

Abstract
Public concerns over the health effects associated with low-level and long-term exposure to tritium released from industrial point sources have generated the demand for better methods to evaluate historical tritium exposure levels for these communities. The cellulose of trees accurately reflects the tritium concentration in the source water and may contain the only historical record of tritium exposure. The tritium activity in the annual rings of a tree was measured using accelerator mass spectrometry to reconstruct historical annual averages of tritium exposure. Milligram-sized samples of the annual tree rings from a Tamarix located at the Nevada Test Site are used for validation of this methodology. The salt cedar was chosen since it had a single source of tritiated water that was well-characterized as it varied over time. The decay-corrected tritium activity of the water in which the salt cedar grew closely agrees with the organically bound tritium activity in its annual rings. This demonstrates that the milligram-sized samples used in tritium accelerator mass spectrometry are suited for reconstructing anthropogenic tritium levels in the environment.
Introduction
Tritium is a radioactive isotope of hydrogen that undergoes β-decay (12.4 yr half-life) to yield a helium-3 atom. Tritium has both natural and anthropogenic sources in the environment. Naturally occurring tritium is primarily produced through the nuclear reaction of atoms in the upper atmosphere with cosmic rays but also through nuclear reactions in the earth’s crust. The greatest anthropogenic production of tritium occurred during the atmospheric testing of nuclear weapons, resulting in global increases of environmental tritium levels (1). In addition, there are local industrial sources of tritium from nuclear power plants, nuclear weapons complexes, biotechnology research using radioactive tracers, and radioluminescent paint (2). Tritium quantities are expressed using a variety of units: 1 tritium atom per 1018 hydrogen atom = 1 tritium unit (TU) = 3.2 pCi/L of water = 120 Bq/m3 of water.
In the environment, tritium (T) may exist in many different chemical forms since it can substitute on any molecule containing stabile hydrogen atoms (H). Tritiated hydrogen gas (HT) is produced in the upper atmosphere and is discharged from nuclear weapons complexes (3). The residence time for HT is approximately 1 yr in the atmosphere or 1 day in soil until it is oxidized to tritiated water (HTO) (2). As HTO, tritium is very mobile in the hydrologic cycle. Tritiated water has been used extensively as an ideal tracer in laboratory and field studies to examine water transport in complex systems (4). Tritiated water is also incorporated through biochemical processes into organic molecules, referred to as organically bound tritium (OBT). In organic molecules, tritium can bond with oxygen, nitrogen, sulfur, or carbon atoms. The bonds with oxygen, sulfur, and nitrogen allow tritium to be exchanged with other hydrogen atoms; in contrast, the carbon–hydrogen bond is not exchangeable (5). Also, OBT can be converted back to HTO by biota that uses OBT molecules for respiration.
Tritium poses no external radiotoxicity to humans since the maximum energy released during tritium decay is very low (0.019 MeV) and cannot penetrate the skin, but when tritium is ingested and organically bound in human cells, it is associated with carcinogenic, genetic, developmental, and reproductive effects (2). The current Federal drinking water standard in the United States, 20 000 pCi/L (750 000 Bq/m3, 6250 TU, or 6.25 × 10−15 T/H), is often used as a reference point for concern over human exposure levels. Communities located near industrial facilities that utilize tritium are demanding better techniques to evaluate exposure levels. Although thorough monitoring is appropriate for determining current exposures, historical exposure data on 50-yr time scales is often missing, sometimes incomplete, and rarely based on environmental measurements. Exposure measurements are necessary for epidemiologists to better assess the health effects of chronic exposure. Also, cross-comparisons between reported releases and environmental tritium measurements provide one method to verify that adequate monitoring has taken place.
In some circumstances, the annual rings from trees are capable of being utilized as a passive monitor of the tritium release history. Photosynthesis combines water with carbon dioxide to synthesize cellulose that is not reutilized as stored energy. Changes in growth conditions result in quantity and density differences of the cellulose that when viewed in cross-section form visible rings. Thus, when growth conditions change as a result of the annual cycle of seasons, the rings formed consist of the cellulose synthesized during a single year. Since other elements or compounds may also become part of the biomass, either as primary constituents or as contaminants that do not play a role in the physiology of the tree, tree rings have been shown in a number of circumstances to accumulate annual records of chemicals in their immediate environment (6, 7). Furthermore, the carbon–hydrogen bonds in the cellulose of trees are strongly bound and can be used to determine the tritium activity in the water used by the tree. Groundwater is the dominant source of water for trees, but in environments where the atmospheric water vapor has a dramatically different tritium activity, it is possible for the cellulose to reflect small contributions of HTO that enter through the leaf stomata.
The use of tree rings to study environmental levels of tritium was originally reported in 1961 (8). Other studies have shown that environmental tritium levels in precipitation can be reconstructed using both tree rings and wines (9–11). Kozak et al. (12) also demonstrated that an unusually high tritium activity in the precipitation of a single year was reflected in the tree ring associated with that year, and no anomaly was seen in the tree rings of adjacent years. Therefore, tree rings appear to have the precision and sensitivity for use as passive monitors in tritium exposure reconstruction. More recently, studies have used trees to monitor anthropogenic sources of tritium in the environment (13–15). In the previous studies (8–15), sample sizes in excess of 10–100 g of woody tissues were needed for decay counting. This required a prohibitive amount of effort in collection and preparation for analysis and destruction of old trees each time.
Traditional methods for quantifying tritium activity involve either the detection of the energy it releases during radioactive decay (liquid scintillation counting/gas proportional counting) or the subsequent accumulation of its decay product (helium-3 in-growth mass spectrometry). In contrast, tritium accelerator mass spectrometry (3H AMS) directly counts individual tritium atoms from each sample. Since the half-life of tritium is 12.4 yr, a 2-mg sample of water with an activity of 104 TU has 0.1 decays/min for decay counting or helium-3 in-growth mass spectrometry as compared to a count rate of 50–100 cpm using 3H AMS. There is a 500–1000-fold improvement in analysis time for 3H AMS as compared to the instrument time required for decay counting or the in-growth time required for helium-3 mass spectrometry. However, the number of decays per minute can be increased proportionally for decay counting or helium-3 mass spectrometry using orders of magnitude larger samples when available, whereas 3H AMS is limited to milligram-sized samples. Typical instrument quantitation limits are 70 TU for 3H AMS using 2 mg of sample as compared to 3 TU for decay methods using 25 g of sample (16) or 0.01 TU for helium-3 in-growth mass spectrometry (17–19) using 0.5–1 kg of sample. Electrolytic tritium enrichment of kilogram-sized samples are able to decrease the quantitation limit of decay counting to 0.1 TU (20) and also has the potential to significantly decrease the 3H AMS quantitation limit for samples that are tens to hundreds of milligrams in size. Thus, for small samples, 3H AMS has great potential for use in examining tritium levels with an improved measurement throughput and greater ease in sample collection and preparation. Currently, 3H AMS has been used in a biomedical tracer study (21), and there are potential applications for environmental research.
Although the first paper reporting 3H AMS measurements was published in 1977 (22), this is the first paper that reports environmental measurements of organically bound tritium using 3H AMS. As has been shown previously for other radioisotopic analyses of environmental samples (23), accelerator mass spectrometry techniques are accurate and relatively rapid at environmental levels. This study demonstrates the capability of 3H AMS for measuring organically bound tritium levels in the annual rings of a tree growing in well-characterized tritiated water. Although the environmental tritium levels in this study are unusually high, the long-term records and the unique environment provided considerable control useful for validating this methodology under field conditions.
Site Description
The tree rings chosen for analysis are from a salt cedar (Tamarix) at the Nevada Test Site (NTS) in Nye County, NV (Figure 1). The climate at the NTS is arid with an average annual precipitation of 10 cm and sparse vegetation. On May 14, 1965, the Cambric device, a small nuclear weapon (0.75 kt), was detonated at a depth of 294 m in tuffaceous alluvium, 73 m below the water table, in an area of NTS referred to locally as western Frenchman Flat. The device released over 1015 Bq of tritium as well as other radionuclides (24). As part of the Department of Energy Hydrology and Radionclide Migration Project, a satellite well was installed 91 m from the Cambric cavity. To induce radionuclide migration, an artificial hydraulic gradient was imposed by pumping the groundwater nearly continuously for 16 yr, from October 1975 to 1991, at flow rates of 1–2 m3/min (25). Approximately 50 m away from the well head, the pumped groundwater was discharged into an unlined ditch that runs for 1.6 km along the desert floor. Some of the water in the ditch infiltrated, and the rest drained into a dry lake.
FIGURE 1.

Map locating Frenchman Flat at the Nevada Test Site and cross-section showing the location of the Cambric device and subsequent well used to pump water for the Radionuclide Migration Project.
The tritium activity in the water pumped from the aquifer into the ditch was measured at least monthly for the 16-yr experiment. The first 2 yr of pumping had only background levels of tritium, then the activity rapidly peaked at 3 × 108 Bq/m3 (~2 000 000 TU) and had a slow decay that is typical of elution processes in pumped aquifers (25) (Figure 2). Over the course of this experiment non-native vegetation grew along the length of the ditch. Even after the end of groundwater pumping the salt cedar continued to produce new leaves, indicating growth up to 1999, and their annual rings are expected to contain the strong tritium signal from the pumped groundwater.
FIGURE 2.
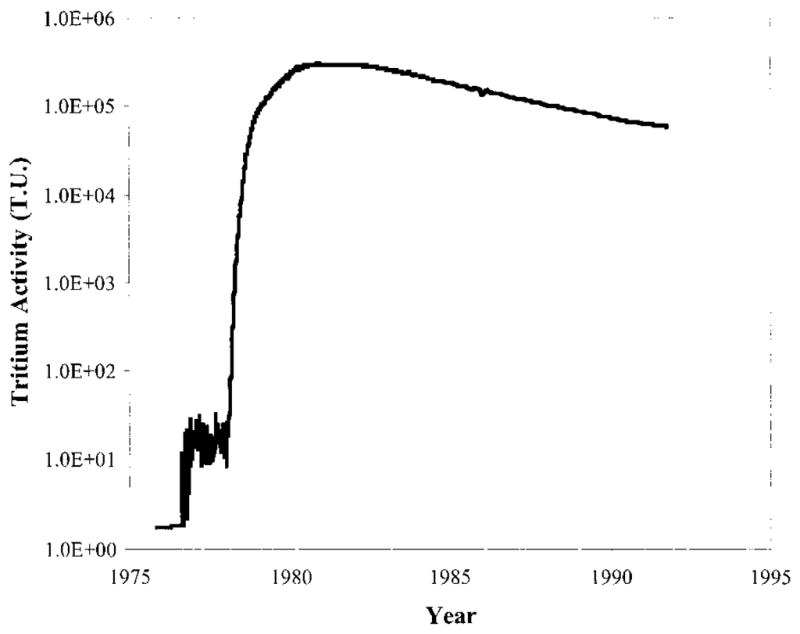
Tritium levels measured in water pumped during the Radionuclide Migration Project and discharged into an unlined ditch at Frenchman Flat, Nevada Test Site. The tritium measurements from each year have been decay corrected to the year 2000.
Methods
A salt cedar was felled in the spring of 1999. The sample of the tree trunk was approximately 1 m above the ground, 0.15 m in diameter, and contained 17 well-defined rings. The sample was divided into each ring using a scalpel. The sub-millimeter outermost ring was lost during removal of the bark. The wood from each ring was chopped with a scalpel and ground using a mortar and pestle. The wood pulp was put in glass vials and repeatedly soaked for 12 h with deionized water at 95 °C. This equilibration removed the easily extractable resins from the wood pulp and the tritium on hydroxyl sites through equilibration (14). A total of 5–12 mg of wet wood from each ring was weighed into 6-mm quartz culture tubes. The quartz culture tubes were placed within larger Pyrex test tubes, and filter paper was inserted in the top to prevent sample loss. The tubes were placed in a centrifugal evaporator and dried for 8 h. The dry weight of the wood ranged from 1.5 to 5.4 mg.
The 3H AMS sample preparation is based on previously established sample preparation techniques used for measuring deuterium-to-hydrogen ratios (D/H) and organically bound deuterium analysis (26, 27). In D/H analyses, hydrogen gas is injected into a stable isotope mass spectrometer, but for 3H AMS the hydrogen must be in a solid form for compatibility with the negative cesium ion source. Therefore, the D/H sample preparation was modified to capture the hydrogen as a solid, in the form of titanium hydride. To summarize, the 6-mm quartz culture tube containing the dried sample was placed into a 9-mm quartz test tube containing 200 mg of cupric oxide. The test tubes were evacuated on a manifold and sealed with an oxygen/acetylene torch. The samples were combusted in a muffle furnace at 900 °C for 2 h to completely oxidize the organic sample. The water that resulted from the sample oxidation was then cryogenically transferred through an evacuated manifold heated to 120 °C into a 9-mm quartz tube containing 200 mg of granular zinc (Indiana Zinc) and into a 6-mm borosilicate culture tube containing 10 mg of titanium powder. The quartz tubes containing the zinc, titanium, and combustion water were then sealed with an oxygen/acetylene torch. The samples were put in the muffle furnace at 530 °C for 4 h, and then the temperature was lowered to 400 °C for 6 h. This heating step results in the reduction of the water by the zinc and the absorption of the resulting hydrogen gas into the titanium metal, forming titanium hydride. The metal hydride powder is then hammered into an aluminum sample holder for analysis. Standards were prepared similarly using tritium-labeled 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) that was serially diluted with tributyrin, a nonvolatile organic liquid. The tritium activities of the standards were determined by performing triplicate measurements on larger sized samples using liquid scintillation counting.
The samples were measured using the tandem Van de Graff accelerator mass spectrometer in the Center for Accelerator Mass Spectrometry at Lawrence Livermore National Laboratory. A diagram of the instrument is shown in Figure 3. The acceleration voltage was 3 MV. Hydrogen currents ranged from 1.5 to over 60 μA. Each sample was measured 3 times for a 5-min duration each time. The standard curve (Figure 4) was linear for the expected range of tree ring samples (108–104 TU). The precision of the measurements on the standards was typically ±15%, based on the scatter of repeated measurements.
FIGURE 3.

Diagram showing how tritium is measured using the accelerator mass spectrometer at Lawrence Livermore National Laboratory.
FIGURE 4.

Tritium AMS standard curve over the expected range of tree ring samples. Error bars for individual measurements are smaller than the data points. The instrument quantitation limit (5σ) was 70 TU, and sample preparation quantitation limit (5σ) was 5000 TU.
Results and Discussion
Results of tree ring measurements are shown in Figure 5. The tritium activity of the water pumped into the unlined ditch (line) has been decay corrected to the year 2000. The organically bound tritium activity in the tree rings (circles) was corrected for the 30% of hydrogen atoms that are exchangeable through equilibration based on the stoichiometry of cellulose. Multiple data points for a given year indicate results from separate wood samples. Error bars represent the uncertainty of the 3H AMS measurement (typically ±20%) due to the combined uncertainty associated with counting statistics and the variance of multiple measurements of the same sample. The sample preparation quantitation limit (5000 TU), determined using process blanks, was significantly greater than the instrument quantitation limit (70 TU), indicating some background tritium in the preparation process. This was not unexpected since the laboratory used for sample preparation is also used for preparing samples from biomedical tracing experiments using tritium-labeled compounds. The outermost ring that was lost is believed to be representative of the year prior to sampling (1998) since it is assumed that there was not enough time in 1999 to set a perceptible ring. The age of all other tree rings were determined by counting back annual rings from this outmost ring.
FIGURE 5.

Tritium levels measured in water pumped into the unlined ditch (line) compared with organically bound tritium levels measured in the annual rings of a Tamarix. The pumped water levels are decay corrected to the year the samples were measured by AMS (2000). The organically bound tritium levels are corrected for the 30% of hydrogen atoms in the wood that exchanged with water with low tritium levels. Error bars represent the error from counting statistics, internal variability from multiple measurements, and variability of external standards.
The results also provide some observations about the tree’s life cycle. The center ring at 1 m high appears to be from 1982, 7 yr after pumping began. Although water was no longer being pumped into the ditch after 1991, the tree seems to have utilized the water retained in the vadose zone from the last year of pumping.
There is good agreement between the organically bound tritium content in the tree rings and the tritium content from the tree’s source water. A correlation of the data is shown in Figure 6 and has a correlation coefficient of 0.88. These results are consistent with previous studies of tritium in tree rings using liquid scintillation counting, but a much smaller sample size was used for these measurements. The decrease in sample size allows rapid and adequate sample collection for reconstructing tritium levels using an increment borer, a standard dendrological tool that does not harm the tree being sampled. Therefore, environmental measurements of organically bound tritium using 3H AMS enable a tree to be used as a passive monitor for HTO in the groundwater without harming the tree, with a less laborious sample collection and preparation, and with greater sample measurement throughput.
FIGURE 6.

Correlation of tritium levels measured in water pumped into the unlined ditch compared with organically bound tritium levels measured in the annual rings of a Tamarix from 1982 to 1992. Y-error bars represent the tritium AMS uncertainty based on counting statistics, internal variability from multiple measurements, and variability of external standards. X-error bars represent the range of tritium levels measured in the pumped water over the calendar year.
As a validation, a tree exposed to a relatively high activity tritium source that slowly changed over time was chosen for the first environmental tree ring measurements using 3H AMS. Although the sample chosen for the validation is unique in origin, it provides a well-characterized sample that has few uncertainties related to tritium exposure pathways. Since none of the processes are fundamentally different at lower tritium activities, these validation results should be easily extended to tree rings with lower activity tritium sources where more relevant facility monitoring scenarios exist.
Complex pathways in environmental systems can be better understood through the use of isotopic labels. Although the focus of this study is on reconstructing tritium exposure, the ability of 3H AMS to rapidly measure small samples with a relatively low tritium activity permits greater spatial and temporal resolution for tritium tracer experiments using lower amounts of radiation in hydrological, chemical, and biological research. 3H AMS has possible environmental applications in (i) understanding water transport in the upper soil zone on millimeter scales without the production of waste that is classified as radioactive, (ii) identifying the fate of labeled contaminants in natural or engineered systems, (iii) measuring the kinetics of microbial processes, and (iv) studying environmental toxicology at environmentally relevant doses and without significant radiation risk. Further improvements to decrease the background tritium contamination during sample preparation and the potential for tritium enrichment of small environmental samples should permit a greater array of environmental tritium applications.
Acknowledgments
Supported by the Center for Accelerator Mass Spectrometry Mini-grant Program and by the Laboratory Directed Research and Development (LDRD) Program (00-ERI-01) at Lawrence Livermore National Laboratory. Additional funding by the National Institute of Environmental Health Sciences, Superfund Basic Research Program, P42 ES04705-15. This work was performed under the auspices of the U.S. Department of Energy by the University of California, Lawrence Livermore National Laboratory under Contract W-7405-Eng-48. Many thanks to Jackie Knealey for collecting samples at the Nevada Test Site.
Literature Cited
- 1.Carter MW, Moghissi AA. Health Phys. 1977;33:55–71. doi: 10.1097/00004032-197707000-00006. [DOI] [PubMed] [Google Scholar]
- 2.Okada S, Momoshima N. Health Phys. 1993;65:595–609. doi: 10.1097/00004032-199312000-00001. [DOI] [PubMed] [Google Scholar]
- 3.Evans MW, Charp P. Public Health Consultation, Tritium Releases and Potential Offsite Exposures at the Lawrence Livermore National Laboratory and the Savannah River Site. Agency for Toxic Substances and Disease Registry; Atlanta, GA: 2001. [Google Scholar]
- 4.Santschi PH, Hoehn E, Lueck A, Farrenkothen K. Environ Sci Technol. 1987;21:909–916. doi: 10.1021/es00170a004. [DOI] [PubMed] [Google Scholar]
- 5.Schimmelmann A, Lewan MD, Wintsch RP. Geochim Cosmochim Acta. 1999;63:3751–3766. [Google Scholar]
- 6.Simonich SL, Hites RA. Environ Sci Technol. 1995;29:2905–2914. doi: 10.1021/es00012a004. [DOI] [PubMed] [Google Scholar]
- 7.Lepp NW. Environ Pollut. 1975;9:49–61. [Google Scholar]
- 8.Kigoshi K, Tomikura Y. B Chem Soc Jpn. 1961;34:1738–1739. [Google Scholar]
- 9.Brown RM. Proceedings Symposium Behavior of Tritium in the Environment; Vienna: International Atomic Energy Agency; 1979. pp. 405–418. [Google Scholar]
- 10.Kozak K, Biro T. Health Phys. 1984;46:193–203. doi: 10.1097/00004032-198401000-00017. [DOI] [PubMed] [Google Scholar]
- 11.Kozak K, Obelic B, Horvatincic N. Radiocarbon. 1989;31:766–770. [Google Scholar]
- 12.Kozak K, Rank D, Biro T, Rajner F, Golder F, Staudner F. J Environ Radioact. 1993;19:67–77. [Google Scholar]
- 13.Yamada Y, Itoh M, Kiriyama N, Komura K, Ueno K. J Radioanal Nucl Chem. 1989;132:59–64. [Google Scholar]
- 14.Fuma S, Inoue Y. Appl Radiat Isot. 1995;46:991–997. [Google Scholar]
- 15.Kalin R, Murphy C, Hall G. Fusion Technol. 1995;28:883–887. [Google Scholar]
- 16.Theodorsson P. Appl Radiat Isot. 1999;50(2):311–316. doi: 10.1016/s0969-8043(97)10153-1. [DOI] [PubMed] [Google Scholar]
- 17.Clarke WB, Jenkins WJ, Top Z. Int J Appl Radiat Isot. 1976;27:515–522. [Google Scholar]
- 18.Surano KA, Hudson GB, Failor RA, Sims JM, Holland RC, MacLean SC, Garrison JC. J Radioanal Nucl Chem. 1992;161:443. [Google Scholar]
- 19.Beyerle U, Aeschbach-Hertig W, Imboden DM, Baur H, Graf T, Kipfer R. Environ Sci Technol. 2000;34(10):2042–2050. [Google Scholar]
- 20.Neary MP. Radioact Radiochem. 1997;8(4):23–35. [Google Scholar]
- 21.Dingley KH, Roberts ML, Velsko CA, Turteltaub KW. Chem Res Toxicol. 1998;11:1217–1222. doi: 10.1021/tx9801458. [DOI] [PubMed] [Google Scholar]
- 22.Muller RA. Science. 1977;196:489–494. doi: 10.1126/science.196.4289.489. [DOI] [PubMed] [Google Scholar]
- 23.Rucklidge J. Analyst. 1995;120:1283–1290. [Google Scholar]
- 24.Hoffman DC, Daniels K, Wolfsberg K, Thompson JL, Rundberg RS, Fraser SL, Daniels KS. Proceedings Radioactive Waste Management. Vol. 5. International Atomic Energy Agency; Vienna: 1984. pp. 241–257. [Google Scholar]
- 25.Hunt JR. Tracers in Hydrology. Proceedings of the Yokohama Symposium; 1993. pp. 135–142. IAHS No. 215. [Google Scholar]
- 26.Wong WW, Klein PD. Mass Spectrom Rev. 1986;5:313–342. [Google Scholar]
- 27.Schimmelmann A. Anal Chem. 1991;63:2456–2459. [Google Scholar]