray H, Wong TW. Purification and identification of subunit structure of the human mitochondrial DNA polymerase. J Biol Chem. 1992;267:5835–5841. [PubMed] [Google Scholar]
- 27.Graves SW, Johnson AA, Johnson KA. Expression, purification, and initial kinetic characterization of the large subunit of the human mitochondrial DNA polymerase. Biochemistry. 1998;37:6050–6058. doi: 10.1021/bi972685u. [DOI] [PubMed] [Google Scholar]
- 28.Longley MJ, Ropp PA, Lim SE, Copeland WC. Characterization of the native and recombinant catalytic subunit of human DNA polymerase gamma: identification of residues critical for exonuclease activity and dideoxynucleotide sensitivity. Biochemistry. 1998;37:10529–10539. doi: 10.1021/bi980772w. [DOI] [PubMed] [Google Scholar]
- 29.Wernette CM, Kaguni LS. A mitochondrial DNA polymerase from embryos of Drosophila melanogaster. Purification, subunit structure, and partial characterization. J Biol Chem. 1986;261:14764–14770. [PubMed] [Google Scholar]
- 30.Tabor S, Huber HE, Richardson CC. Escherichia coli thioredoxin confers processivity on the DNA polymerase activity of the gene 5 protein of bacteriophage T7. J Biol Chem. 1987;262:16212–16223. [PubMed] [Google Scholar]
- 31.Carrodeguas JA, Kobayashi R, Lim SE, Copeland WC, Bogenhagen DF. The accessory subunit of Xenopus laevis mitochondrial DNA polymerase gamma increases processivity of the catalytic subunit of human DNA polymerase gamma and is related to class II aminoacyl-tRNA synthetases. Mol Cell Biol. 1999;19:4039–4046. doi: 10.1128/mcb.19.6.4039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carrodeguas JA, Bogenhagen DF. Protein sequences conserved in prokaryotic aminoacyl-tRNA synthetases are important for the activity of the processivity factor of human mitochondrial DNA polymerase. Nucleic Acids Res. 2000;28:1237–1244. doi: 10.1093/nar/28.5.1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lim SE, Longley MJ, Copeland WC. The mitochondrial p55 accessory subunit of human DNA polymerase gamma enhances DNA binding, promotes processive DNA synthesis, and confers N-ethylmaleimide resistance. J Biol Chem. 1999;274:38197–38203. doi: 10.1074/jbc.274.53.38197. [DOI] [PubMed] [Google Scholar]
- 34.Johnson AA, Tsai Y, Graves SW, Johnson KA. Human mitochondrial DNA polymerase holoenzyme: reconstitution and characterization. Biochemistry. 2000;39:1702–1708. doi: 10.1021/bi992104w. [DOI] [PubMed] [Google Scholar]
- 35.Olson MW, Wang Y, Elder RH, Kaguni LS. Subunit structure of mitochondrial DNA polymerase from Drosophila embryos. Physical and immunological studies. J Biol Chem. 1995;270:28932–28937. doi: 10.1074/jbc.270.48.28932. [DOI] [PubMed] [Google Scholar]
- 36.Fan L, Sanschagrin PC, Kaguni LS, Kuhn LA. The accessory subunit of mtDNA polymerase shares structural homology with aminoacyl-tRNA synthetases: implications for a dual role as a primer recognition factor and processivity clamp. Proc Natl Acad Sci U S A. 1999;96:9527–9532. doi: 10.1073/pnas.96.17.9527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ibba M, Soll D. Aminoacyl-tRNA synthesis. Annu Rev Biochem. 2000;69:617–650. doi: 10.1146/annurev.biochem.69.1.617. [DOI] [PubMed] [Google Scholar]
- 38.Carrodeguas JA, Theis K, Bogenhagen DF, Kisker C. Crystal structure and deletion analysis show that the accessory subunit of mammalian DNA polymerase gamma, Pol gamma B, functions as a homodimer. Mol Cell. 2001;7:43–54. doi: 10.1016/s1097-2765(01)00153-8. [DOI] [PubMed] [Google Scholar]
- 39.Yakubovskaya E, Chen Z, Carrodeguas JA, Kisker C, Bogenhagen DF. Functional human mitochondrial DNA polymerase gamma forms a heterotrimer. J Biol Chem. 2006;281:374–382. doi: 10.1074/jbc.M509730200. [DOI] [PubMed] [Google Scholar]
- 40.Yakubovskaya E, Lukin M, Chen Z, Berriman J, Wall JS, Kobayashi R, Kisker C, Bogenhagen DF. The EM structure of human DNA polymerase gamma reveals a localized contact between the catalytic and accessory subunits. Embo J. 2007;26:4283–4291. doi: 10.1038/sj.emboj.7601843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fan L, Kim S, Farr CL, Schaefer KT, Randolph KM, Tainer JA, Kaguni LS. A novel processive mechanism for DNA synthesis revealed by structure, modeling and mutagenesis of the accessory subunit of human mitochondrial DNA polymerase. J Mol Biol. 2006;358:1229–1243. doi: 10.1016/j.jmb.2006.02.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee YS, Kennedy WD, Yin YW. Structural insight into processive human mitochondrial DNA synthesis and disease-related polymerase mutations. Cell. 2009;139:312–324. doi: 10.1016/j.cell.2009.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee YS, Lee S, Demeler B, Molineux IJ, Johnson KA, Yin YW. Each monomer of the dimeric accessory protein for human mitochondrial DNA polymerase has a distinct role in conferring processivity. J Biol Chem. 2009 doi: 10.1074/jbc.M109.062752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Carrodeguas JA, Pinz KG, Bogenhagen DF. DNA binding properties of human pol gammaB. J Biol Chem. 2002;277:50008–50014. doi: 10.1074/jbc.M207030200. [DOI] [PubMed] [Google Scholar]
- 45.Di Re M, Sembongi H, He J, Reyes A, Yasukawa T, Martinsson P, Bailey LJ, Goffart S, Boyd-Kirkup JD, Wong TS, Fersht AR, Spelbrink JN, Holt IJ. The accessory subunit of mitochondrial DNA polymerase {gamma} determines the DNA content of mitochondrial nucleoids in human cultured cells. Nucleic Acids Res. 2009 doi: 10.1093/nar/gkp614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Johnson AA, Johnson KA. Fidelity of nucleotide incorporation by human mitochondrial DNA polymerase. J Biol Chem. 2001;276:38090–38096. doi: 10.1074/jbc.M106045200. [DOI] [PubMed] [Google Scholar]
- 47.Lee HR, Johnson KA. Fidelity of the human mitochondrial DNA polymerase. J Biol Chem. 2006;281:36236–36240. doi: 10.1074/jbc.M607964200. [DOI] [PubMed] [Google Scholar]
- 48.Donlin MJ, Patel SS, Johnson KA. Kinetic partitioning between the exonuclease and polymerase sites in DNA error correction. Biochemistry. 1991;30:538–546. doi: 10.1021/bi00216a031. [DOI] [PubMed] [Google Scholar]
- 49.Johnson AA, Johnson KA. Exonuclease proofreading by human mitochondrial DNA polymerase. J Biol Chem. 2001;276:38097–38107. doi: 10.1074/jbc.M106046200. [DOI] [PubMed] [Google Scholar]
- 50.Murakami E, Feng JY, Lee H, Hanes J, Johnson KA, Anderson KS. Characterization of novel reverse transcriptase and other RNA-associated catalytic activities by human DNA polymerase gamma: importance in mitochondrial DNA replication. J Biol Chem. 2003;278:36403–36409. doi: 10.1074/jbc.M306236200. [DOI] [PubMed] [Google Scholar]
- 51.Lee HR, Johnson KA. Fidelity and processivity of reverse transcription by the human mitochondrial DNA polymerase. J Biol Chem. 2007;282:31982–31989. doi: 10.1074/jbc.M705392200. [DOI] [PubMed] [Google Scholar]
- 52.De Clercq E. Strategies in the design of antiviral drugs. Nat Rev Drug Discov. 2002;1:13–25. doi: 10.1038/nrd703. [DOI] [PubMed] [Google Scholar]
- 53.Goody RS, Muller B, Restle T. Factors contributing to the inhibition of HIV reverse transcriptase by chain-terminating nucleotides in vitro and in vivo. FEBS Lett. 1991;291:1–5. doi: 10.1016/0014-5793(91)81089-q. [DOI] [PubMed] [Google Scholar]
- 54.Starnes MC, Cheng YC. Cellular metabolism of 2′,3′-dideoxycytidine, a compound active against human immunodeficiency virus in vitro. J Biol Chem. 1987;262:988–991. [PubMed] [Google Scholar]
- 55.Chen CH, Cheng YC. Delayed cytotoxicity and selective loss of mitochondrial DNA in cells treated with the anti-human immunodeficiency virus compound 2′,3′-dideoxycytidine. J Biol Chem. 1989;264:11934–11937. [PubMed] [Google Scholar]
- 56.Kakuda TN. Pharmacology of nucleoside and nucleotide reverse transcriptase inhibitor-induced mitochondrial toxicity. Clin Ther. 2000;22:685–708. doi: 10.1016/S0149-2918(00)90004-3. [DOI] [PubMed] [Google Scholar]
- 57.Richman DD, Fischl MA, Grieco MH, Gottlieb MS, Volberding PA, Laskin OL, Leedom JM, Groopman JE, Mildvan D, Hirsch MS, et al. The toxicity of azidothymidine (AZT) in the treatment of patients with AIDS and AIDS-related complex. A double-blind, placebo-controlled trial. N Engl J Med. 1987;317:192–197. doi: 10.1056/NEJM198707233170402. [DOI] [PubMed] [Google Scholar]
- 58.Dalakas MC, Illa I, Pezeshkpour GH, Laukaitis JP, Cohen B, Griffin JL. Mitochondrial myopathy caused by long-term zidovudine therapy. N Engl J Med. 1990;322:1098–1105. doi: 10.1056/NEJM199004193221602. [DOI] [PubMed] [Google Scholar]
- 59.Arnaudo E, Dalakas M, Shanske S, Moraes CT, DiMauro S, Schon EA. Depletion of muscle mitochondrial DNA in AIDS patients with zidovudine-induced myopathy. Lancet. 1991;337:508–510. doi: 10.1016/0140-6736(91)91294-5. [DOI] [PubMed] [Google Scholar]
- 60.Herskowitz A, Willoughby SB, Baughman KL, Schulman SP, Bartlett JD. Cardiomyopathy associated with antiretroviral therapy in patients with HIV infection: a report of six cases. Ann Intern Med. 1992;116:311–313. doi: 10.7326/0003-4819-116-4-311. [DOI] [PubMed] [Google Scholar]
- 61.Yarchoan R, Mitsuya H, Myers CE, Broder S. Clinical pharmacology of 3′-azido-2′,3′-dideoxythymidine (zidovudine) and related dideoxynucleosides. N Engl J Med. 1989;321:726–738. doi: 10.1056/NEJM198909143211106. [DOI] [PubMed] [Google Scholar]
- 62.Keilbaugh SA, Prusoff WH, Simpson MV. The PC12 cell as a model for studies of the mechanism of induction of peripheral neuropathy by anti-HIV-1 dideoxynucleoside analogs. Biochem Pharmacol. 1991;42:R5–8. doi: 10.1016/0006-2952(91)90672-r. [DOI] [PubMed] [Google Scholar]
- 63.Chen CH, Vazquez-Padua M, Cheng YC. Effect of anti-human immunodeficiency virus nucleoside analogs on mitochondrial DNA and its implication for delayed toxicity. Mol Pharmacol. 1991;39:625–628. [PubMed] [Google Scholar]
- 64.Parker WB, Cheng YC. Mitochondrial Toxicity of Antiviral Nucleoside Analogs. Journal of NIH Research. 1994;6:57–61. [Google Scholar]
- 65.Lewis W, Dalakas MC. Mitochondrial toxicity of antiviral drugs. Nat Med. 1995;1:417–422. doi: 10.1038/nm0595-417. [DOI] [PubMed] [Google Scholar]
- 66.Tabor S, Richardson CC. A single residue in DNA polymerases of the Escherichia coli DNA polymerase I family is critical for distinguishing between deoxy- and dideoxyribonucleotides. Proc Natl Acad Sci U S A. 1995;92:6339–6343. doi: 10.1073/pnas.92.14.6339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Astatke M, Grindley ND, Joyce CM. How E. coli DNA polymerase I (Klenow fragment) distinguishes between deoxy- and dideoxynucleotides. J Mol Biol. 1998;278:147–165. doi: 10.1006/jmbi.1998.1672. [DOI] [PubMed] [Google Scholar]
- 68.Lim SE, Ponamarev MV, Longley MJ, Copeland WC. Structural determinants in human DNA polymerase gamma account for mitochondrial toxicity from nucleoside analogs. J Mol Biol. 2003;329:45–57. doi: 10.1016/s0022-2836(03)00405-4. [DOI] [PubMed] [Google Scholar]
- 69.Brahams D. Deaths in US fialuridine trial. Lancet. 1994;343:1494–1495. doi: 10.1016/s0140-6736(94)92599-2. [DOI] [PubMed] [Google Scholar]
- 70.McKenzie R, Fried MW, Sallie R, Conjeevaram H, Di Bisceglie AM, Park Y, Savarese B, Kleiner D, Tsokos M, Luciano C, et al. Hepatic failure and lactic acidosis due to fialuridine (FIAU), an investigational nucleoside analogue for chronic hepatitis B. N Engl J Med. 1995;333:1099–1105. doi: 10.1056/NEJM199510263331702. [DOI] [PubMed] [Google Scholar]
- 71.Cui L, Yoon S, Schinazi RF, Sommadossi JP. Cellular and molecular events leading to mitochondrial toxicity of 1-(2-deoxy-2-fluoro-1-beta-D-arabinofuranosyl)-5-iodouracil in human liver cells. J Clin Invest. 1995;95:555–563. doi: 10.1172/JCI117698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lewis W, Levine ES, Griniuviene B, Tankersley KO, Colacino JM, Sommadossi JP, Watanabe KA, Perrino FW. Fialuridine and its metabolites inhibit DNA polymerase gamma at sites of multiple adjacent analog incorporation, decrease mtDNA abundance, and cause mitochondrial structural defects in cultured hepatoblasts. Proc Natl Acad Sci U S A. 1996;93:3592–3597. doi: 10.1073/pnas.93.8.3592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cherrington JM, Allen SJ, McKee BH, Chen MS. Kinetic analysis of the interaction between the diphosphate of (S)-1-(3-hydroxy-2-phosphonylmethoxypropyl)cytosine, ddCTP, AZTTP, and FIAUTP with human DNA polymerases beta and gamma. Biochem Pharmacol. 1994;48:1986–1988. doi: 10.1016/0006-2952(94)90600-9. [DOI] [PubMed] [Google Scholar]
- 74.Lewis W, Meyer RR, Simpson JF, Colacino JM, Perrino FW. Mammalian DNA polymerases alpha, beta, gamma, delta, and epsilon incorporate fialuridine (FIAU) monophosphate into DNA and are inhibited competitively by FIAU Triphosphate. Biochemistry. 1994;33:14620–14624. doi: 10.1021/bi00252a030. [DOI] [PubMed] [Google Scholar]
- 75.Klecker RW, Katki AG, Collins JM. Toxicity, metabolism, DNA incorporation with lack of repair, and lactate production for 1-(2′-fluoro-2′-deoxy-beta-D-arabinofuranosyl)-5-iodouracil in U-937 and MOLT-4 cells. Mol Pharmacol. 1994;46:1204–1209. [PubMed] [Google Scholar]
- 76.Colacino JM, Malcolm SK, Jaskunas SR. Effect of fialuridine on replication of mitochondrial DNA in CEM cells and in human hepatoblastoma cells in culture. Antimicrob Agents Chemother. 1994;38:1997–2002. doi: 10.1128/aac.38.9.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Feng JY, Johnson AA, Johnson KA, Anderson KS. Insights into the molecular mechanism of mitochondrial toxicity by AIDS drugs. J Biol Chem. 2001;276:23832–23837. doi: 10.1074/jbc.M101156200. [DOI] [PubMed] [Google Scholar]
- 78.Lim SE, Copeland WC. Differential incorporation and removal of antiviral deoxynucleotides by human DNA polymerase gamma. J Biol Chem. 2001;276:23616–23623. doi: 10.1074/jbc.M101114200. [DOI] [PubMed] [Google Scholar]
- 79.Johnson AA, Ray AS, Hanes J, Suo Z, Colacino JM, Anderson KS, Johnson KA. Toxicity of antiviral nucleoside analogs and the human mitochondrial DNA polymerase. J Biol Chem. 2001;276:40847–40857. doi: 10.1074/jbc.M106743200. [DOI] [PubMed] [Google Scholar]
- 80.Lee H, Hanes J, Johnson KA. Toxicity of nucleoside analogues used to treat AIDS and the selectivity of the mitochondrial DNA polymerase. Biochemistry. 2003;42:14711–14719. doi: 10.1021/bi035596s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Feng JY, Murakami E, Zorca SM, Johnson AA, Johnson KA, Schinazi RF, Furman PA, Anderson KS. Relationship between antiviral activity and host toxicity: comparison of the incorporation efficiencies of 2′,3′-dideoxy-5-fluoro-3′-thiacytidine-triphosphate analogs by human immunodeficiency virus type 1 reverse transcriptase and human mitochondrial DNA polymerase. Antimicrob Agents Chemother. 2004;48:1300–1306. doi: 10.1128/AAC.48.4.1300-1306.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Feng JY, Anderson KS. Mechanistic studies comparing the incorporation of (+) and (-) isomers of 3TCTP by HIV-1 reverse transcriptase. Biochemistry. 1999;38:55–63. doi: 10.1021/bi982340r. [DOI] [PubMed] [Google Scholar]
- 83.Feng JY, Shi J, Schinazi RF, Anderson KS. Mechanistic studies show that (-)-FTC-TP is a better inhibitor of HIV-1 reverse transcriptase than 3TC-TP. Faseb J. 1999;13:1511–1517. doi: 10.1096/fasebj.13.12.1511. [DOI] [PubMed] [Google Scholar]
- 84.Fowler JD, Brown JA, Johnson KA, Suo Z. Kinetic investigation of the inhibitory effect of gemcitabine on DNA polymerization catalyzed by human mitochondrial DNA polymerase. J Biol Chem. 2008;283:15339–15348. doi: 10.1074/jbc.M800310200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hanes JW, Zhu Y, Parris DS, Johnson KA. Enzymatic therapeutic index of acyclovir. Viral versus human polymerase gamma specificity. J Biol Chem. 2007;282:25159–25167. doi: 10.1074/jbc.M703972200. [DOI] [PubMed] [Google Scholar]
- 86.Kim HO, Shanmuganathan K, Alves AJ, Jeong LS, Beach JW, Schinazi RF, Chang CN, Cheng YC, Chu CK. Potent anti-HIV and anti-HBV activities of (-)-L-beta-dioxolane-C and (+)-L-beta-dioxolane-T and their asymmetric syntheses. Tetrahedron Lett. 1992;33:6899–6902. [Google Scholar]
- 87.Grove KL, Guo X, Liu SH, Gao Z, Chu CK, Cheng YC. Anticancer activity of beta-L-dioxolane-cytidine, a novel nucleoside analogue with the unnatural L configuration. Cancer Res. 1995;55:3008–3011. [PubMed] [Google Scholar]
- 88.Seok JI, Lee DK, Lee CH, Park MS, Kim SY, Kim HS, Jo HY, Kim DS. Long-term therapy with clevudine for chronic hepatitis B can be associated with myopathy characterized by depletion of mitochondrial DNA. Hepatology. 2009;49:2080–2086. doi: 10.1002/hep.22959. [DOI] [PubMed] [Google Scholar]
- 89.Kim BK, Oh J, Kwon SY, Choe WH, Ko SY, Rhee KH, Seo TH, Lim SD, Lee CH. Clevudine myopathy in patients with chronic hepatitis B. J Hepatol. 2009;51:829–834. doi: 10.1016/j.jhep.2009.04.019. [DOI] [PubMed] [Google Scholar]
- 90.Tak WY, Park SY, Jung MK, Jeon SW, Cho CM, Kweon YO, Kim SK, Choi YH. Mitochondrial myopathy caused by clevudine therapy in chronic hepatitis B patients. Hepatol Res. 2009;39:944–947. doi: 10.1111/j.1872-034X.2009.00515.x. [DOI] [PubMed] [Google Scholar]
- 91.Chu CK, Ma T, Shanmuganathan K, Wang C, Xiang Y, Pai SB, Yao GQ, Sommadossi JP, Cheng YC. Use of 2′-fluoro-5-methyl-beta-L-arabinofuranosyluracil as a novel antiviral agent for hepatitis B virus and Epstein-Barr virus. Antimicrob Agents Chemother. 1995;39:979–981. doi: 10.1128/aac.39.4.979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pai S Balakrishna, Liu SH, Zhu YL, Chu CK, Cheng YC. Inhibition of hepatitis B virus by a novel L-nucleoside, 2′-fluoro-5-methyl-beta-L-arabinofuranosyl uracil. Antimicrob Agents Chemother. 1996;40:380–386. doi: 10.1128/aac.40.2.380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yao GQ, Liu SH, Chou E, Kukhanova M, Chu CK, Cheng YC. Inhibition of Epstein-Barr virus replication by a novel L-nucleoside, 2′-fluoro-5-methyl-beta-L-arabinofuranosyluracil. Biochem Pharmacol. 1996;51:941–947. doi: 10.1016/0006-2952(96)00049-4. [DOI] [PubMed] [Google Scholar]
- 94.Yamanaka H, Gatanaga H, Kosalaraksa P, Matsuoka-Aizawa S, Takahashi T, Kimura S, Oka S. Novel mutation of human DNA polymerase gamma associated with mitochondrial toxicity induced by anti-HIV treatment. J Infect Dis. 2007;195:1419–1425. doi: 10.1086/513872. [DOI] [PubMed] [Google Scholar]
- 95.Bailey CM, Kasiviswanathan R, Copeland WC, Anderson KS. R964C mutation of DNA polymerase gamma imparts increased stavudine toxicity by decreasing nucleoside analog discrimination and impairing polymerase activity. Antimicrob Agents Chemother. 2009;53:2610–2612. doi: 10.1128/AAC.01659-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chiappini F, Teicher E, Saffroy R, Debuire B, Vittecoq D, Lemoine A. Relationship between polymerase gamma (POLG) polymorphisms and antiretroviral therapy-induced lipodystrophy in HIV-1 infected patients: a case-control study. Curr HIV Res. 2009;7:244–253. doi: 10.2174/157016209787581409. [DOI] [PubMed] [Google Scholar]
- 97.Van Goethem G, Luoma P, Rantamaki M, Al Memar A, Kaakkola S, Hackman P, Krahe R, Lofgren A, Martin JJ, De Jonghe P, Suomalainen A, Udd B, Van Broeckhoven C. POLG mutations in neurodegenerative disorders with ataxia but no muscle involvement. Neurology. 2004;63:1251–1257. doi: 10.1212/01.wnl.0000140494.58732.83. [DOI] [PubMed] [Google Scholar]
- 98.Hakonen AH, Heiskanen S, Juvonen V, Lappalainen I, Luoma PT, Rantamaki M, Goethem GV, Lofgren A, Hackman P, Paetau A, Kaakkola S, Majamaa K, Varilo T, Udd B, Kaariainen H, Bindoff LA, Suomalainen A. Mitochondrial DNA polymerase W748S mutation: a common cause of autosomal recessive ataxia with ancient European origin. Am J Hum Genet. 2005;77:430–441. doi: 10.1086/444548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chan SS, Longley MJ, Copeland WC. Modulation of the W748S mutation in DNA polymerase gamma by the E1143G polymorphismin mitochondrial disorders. Hum Mol Genet. 2006;15:3473–3483. doi: 10.1093/hmg/ddl424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chan SS, Copeland WC. DNA polymerase gamma and mitochondrial disease: Understanding the consequence of POLG mutations. Biochim Biophys Acta. 2008 doi: 10.1016/j.bbabio.2008.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zeviani M, Servidei S, Gellera C, Bertini E, DiMauro S, DiDonato S. An autosomal dominant disorder with multiple deletions of mitochondrial DNA starting at the D-loop region. Nature. 1989;339:309–311. doi: 10.1038/339309a0. [DOI] [PubMed] [Google Scholar]
- 102.Zeviani M, Bresolin N, Gellera C, Bordoni A, Pannacci M, Amati P, Moggio M, Servidei S, Scarlato G, DiDonato S. Nucleus-driven multiple large-scale deletions of the human mitochondrial genome: a new autosomal dominant disease. Am J Hum Genet. 1990;47:904–914. [PMC free article] [PubMed] [Google Scholar]
- 103.Servidei S, Zeviani M, Manfredi G, Ricci E, Silvestri G, Bertini E, Gellera C, Di Mauro S, Di Donato S, Tonali P. Dominantly inherited mitochondrial myopathy with multiple deletions of mitochondrial DNA: clinical, morphologic, and biochemical studies. Neurology. 1991;41:1053–1059. doi: 10.1212/wnl.41.7.1053. [DOI] [PubMed] [Google Scholar]
- 104.Suomalainen A, Majander A, Haltia M, Somer H, Lonnqvist J, Savontaus ML, Peltonen L. Multiple deletions of mitochondrial DNA in several tissues of a patient with severe retarded depression and familial progressive external ophthalmoplegia. J Clin Invest. 1992;90:61–66. doi: 10.1172/JCI115856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Napoli L, Bordoni A, Zeviani M, Hadjigeorgiou GM, Sciacco M, Tiranti V, Terentiou A, Moggio M, Papadimitriou A, Scarlato G, Comi GP. A novel missense adenine nucleotide translocator-1 gene mutation in a Greek adPEO family. Neurology. 2001;57:2295–2298. doi: 10.1212/wnl.57.12.2295. [DOI] [PubMed] [Google Scholar]
- 106.Spelbrink JN, Li FY, Tiranti V, Nikali K, Yuan QP, Tariq M, Wanrooij S, Garrido N, Comi G, Morandi L, Santoro L, Toscano A, Fabrizi GM, Somer H, Croxen R, Beeson D, Poulton J, Suomalainen A, Jacobs HT, Zeviani M, Larsson C. Human mitochondrial DNA deletions associated with mutations in the gene encoding Twinkle, a phage T7 gene 4-like protein localized in mitochondria. Nat Genet. 2001;28:223–231. doi: 10.1038/90058. [DOI] [PubMed] [Google Scholar]
- 107.Van Goethem G, Dermaut B, Lofgren A, Martin JJ, Van Broeckhoven C. Mutation of POLG is associated with progressive external ophthalmoplegia characterized by mtDNA deletions. Nat Genet. 2001;28:211–212. doi: 10.1038/90034. [DOI] [PubMed] [Google Scholar]
- 108.Longley MJ, Clark S, Yu Wai Man C, Hudson G, Durham SE, Taylor RW, Nightingale S, Turnbull DM, Copeland WC, Chinnery PF. Mutant POLG2 disrupts DNA polymerase gamma subunits and causes progressive external ophthalmoplegia. Am J Hum Genet. 2006;78:1026–1034. doi: 10.1086/504303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Suomalainen A, Kaukonen J. Diseases caused by nuclear genes affecting mtDNA stability. Am J Med Genet. 2001;106:53–61. doi: 10.1002/ajmg.1379. [DOI] [PubMed] [Google Scholar]
- 110.Doublie S, Tabor S, Long AM, Richardson CC, Ellenberger T. Crystal structure of a bacteriophage T7 DNA replication complex at 2.2 A resolution. Nature. 1998;391:251–258. doi: 10.1038/34593. [DOI] [PubMed] [Google Scholar]
- 111.Ponamarev MV, Longley MJ, Nguyen D, Kunkel TA, Copeland WC. Active site mutation in DNA polymerase gamma associated with progressive external ophthalmoplegia causes error-prone DNA synthesis. J Biol Chem. 2002;277:15225–15228. doi: 10.1074/jbc.C200100200. [DOI] [PubMed] [Google Scholar]
- 112.Graziewicz MA, Longley MJ, Bienstock RJ, Zeviani M, Copeland WC. Structure-function defects of human mitochondrial DNA polymerase in autosomal dominant progressive external ophthalmoplegia. Nat Struct Mol Biol. 2004;11:770–776. doi: 10.1038/nsmb805. [DOI] [PubMed] [Google Scholar]
- 113.Shadel GS. Yeast as a model for human mtDNA replication. Am J Hum Genet. 1999;65:1230–1237. doi: 10.1086/302630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Stuart GR, Santos JH, Strand MK, Van Houten B, Copeland WC. Mitochondrial and nuclear DNA defects in Saccharomyces cerevisiae with mutations in DNA polymerase gamma associated with progressive external ophthalmoplegia. Hum Mol Genet. 2006;15:363–374. doi: 10.1093/hmg/ddi454. [DOI] [PubMed] [Google Scholar]
- 115.Luo N, Kaguni LS. Mutations in the spacer region of Drosophila mitochondrial DNA polymerase affect DNA binding, processivity, and the balance between Pol and Exo function. J Biol Chem. 2005;280:2491–2497. doi: 10.1074/jbc.M411447200. [DOI] [PubMed] [Google Scholar]
- 116.Chan SS, Longley MJ, Copeland WC. The common A467T mutation in the human mitochondrial DNA polymerase (POLG) compromises catalytic efficiency and interaction with the accessory subunit. J Biol Chem. 2005;280:31341–31346. doi: 10.1074/jbc.M506762200. [DOI] [PubMed] [Google Scholar]
- 117.Luoma PT, Luo N, Loscher WN, Farr CL, Horvath R, Wanschitz J, Kiechl S, Kaguni LS, Suomalainen A. Functional defects due to spacer-region mutations of human mitochondrial DNA polymerase in a family with an ataxiamyopathy syndrome. Hum Mol Genet. 2005;14:1907–1920. doi: 10.1093/hmg/ddi196. [DOI] [PubMed] [Google Scholar]
- 118.Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, Bohlooly YM, Gidlof S, Oldfors A, Wibom R, Tornell J, Jacobs HT, Larsson NG. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429:417–423. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- 119.Pfeffer G, Cote HC, Montaner JS, Li CC, Jitratkosol M, Mezei MM. Ophthalmoplegia and ptosis: mitochondrial toxicity in patients receiving HIV therapy. Neurology. 2009;73:71–72. doi: 10.1212/WNL.0b013e3181aae814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Li Y, Korolev S, Waksman G. Crystal structures of open and closed forms of binary and ternary complexes of the large fragment of Thermus aquaticus DNA polymerase I: structural basis for nucleotide incorporation. EMBO J. 1998;17:7514–7525. doi: 10.1093/emboj/17.24.7514. [DOI] [PMC free article] [PubMed] [Google Scholar]