

Abstract
Rett syndrome is a human neurodevelopmental disorder presenting almost exclusively in female infants; it is the second most common cause of mental retardation in girls, after Down’s syndrome. The identification in 1999 that mutation of the methyl-CpG-binding protein 2 (MECP2) gene on the X chromosome causes Rett syndrome has led to a rapid increase in understanding of the neurobiological basis of the disorder. However, much about the functional role of MeCP2, and the cellular phenotype of both patients with Rett syndrome and mutant Mecp2 mouse models, remains unclear.
Building on prior work in which we demonstrated that cortical layer 2/3 pyramidal neurons (primarily interhemispheric “callosal projection neurons” (CPN)) have reduced dendritic complexity and smaller somata in Mecp2-null mice, here we investigate whether Mecp2 loss-of-function affects neuronal maturation cell-autonomously and/or non-cell-autonomously by creating physical chimeras. We transplanted Mecp2-null or wild-type (wt) E17-18 cortical neuroblasts and immature neurons from mice constitutively expressing enhanced green fluorescent protein (eGFP) into wt P2-3 mouse cortices to generate chimeric cortices. Mecp2-null layer 2/3 pyramidal neurons in both Mecp2-null and wt neonatal cortices exhibit equivalent reduction in dendritic complexity, and are smaller than transplanted wt neurons, independent of recipient environment. These results indicate that the phenotype of Mecp2-null pyramidal neurons results largely from cell-autonomous mechanisms, with additional non-cell-autonomous effects. Dysregulation of MeCP2 target genes in individual neuronal populations such as CPN is likely centrally involved in Rett syndrome pathogenesis. Our results indicating MeCP2 function in the centrally affected projection neuron population of CPN themselves provide a foundation and motivation for identification of transcriptionally regulated MeCP2 target genes in developing CPN.
Keywords: MeCP2, Rett syndrome, Mecp2-null mice, neuronal maturation, physical chimeras, dendritic complexity, callosal projection neurons
Introduction
Rett syndrome is a neurodevelopmental disorder presenting almost exclusively in girls, with a prevalence rate of one in 10,000 to 15,000 female births (Bienvenu and Chelly, 2006; Francke, 2006; Hagberg, 2002; Kishi and Macklis, 2005; Kriaucionis and Bird, 2003; Moretti and Zoghbi, 2006; Rett, 1966). After Down’s syndrome, Rett syndrome is the second most common cause of mental retardation in girls (Ellaway and Christodoulou, 2001). Children with Rett syndrome exhibit mental retardation, autistic features, and a breathing disorder after 6-18 months of relatively normal development. In 1999, it was discovered that loss-of-function mutation of the methyl-CpG-binding protein 2 (MECP2) gene on the X chromosome causes Rett syndrome (Amir et al., 1999). MeCP2 is typically referred to as a transcriptional repressor that selectively binds methyl CpG dinucleotides in the mammalian genome, mediating transcriptional repression, though a recent report describes transcriptional activator activity (Chahrour et al., 2008). It is becoming widely accepted that many transcriptional regulators can function as both repressors and activators, depending on context and co-factors.
Mecp2-deficient mice display several phenotypic characteristics that mimic symptoms of Rett syndrome patients, offering a valuable investigative tool as a murine disease model (Chen et al., 2001; Guy et al., 2001; Lawson-Yuen et al., 2007; Pelka et al., 2006; Shahbazian et al., 2002). These phenotypes are known to be due to the loss of MeCP2 function in the central nervous system (CNS), because specific deletion of Mecp2 in the brain mimics the germline loss of Mecp2 (Chen et al., 2001; Guy et al., 2001). Importantly, recent evidence clearly indicates that gene silencing mechanisms and target genes of MeCP2 are quite different between neural and non-neural cells (Lunyak et al., 2002). MeCP2 expression increases with maturity in the developing cortex, and MeCP2 is broadly expressed in the adult mouse brain. While MeCP2 begins expression as neurons emerge from progenitors, and is strongly expressed in mature neurons (Kishi and Macklis, 2004), recent studies report that MeCP2 is also expressed in astroglia and oligodendroglia at low levels (Ballas et al., 2009; Maezawa et al., 2009). Despite the broad expression of MeCP2 at high levels in neurons, and at lower levels in glia of the CNS, recent results indicate that loss of Mecp2 function has distinct influences on different cell types (Adachi et al., 2009; Fyffe et al., 2008; Kishi and Macklis, 2004). Therefore, in order to understand the pathogenesis of Rett syndrome, it is critical to investigate MeCP2 function specifically in affected populations of CNS neurons. This approach has provided insight into MeCP2 function, including MeCP2 target genes in neurons (Bienvenu and Chelly, 2006), as well as activity-dependent Bdnf regulation by MeCP2 (Chen et al., 2003; Martinowich et al., 2003). However, due to lack of accurate in vitro models, and the subtlety of the in vivo phenotype, it has remained unclear whether the neuronal phenotype in the absence of MeCP2 function is due to lack of Mecp2 in the affected neurons themselves (cell-autonomous mechanisms) and/or due to lack of Mecp2 in surrounding cells (non-cell-autonomous mechanisms).
In the experiments reported here, we investigate whether loss of Mecp2 function affects neuronal maturation largely cell-autonomously or non-cell-autonomously. Previous studies from our lab and others have shown that MeCP2 functions in neocortical neuronal maturation and dendritic development (Fukuda et al., 2005; Kishi and Macklis, 2004). In particular, Golgi staining, which allows visualization of the morphology of individual neurons, showed that Mecp2-null layer 2/3 pyramidal neurons (predominantly interhemispheric CPN) specifically exhibit reduced dendritic complexity and smaller somata than those in wt mice (Kishi and Macklis, 2004). Similar findings have been reported in human postmortem Rett syndrome brains (Armstrong et al., 1995; Bauman et al., 1995; Schule et al., 2008). Appropriate callosal connectivity is thought to underlie associative aspects of higher cognition, and has been centrally implicated in autism spectrum disorders (Egaas et al., 1995). To investigate whether these deficits in CPN development are due to MeCP2 transcriptional dysregulation within the neurons themselves, we perfomed a series of neuronal transplantation experiments using eGFP-labeled wt and Mecp2-null neurons, generating physical chimeras in both wt and Mecp2-null recipients. Our results demonstrate that transplanted Mecp2-null layer 2/3 pyramidal neurons are smaller than and have reduced dendritic complexity compared with transplanted wt neurons in the wt environment, indicating that the mechanisms underlying abnormal dendritic development of CPN in Mecp2-null mice are largely cell-autonomous.
Materials and Methods
Animals
All mouse experimental protocols were approved by the institutional animal care and use committee, and adhere to NIH guidelines. Wild-type C57BL/6 mice were purchased from Charles River Laboratories (Wilmington, MA). GFP-expressing transgenic mice (C57BL/6-Tg(ACTB-EGFP)1Osb/J) (Okabe et al., 1997) were purchased from the Jackson Laboratories (Bar Harbor, ME). This transgenic mouse line with an “enhanced” GFP (eGFP) cDNA under the control of a chicken beta-actin promoter and cytomegalovirus enhancer expresses eGFP in all tissues except erythrocytes and hair (Okabe et al., 1997). Female Mecp2 heterozygous mice were generously provided by the laboratory of Dr. Rudolf Jaenisch (Chen et al., 2001). To obtain Mecp2-null hemizygous male mice (non-eGFP or eGFP), we crossed female heterozygotes with wt male C57BL/6 or male eGFP transgenic mice. For timed gestations, midnight prior to the morning upon which the vaginal plug was observed was defined as day 0. The genotypes of Mecp2 mutant mice were identified by PCR on tail genomic DNA as described previously (Chen et al., 2001; Kishi and Macklis, 2004). The genotypes of GFP transgenic mice were determined using fluorescence excitation.
Transplantation to produce physical chimeras
We crossed female Mecp2 heterozygotes with wt male C57BL/6 or male eGFP transgenic mice and obtained embryonic day 17-18 (E17-18) eGFP-labeled wt or eGFP-labeled Mecp2-null embryos as donor cell sources, and postnatal day 2-3 (P2-3) wt or Mecp2-null neonatal mice as recipients on the same day. On the day of transplantation, eGFP-positive E17-18 embryos were selected by fluorescence excitation, tails were collected for genotyping, and individual cortices were dissected out for donor neuroblast dissociation. Cortices were dissected in cold dissociation medium (20 mM glucose, 0.8 mM kynurenic acid, 0.05 mM DL-2-Amino-5-phosphonopentanoic acid (APV), 50 U/ml penicillin–0.05 mg/ml streptomycin, 0.09 M Na2SO4, 0.03 M K2SO4, and 0.014 M MgCl2), and enzymatically digested in dissociation medium containing 0.16 mg/l L-cysteine HCl and 10 U/ml papain (Worthington, Lakewood, NJ) at 37°C for 15 min, followed by rinsing with iced OptiMem (Life Technologies, Gaithersburg, MD) containing 20 mM glucose, 2.5% fetal bovine serum (Invitrogen, Carlsbad, CA), and both 0.4 mM kynurenic acid and 0.025 mM APV to protect against glutamate-induced neurotoxicity (Catapano et al., 2001). Cortices were mechanically dissociated by gentle trituration to create a single cell suspension. Dissociated cortical cells were resuspended at 5×106 cells/ml. P2-3 wt or Mecp2-null mice were anesthetized by hypothermia, and ~10,000 total dissociated cortical cells were transplanted into the neocortex at 10 sites (~1,000 cells/site) with a digitally controlled oocyte injector (Nanoject II; Drummond Scientific, Broomall, PA) using pulled glass micropipettes with beveled tip diameter of 50-60 μm. Dissociated cells were microinjected in ten “columns” from 300 μm deep to the pial surface, with the 10 transplantation sites spaced evenly throughout the parietal cortex. Following monitored recovery, the pups were returned to their mothers. At 8 weeks of age, the transplanted (physical chimera) mice were perfused, and brains were sectioned for immunocytochemistry.
Histology, immunocytochemistry, and Sholl analysis
Following deep anesthesia with Avertin, brains were fixed by transcardial perfusion with 0.1 M phosphate-buffered saline (PBS) with 10 U/ml heparin, pH 7.4, followed by 4% paraformaldehyde (PFA) in PBS buffer. Brains were postfixed in 4% PFA overnight and sectioned at a thickness of 50 μm with a vibrating microtome (Leica). Vibrating microtome sections were collected into PBS, incubated in blocking solution (PBS, 1% BSA, 3% goat serum, and 0.3% TritonX-100) for 30 min at room temperature, then incubated in primary antibody, chick anti-GFP IgG (1:1,500, Chemicon, Temecula, CA), overnight at 4°C. Sections were washed three times in PBS for 10 min, and incubated in Alexa 488 goat anti-chick IgG antibody (1:750, Invitrogen) for 2 h at room temperature. Sections were mounted on glass slides after washing with PBS and deionized water.
While most of the transplanted neurons remained in the injection sites and extended their dendrites locally, a subpopulation of transplanted neurons migrated away from the injection sites and extended their dendrites, integrating within the recipient brains. To avoid assessing transplanted neurons that remained in the injection sites as heterotopias, only GFP-positive transplanted layer 2/3 pyramidal neurons more than 200 μm from the injection sites were systematically selected for Sholl analysis (Kishi and Macklis, 2004; Sholl, 1953). Images were captured using a fluorescence microscope (Nikon E1000) with high numerical aperture optics and a cooled CCD camera (Q-Imaging) running Openlab software (Improvision, Lexington, MA) to cover entire dendritic arbors. Series of images were reconstructed using Photoshop (Adobe, San Jose, CA). Concentric circles with 20 μm radius increments were superimposed around the center of reconstructed somas, and the number of dendrites crossing each circle was counted. Soma sizes were measured using Openlab software. Statistical analysis was performed using an unpaired one-tailed t-test. All results are presented as the mean ± standard error of the mean (SEM).
Results
Our previous data showed that layer 2/3 pyramidal neurons (predominantly interhemispheric CPN) in Mecp2-null mice have reduced dendritic complexity and smaller somata than those in wt mice (Kishi and Macklis, 2004). These results indicate that Mecp2 is involved in neuronal maturation and/or maintenance of CPN. The specific CPN phenotype indicates that layer 2/3 pyramidal/callosal projection neurons are an ideal target cell population in which to investigate MeCP2 function.
To further investigate whether these abnormalities arise from cell-autonomous mechanisms, we performed neuronal transplantation experiments using eGFP-labeled neurons to generate physical chimeras and analyzed the neuronal dendritic complexity of layer 2/3 pyramidal neurons (Fig. 1). We crossed female Mecp2 heterozygous mice with male eGFP-expressing transgenic mice to obtain Mecp2-null;eGFP+/- E17-18 neuroblasts for transplantation into P2-3 wt cortex, and wt;eGFP+/- neuroblasts for transplantation into Mecp2-null cortex. Transplantation of wt;eGFP+/- neuroblasts into wt cortex and Mecp2-null;eGFP+/- neuroblasts into Mecp2-null cortex served as controls. E17-18 corresponds to the peak of layer 2/3 neuron migration, and early differentiation (Floeter and Jones, 1985), and neonatal P2-3 cortex readily integrates exogenously transplanted neuroblasts and immature neurons into cortical circuitry (Brustle et al., 1998; Gates et al., 2000). Because our previous results demonstrate that reduction in dendritic complexity of Mecp2-null layer 2/3 pyramidal neurons is statistically evident at 8 weeks of age, but not at 2 weeks of age (Kishi and Macklis, 2004), we imaged the extent of dendritic arborization of the GFP-labeled pyramidal neurons in layer 2/3, and reconstructed the entire dendritic morphology at 8 weeks of age (Figs. 2A and B). We found no substantial difference in survival of transplanted neurons among the four different transplantation experiments (WT-to-WT, n=3; KO-to-WT, n=5; WT-to-KO n= 4; KO-to-KO, n= 4). While most of the transplanted neurons remained in the injection sites and formed heterotopias, a subpopulation of transplanted neurons migrated away from the injection sites, and extended their dendrites, integrating within the recipient brains. To avoid assessing transplanted neurons that remained as heterotopias at the injection sites, we selected for analysis only GFP-positive transplanted layer 2/3 pyramidal neurons more than 200 μm from the injection sites. Using the reconstructed images, we analyzed dendritic arborization via Sholl analysis by counting the number of dendrites crossing concentric circles originating from the cell body, and we analyzed soma size by measuring cell body area (Figs. 2C and D).
Fig. 1. Schematic of generation and analysis of physical chimeras; transplantation of GFP-labeled wt or Mecp2-null neuroblasts into wt or Mecp2-null cortex.
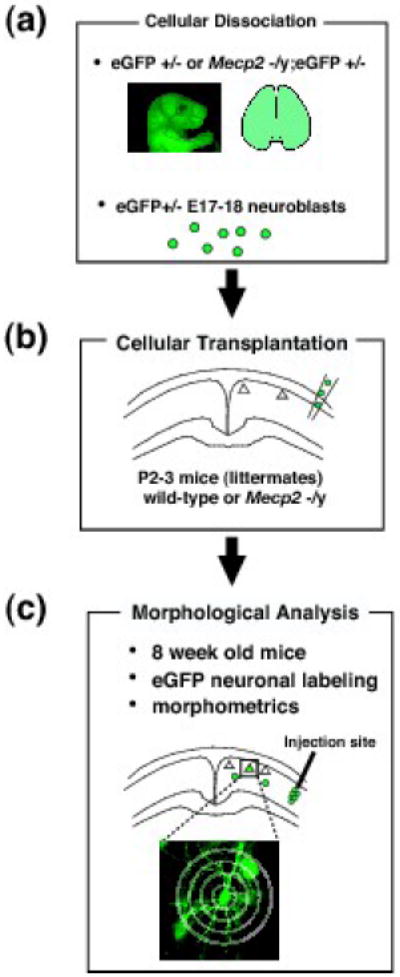
To investigate whether Mecp2-null abnormalities are cell-autonomous, neuronal transplantation experiments using eGFP labeled neurons were performed, generating physical chimeric cortices. (a): Mecp2 heterozygous mice were crossed with eGFP-expressing transgenic mice to generate either Mecp2-null;eGFP+/- or wt;eGFP+/- E17-18 neuroblasts. (b): These eGFP-expressing neuroblasts were transplanted into either wt or Mecp2-null cortex at P2-3, respectively. Wt;eGFP+/- neuroblasts transplanted into wt cortex and Mecp2-null;eGFP+/- neuroblasts into Mecp2-null cortex served as controls. E17-18 is the peak of layer 2/3 neuron birth, migration, and early differentiation. Neonatal P2-3 cortex readily integrates exogenously transplanted neuroblasts and immature neurons into brain circuitry. (c): Neuronal phenotype was examined at 8 weeks, an age when the Mecp2-null phenotype of decreased dendritic complexity in layer 2/3 pyramidal neurons is readily apparent.
Fig. 2. Reduced dendritic complexity of Mecp2-null pyramidal neurons is largely due to cell-autonomous mechanisms.
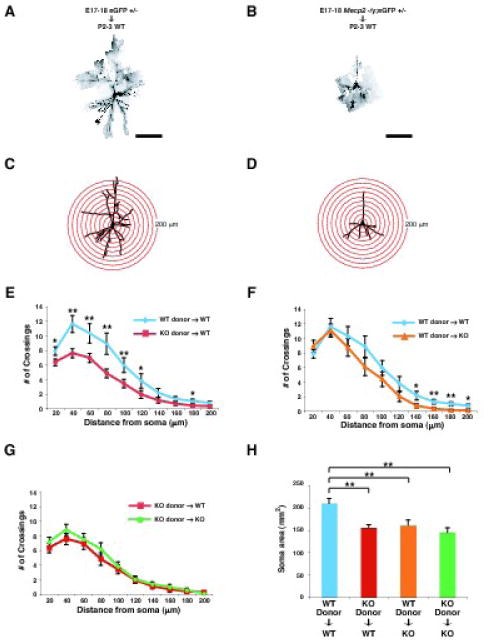
(A - D) Examples of Sholl analysis of transplanted layer 2/3 pyramidal neurons. GFP-labeled E17-18 wt or Mecp2-null neuroblasts were transplanted into P2-3 wt cortex. Eight weeks later, the mice were perfused, and the transplanted GFP-labeled layer 2/3 wt (A) or Mecp2-null (B) pyramidal neurons were immunocytochemically visualized using an anti-GFP antibody. (C and D) Drawings of layer 2/3 pyramidal neurons from A in (C) and from B in (D). Concentric circles in 20 μm radius increments for Sholl analysis are superimposed. (E – G) Dendritic arborization was measured by Sholl analysis of transplanted GFP-labeled layer 2/3 pyramidal neurons. Transplanted Mecp2-null layer 2/3 pyramidal neurons have less complex dendritic arborization (E) in the wt recipient environment than that of transplanted wt neurons in the wt recipient environment (WT neurons, n=14 from 3 mice; KO neurons, n=25 from 5 mice). We also analyzed dendritic arborization (F) of wt GFP-labeled layer 2/3 pyramidal neurons transplanted into either wt or Mecp2-null cortex (WT cortex, n=14 from 3 mice; KO cortex, n=15 from 4 mice), and dendritic arborization (G) of Mecp2-null layer 2/3 pyramidal neurons transplanted into either wt or Mecp2-null environment (WT cortex, n=25 from 5 mice; KO cortex, n=22 from 4 mice). (H) Soma size was measured in transplanted GFP labeled layer 2/3 pyramidal neurons in the four transplantation paradigms (WT-to-WT, n=14 from 3 mice; KO-to-WT, n=25 from 5 mice; WT-to-KO, n=15 from 4 mice; KO-to-KO, n=22 from 4 mice). Scale bars, 100 μm in A and B. Values represent the mean ± standard error of the mean (SEM). *p < 0.05, **p < 0.01, Student’s t-Test. WT; wild type, KO; Mecp2 -/y.
After eGFP-labeled wt or Mecp2-null neuroblasts were transplanted into wt cortex, transplanted Mecp2-null layer 2/3 pyramidal neurons displayed significantly less complex dendritic arborization (Fig. 2E) than transplanted wt neurons (wt, n=14 from 3 mice; Mecp2-null, n=25 from 5 mice). These results indicate that the wt environment does not rescue the phenotype of Mecp2-null pyramidal neurons, and that these deficits result predominantly via cell-autonomous mechanisms.
In contrast, to investigate a potential non-cell-autonomous contribution of Mecp2 function, we analyzed dendritic arborization at 8 weeks of age after transplantation of wt E17-18 eGFP-labeled neuroblasts into either wt or Mecp2-null cortex (Fig. 2F). While there were no significant differences in proximal dendritic complexity (20-120 μm from soma) between wt layer 2/3 pyramidal neurons in wt or Mecp2-null cortex, we found that distal dendritic complexity (120-200 μm from soma) of wt neurons in Mecp2-null cortex is slightly reduced compared with that of wt neurons in wt cortex (wt, n=14 from 3 mice; Mecp2-null, n=15 from 4 mice) (Fig. 2F). These results indicate that the Mecp2-null environment contributes in part to the neuronal phenotype.
To further investigate the relative contributions of cell-autonomous vs. non-cell-autonomous mechanisms in affecting dendritic complexity in Mecp2-null CPN, we compared the dendritic arborization of Mecp2-null layer 2/3 pyramidal neurons in Mecp2-null cortex (n=22 from 4 mice) to that of Mecp2-null neurons in wt cortex (n=25 from 5 mice). We found that there was no additional contribution of the Mecp2-null environment to the phenotype of Mecp2-null neurons (Fig. 2G). These three paired comparisons indicate that lack of Mecp2 in neurons themselves is the predominant cause of the Mecp2 mutant phenotype in cortical layer 2/3 projection neurons, though the Mecp2-null environment also contributes to the phenotype.
In addition to the dendritic phenotype of Mecp2-null pyramidal neurons, we also analyzed soma size of transplanted neurons in the four transplantation paradigms (Fig. 2H). In contrast to the dendritic phenotype, soma size of wt neurons in the Mecp2-null cortex, and of Mecp2-null neurons in wt cortex, was significantly reduced compared to that of wt neurons in wt cortex, and was indistinguishable from that of Mecp2-null neurons in Mecp2-null cortex (WT-to-WT, n=14 from 3 mice; KO-to-WT, n=25 from 5 mice; WT-to-KO, n=15 from 4 mice; KO-to-KO, n=22 from 4 mice). Unlike the dendritic morphology results indicating predominantly cell-autonomous MeCP2 function in dendritic arborization, these data indicate that Mecp2 mutation affects soma size via a combination of cell-autonomous and non-cell-autonomous mechanisms.
To further examine the nature of the reduction in dendritic complexity in Mecp2-null neurons in detail, we analyzed the number of primary and secondary dendrites (Fig. 3), as well as the dendritic complexity of apical and basal dendrites of transplanted layer 2/3 pyramidal neurons (Fig. 4). The dendritic tree of a pyramidal neuron can be broadly subdivided into two distinct classes of dendrites: basal and apical dendrites, which extend from the base and the apex of the soma, respectively (Spruston, 2008). Basal and proximal apical dendrites receive input from layer 4 neurons, and also receive local-circuit excitation, while the apical tufts of layer 2/3 pyramidal neurons receive input from other cortical areas, and also receive non-specific thalamic input. Consistent with and further extending the results above, Mecp2-null neurons in wt cortex have significantly fewer primary and secondary dendrites (Figs. 3A and B), and significantly less complex apical and basal dendrites than transplanted wt neurons in wt cortex (Figs 4A and B). These results indicate a major cell-autonomous component to the observed abnormalities of dendritic complexity in the absence of MeCP2 function. Interestingly, however, dendritic arborization of wt neurons in Mecp2-null cortex is modestly reduced compared with that of wt neurons in wt cortex (Figs. 4C and D), indicating an additional (but more limited) non-cell-autonomous effect on dendritic complexity. We also detected a modest reduction of secondary dendrites (Fig. 3B) and apical dendritic complexity of Mecp2-null neurons transplanted into wt cortex (Fig. 4E), compared with those of Mecp2-null neurons in Mecp2-null cortex, indicating that their presence in a wt environment is detrimental to the dendritic complexity of Mecp2-null neurons under certain conditions. In combination with the observed modest increase in dendritic arborization of wt neurons in Mecp2-null cortex 20 μm from the soma compared with that of wt neurons in wt cortex (Fig. 4C), this finding suggests the possibility that competition between transplanted Mecp2-null neurons and neighboring wt neurons might further detrimentally affect the dendritic morphology of transplanted Mecp2-null neurons. While the substantial (cell-autonomous) reduction in the ability of Mecp2-null transplanted neurons to form and/or mature dendrites might not be a competitive disadvantage when surrounded by similarly affected Mecp2-null neurons in Mecp2-null mice, such cell-autonomous reduction in dendritic complexity might be predicted to be a competitive disadvantage compared to neighboring wt neurons in wt mice.
Fig. 3. Reduction in primary and secondary dendrites of Mecp2-null pyramidal neurons cell-autonomously contributes to the reduced dendritic phenotype.
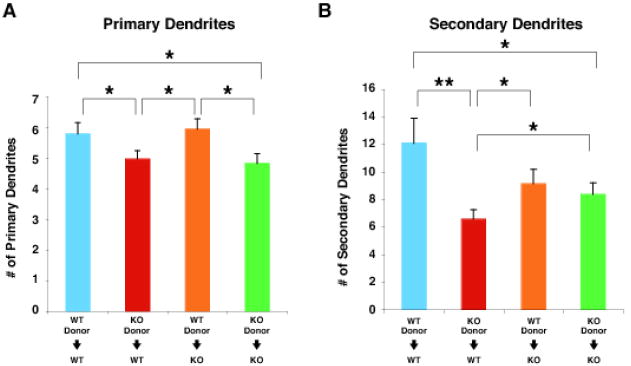
(A and B) The numbers of primary (A) and secondary (B) dendrites per transplanted layer 2/3 neuron were analyzed in the four transplantation paradigms at 8 weeks of age: wt neurons in wt cortex (blue, n=14 from 3 mice); Mecp2-null neurons in wt cortex (red, n=25 from 5 mice); wt neurons in Mecp2-null cortex (orange, n=15 from 4 mice); and Mecp2-null neurons in Mecp2-null cortex (green, n=22 from 4 mice). The numbers of both primary and secondary dendrites of Mecp2-null neurons in wt cortex are significantly reduced compared to those of wt neurons in wt cortex, reinforcing that the phenotype caused by Mecp2 mutations is through predominantly cell-autonomous mechanisms. However, we also found that the number of secondary dendrites of Mecp2-null neurons in wt cortex is modestly less than that of Mecp2-null neurons in Mecp2-null cortex, suggesting the possibility that competition between transplanted Mecp2-null neurons and neighboring wt neurons might further detrimentally affect the dendritic morphology of transplanted Mecp2-null neurons. Values represent the mean ± SEM. *p < 0.05, **p < 0.01, Student’s t-Test. WT; wild type, KO; Mecp2 -/y.
Fig. 4. Mutation of Mecp2 cell-autonomously reduces both apical and basal dendritic complexity.
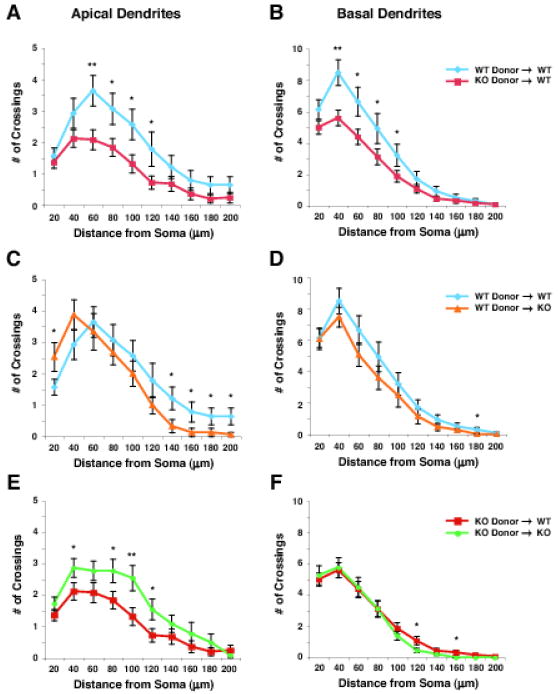
Apical (A, C, and E) and basal (B, D, and F) dendritic arborization of transplanted layer 2/3 neurons were analyzed in the four transplantation paradigms at 8 weeks of age: wt neurons in wt cortex (blue, n=14 from 3 mice); Mecp2-null neurons in wt cortex (red, n=25 from 5 mice); wt neurons in Mecp2-null cortex (orange, n=15 from 4 mice); and Mecp2-null neurons in Mecp2-null cortex (green, n=22 from 4 mice). Consistent with the analysis of global dendritic arborization, mutation of Mecp2 reduced both apical (A) and basal (B) dendritic arborization even in wt cortex, reconfirming that the deficits result largely via cell-autonomous mechanisms. Interestingly, we found that apical dendritic arborization of Mecp2-null neurons in wt cortex is reduced compared with that of Mecp2-null neurons in Mecp2-null cortex (E), again suggesting the possibility that competition between transplanted Mecp2-null neurons and neighboring wt neurons might further detrimentally affect the dendritic morphology of transplanted Mecp2-null neurons. Values represent the mean ± SEM. *p < 0.05, **p < 0.01, Student’s t-Test. WT; wild type, KO; Mecp2 -/y.
Taken together, these results indicate that the striking decrease in CPN dendritic complexity in Mecp2-null cerebral cortex is caused predominantly by cell-autonomous mechanisms, with additional non-cell-autonomous effects, and that loss of Mecp2 function reduces dendritic complexity of both apical and basal dendrites in vivo.
Discussion
These experiments using neuroblast transplantation to produce physical chimeras demonstrate that the decreased dendritic arborization that we previously observed in Mecp2-null callosal projection neurons is largely due to cell-autonomous mechanisms, indicating that Mecp2 acts predominantly cell-autonomously during maturation of these neurons.
Multiple studies using Mecp2 heterozygous female mice are consistent with our results and interpretation (Belichenko et al., 2009; Braunschweig et al., 2004; Young and Zoghbi, 2004). A previous study on X chromosome inactivation patterns in the brain showed that truncated Mecp2 heterozygous mice have a predominant population of Purkinje cells in which the X chromosome bearing the wt allele is the active one (Young and Zoghbi, 2004). The authors confirmed these results using cultured hippocampal neurons, and suggested that the skewed X chromosome inactivation patterns are due to selective survival of neurons carrying the wt Mecp2 allele. The interpretation that wt Mecp2 expression confers survival advantage in neurons is consistent with its effect being largely cell-autonomous. However, another study of female Mecp2 heterozygous mice using laser scanning cytometry suggests that Mecp2-null neurons alter the expression level of MeCP2 in neighboring wt Mecp2-expressing neurons in a non-cell-autonomous manner (Braunschweig et al., 2004). However, this population-based conclusion is uncertain, since each neuron in the brains of Mecp2 heterozygous mice expresses either wt or mutant MeCP2 at random.
In the experiments we report here, we used either wt or Mecp2-null recipient cortex as an environment in which to directly and clearly address the possibility and relative contribution of non-cell-autonomous effects, thus avoiding difficulties associated with Mecp2 heterozygosity. In addition, because we specifically analyzed layer 2/3 pyramidal neurons, which are the most severely affected cell population in the cortex of Mecp2-null mice in vivo, we can interpret the results in a relevant neuronal population, without confounding factors often associated with in vitro systems.
The size and shape of a neuron’s dendritic arbors are influenced by both cell-intrinsic and cell-extrinsic factors (Miller and Kaplan, 2003). It is known that neuronal activity is a key signal for dendrite formation. Neuronal activation by stimuli such as neurotransmitters or neurotrophins (e.g. BDNF) directly or indirectly activates the Ras-MEK-ERK signaling pathway by increasing local Ca2+ concentration, leading to CaMKII activation. Simultaneous activation of CaMKII and ERK enhances MAP2-microtubule interactions and stabilizes dendritic formation. Interestingly, previous studies showed that MeCP2 binds to the promoter region of the Bdnf gene, and MeCP2 dissociates from the promoter upon stimulation, via either phosphorylation of MeCP2 or demethylation of methyl-CpG in the promoter region, indicating a tight relationship between dendrite formation and MeCP2-regulated Bdnf expression (Chen et al., 2003; Martinowich et al., 2003). A recent study showed that neuronal activity and subsequent calcium influx trigger the de novo phosphorylation of MeCP2 at serine 421 by a CaMKII-dependent mechanism (Zhou et al., 2006). These findings further support the existing interpretation that MeCP2 is involved in dendrite and synapse formation and maturation (Asaka et al., 2006; Chao et al., 2007; Fukuda et al., 2005; Kishi and Macklis, 2004; Nelson et al., 2006; Smrt et al., 2007), and the idea that dysregulation of BDNF might be one non-cell-autonomous cause of reduced dendritic complexity in Mecp2-null cerebral cortex.
Very recently, two studies using in vitro co-culture report that MeCP2 is also expressed in astroglia and oligodendroglia at low levels, and that Mecp2-deficient astroglia non-cell-autonomously affect neuronal dendritic growth via aberrant secretion of soluble factors (Ballas et al., 2009; Maezawa et al., 2009). These in vitro results are consistent with the partially non-cell-autonomous effects of Mecp2 observed in our in vivo experiments, suggesting that potentially negative effects of Mecp2-null astroglia and/or other elements might be responsible for the observed reduction in dendritic complexity and soma size of wt neurons in Mecp2-null cortex. While the in vitro studies noted above emphasize non-cell-autonomous MeCP2 function, they do not show evidence of cell-autonomous MeCP2 function. While our in vivo experiments clearly show that integration within a wt environment does not improve the dendritic phenotype of transplanted Mecp2-null neurons, one of the studies noted above reported that conditioned medium from wt astroglia improved the dendritic morphology of Mecp2-null neurons in their co-culture system (Ballas et al., 2009). While physical chimeric analysis by transplantation (widely and successfully used as a developmental biology approach that yields complex, in vivo insights) can directly address MeCP2 function in vivo, in vitro co-culture systems can also potentially offer additional insights into MeCP2 function by eliminating additional complexity present in vivo (e.g. functional compensation). Although in vivo chimeric analysis is likely to mimic real in vivo conditions more closely than in vitro co-culture systems, we think it likely that the transplantation procedures themselves and/or the environment of donor cells before E17 will also affect dendritic maturation later (thus the four distinct and inter-controlled transplantation experiments). Further studies will be useful to further elucidate the relative contributions of cell-autonomous and non-cell-autonomous mechanisms.
Taken together, the data presented here indicate that Mecp2 functions largely cell-autonomously in neuronal maturation and dendritic arborization of the centrally affected projection neuron population of layer 2/3 CPN. These findings provide both foundation and motivation for identification of MeCP2 regulated target genes that might be centrally involved in Rett syndrome pathogenesis. Previous studies have revealed that, although Mecp2 is expressed in all layers of the neocortex (Fukuda et al., 2005; Kishi and Macklis, 2004), layer 2/3 cortical neurons are most severely affected by mutation of Mecp2. Additionally, recent studies reveal that each neuronal subtype has a unique and specific molecular developmental program (Alcamo et al., 2008; Arlotta et al., 2005; Azim et al., 2009a; Azim et al., 2009b; Britanova et al., 2008; Chen et al., 2004; Chen et al., 2005; Joshi et al., 2008; Lai et al., 2008; Molyneaux et al., 2005; Molyneaux et al., 2007; Ozdinler and Macklis, 2006), suggesting the likelihood that loss of Mecp2 function might specifically, subtly, and neuron type-specifically disrupt molecular control of CPN maturation (and, by extension, of other neuron populations). Further investigation can now be pursued to identify new MeCP2 target genes that function cell-autonomously and specifically in the differentiation and maturation of the CPN subtype (in particular, in dendritic development, arborization, and maintenance), in order to further elucidate the cellular and downstream molecular bases and neuropathogenesis of Rett syndrome.
Acknowledgments
We thank Dr. Bartley Mitchell for his critical advice on cellular transplantation, and his helpful comments on this study. We thank Eiman Azim, Mollie Woodworth and Drs. Tina Lai and Jessica MacDonald for critical reading of the manuscript. We also thank Kyle MacQuarrie, Alexander Eswar, Kathryn Quinn, Karen Billmers, and Ashley Palmer for excellent technical assistance. We thank Dr. Rudolf Jaenisch for generously sharing the Mecp2 mutant mice generated in his laboratory. This work was partially supported by fellowships from the Rett Syndrome Research Foundation/International Rett Syndrome Foundation, the Japan Society for the Promotion of Science, the Uehara Memorial Foundation, and the Sumitomo Life Social Welfare Services Foundation to N.K., by grants from the Rett Syndrome Research Foundation and International Rett Syndrome Foundation to JDM, by infrastructure supported by NINDS grants NS41590, NS45523, and NS49553 to JDM, and by the Jane and Lee Seidman Fund for CNS Research, and the Emily and Robert Pearlstein Fund for Nervous System Repair.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adachi M, Autry AE, Covington HE, 3rd, Monteggia LM. MeCP2-mediated transcription repression in the basolateral amygdala may underlie heightened anxiety in a mouse model of Rett syndrome. J Neurosci. 2009;29:4218–4227. doi: 10.1523/JNEUROSCI.4225-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcamo EA, Chirivella L, Dautzenberg M, Dobreva G, Farinas I, Grosschedl R, McConnell SK. Satb2 regulates callosal projection neuron identity in the developing cerebral cortex. Neuron. 2008;57:364–377. doi: 10.1016/j.neuron.2007.12.012. [DOI] [PubMed] [Google Scholar]
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- Arlotta P, Molyneaux BJ, Chen J, Inoue J, Kominami R, Macklis JD. Neuronal subtype-specific genes that control corticospinal motor neuron development in vivo. Neuron. 2005;45:207–221. doi: 10.1016/j.neuron.2004.12.036. [DOI] [PubMed] [Google Scholar]
- Armstrong D, Dunn JK, Antalffy B, Trivedi R. Selective dendritic alterations in the cortex of Rett syndrome. J Neuropathol Exp Neurol. 1995;54:195–201. doi: 10.1097/00005072-199503000-00006. [DOI] [PubMed] [Google Scholar]
- Asaka Y, Jugloff DG, Zhang L, Eubanks JH, Fitzsimonds RM. Hippocampal synaptic plasticity is impaired in the Mecp2-null mouse model of Rett syndrome. Neurobiol Dis. 2006;21:217–227. doi: 10.1016/j.nbd.2005.07.005. [DOI] [PubMed] [Google Scholar]
- Azim E, Jabaudon D, Fame RM, Macklis JD. SOX6 controls dorsal progenitor identity and interneuron diversity during neocortical development. Nat Neurosci. 2009a;12:1238–1247. doi: 10.1038/nn.2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azim E, Shnider SJ, Cederquist GY, Sohur US, Macklis JD. Lmo4 and Clim1 progressively delineate cortical projection neuron subtypes during development. Cereb Cortex. 2009b;19(Suppl 1):i62–69. doi: 10.1093/cercor/bhp030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballas N, Lioy DT, Grunseich C, Mandel G. Non-cell autonomous influence of MeCP2-deficient glia on neuronal dendritic morphology. Nat Neurosci. 2009;12:311–317. doi: 10.1038/nn.2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauman ML, Kemper TL, Arin DM. Pervasive neuroanatomic abnormalities of the brain in three cases of Rett’s syndrome. Neurology. 1995;45:1581–1586. doi: 10.1212/wnl.45.8.1581. [DOI] [PubMed] [Google Scholar]
- Belichenko NP, Belichenko PV, Mobley WC. Evidence for both neuronal cell autonomous and nonautonomous effects of methyl-CpG-binding protein 2 in the cerebral cortex of female mice with Mecp2 mutation. Neurobiol Dis. 2009 doi: 10.1016/j.nbd.2008.12.016. [DOI] [PubMed] [Google Scholar]
- Bienvenu T, Chelly J. Molecular genetics of Rett syndrome: when DNA methylation goes unrecognized. Nat Rev Genet. 2006;7:415–426. doi: 10.1038/nrg1878. [DOI] [PubMed] [Google Scholar]
- Braunschweig D, Simcox T, Samaco RC, LaSalle JM. X-Chromosome inactivation ratios affect wild-type MeCP2 expression within mosaic Rett syndrome and Mecp2-/+ mouse brain. Hum Mol Genet. 2004;13:1275–1286. doi: 10.1093/hmg/ddh142. [DOI] [PubMed] [Google Scholar]
- Britanova O, de Juan Romero C, Cheung A, Kwan KY, Schwark M, Gyorgy A, Vogel T, Akopov S, Mitkovski M, Agoston D, Sestan N, Molnar Z, Tarabykin V. Satb2 is a postmitotic determinant for upper-layer neuron specification in the neocortex. Neuron. 2008;57:378–392. doi: 10.1016/j.neuron.2007.12.028. [DOI] [PubMed] [Google Scholar]
- Brustle O, Choudhary K, Karram K, Huttner A, Murray K, Dubois-Dalcq M, McKay RD. Chimeric brains generated by intraventricular transplantation of fetal human brain cells into embryonic rats. Nat Biotechnol. 1998;16:1040–1044. doi: 10.1038/3481. [DOI] [PubMed] [Google Scholar]
- Catapano LA, Arnold MW, Perez FA, Macklis JD. Specific neurotrophic factors support the survival of cortical projection neurons at distinct stages of development. J Neurosci. 2001;21:8863–8872. doi: 10.1523/JNEUROSCI.21-22-08863.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chahrour M, Jung SY, Shaw C, Zhou X, Wong ST, Qin J, Zoghbi HY. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008;320:1224–1229. doi: 10.1126/science.1153252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao HT, Zoghbi HY, Rosenmund C. MeCP2 controls excitatory synaptic strength by regulating glutamatergic synapse number. Neuron. 2007;56:58–65. doi: 10.1016/j.neuron.2007.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Magavi SS, Macklis JD. Neurogenesis of corticospinal motor neurons extending spinal projections in adult mice. Proc Natl Acad Sci U S A. 2004;101:16357–16362. doi: 10.1073/pnas.0406795101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JG, Rasin MR, Kwan KY, Sestan N. Zfp312 is required for subcortical axonal projections and dendritic morphology of deep-layer pyramidal neurons of the cerebral cortex. Proc Natl Acad Sci U S A. 2005;102:17792–17797. doi: 10.1073/pnas.0509032102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen RZ, Akbarian S, Tudor M, Jaenisch R. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat Genet. 2001;27:327–331. doi: 10.1038/85906. [DOI] [PubMed] [Google Scholar]
- Chen WG, Chang Q, Lin Y, Meissner A, West AE, Griffith EC, Jaenisch R, Greenberg ME. Depression of BDNF Transcription involves calcium-dependent phosphorylation of MECP2. Science. 2003;302:885–889. doi: 10.1126/science.1086446. [DOI] [PubMed] [Google Scholar]
- Egaas B, Courchesne E, Saitoh O. Reduced size of corpus callosum in autism. Arch Neurol. 1995;52:794–801. doi: 10.1001/archneur.1995.00540320070014. [DOI] [PubMed] [Google Scholar]
- Ellaway C, Christodoulou J. Rett syndrome: clinical characteristics and recent genetic advances. Disabil Rehabil. 2001;23:98–106. doi: 10.1080/09638280150504171. [DOI] [PubMed] [Google Scholar]
- Floeter MK, Jones EG. The morphology and phased outgrowth of callosal axons in the fetal rat. Brain Res. 1985;354:7–18. doi: 10.1016/0165-3806(85)90064-1. [DOI] [PubMed] [Google Scholar]
- Francke U. Mechanisms of Disease: neurogenetics of MeCP2 deficiency. Nat Clin Pract Neurol. 2006;2:212–221. doi: 10.1038/ncpneuro0148. [DOI] [PubMed] [Google Scholar]
- Fukuda T, Itoh M, Ichikawa T, Washiyama K, Goto Y. Delayed maturation of neuronal architecture and synaptogenesis in cerebral cortex of Mecp2-deficient mice. J Neuropathol Exp Neurol. 2005;64:537–544. doi: 10.1093/jnen/64.6.537. [DOI] [PubMed] [Google Scholar]
- Fyffe SL, Neul JL, Samaco RC, Chao HT, Ben-Shachar S, Moretti P, McGill BE, Goulding EH, Sullivan E, Tecott LH, Zoghbi HY. Deletion of Mecp2 in Sim1-expressing neurons reveals a critical role for MeCP2 in feeding behavior, aggression, and the response to stress. Neuron. 2008;59:947–958. doi: 10.1016/j.neuron.2008.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gates MA, Tai CC, Macklis JD. Neocortical neurons lacking the protein-tyrosine kinase B receptor display abnormal differentiation and process elongation in vitro and in vivo. Neuroscience. 2000;98:437–447. doi: 10.1016/s0306-4522(00)00106-8. [DOI] [PubMed] [Google Scholar]
- Guy J, Hendrich B, Holmes M, Martin JE, Bird A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat Genet. 2001;27:322–326. doi: 10.1038/85899. [DOI] [PubMed] [Google Scholar]
- Hagberg B. Clinical manifestations and stages of Rett syndrome. Ment Retard Dev Disabil Res Rev. 2002;8:61–65. doi: 10.1002/mrdd.10020. [DOI] [PubMed] [Google Scholar]
- Joshi PS, Molyneaux BJ, Feng L, Xie X, Macklis JD, Gan L. Bhlhb5 regulates the postmitotic acquisition of area identities in layers II-V of the developing neocortex. Neuron. 2008;60:258–272. doi: 10.1016/j.neuron.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishi N, Macklis JD. MECP2 is progressively expressed in post-migratory neurons and is involved in neuronal maturation rather than cell fate decisions. Mol Cell Neurosci. 2004;27:306–321. doi: 10.1016/j.mcn.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Kishi N, Macklis JD. Dissecting MECP2 function in the CNS. J Child Neurol. 2005;20:753–759. doi: 10.1177/08830738050200091001. [DOI] [PubMed] [Google Scholar]
- Kriaucionis S, Bird A. DNA methylation and Rett syndrome. Hum Mol Genet. 2003;12(Suppl 2):R221–227. doi: 10.1093/hmg/ddg286. [DOI] [PubMed] [Google Scholar]
- Lai T, Jabaudon D, Molyneaux BJ, Azim E, Arlotta P, Menezes JR, Macklis JD. SOX5 controls the sequential generation of distinct corticofugal neuron subtypes. Neuron. 2008;57:232–247. doi: 10.1016/j.neuron.2007.12.023. [DOI] [PubMed] [Google Scholar]
- Lawson-Yuen A, Liu D, Han L, Jiang ZI, Tsai GE, Basu AC, Picker J, Feng J, Coyle JT. Ube3a mRNA and protein expression are not decreased in Mecp2R168X mutant mice. Brain Res. 2007;1180:1–6. doi: 10.1016/j.brainres.2007.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunyak VV, Burgess R, Prefontaine GG, Nelson C, Sze SH, Chenoweth J, Schwartz P, Pevzner PA, Glass C, Mandel G, Rosenfeld MG. Corepressor-dependent silencing of chromosomal regions encoding neuronal genes. Science. 2002;298:1747–1752. doi: 10.1126/science.1076469. [DOI] [PubMed] [Google Scholar]
- Maezawa I, Swanberg S, Harvey D, LaSalle JM, Jin LW. Rett syndrome astrocytes are abnormal and spread MeCP2 deficiency through gap junctions. J Neurosci. 2009;29:5051–5061. doi: 10.1523/JNEUROSCI.0324-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinowich K, Hattori D, Wu H, Fouse S, He F, Hu Y, Fan G, Sun YE. DNA-methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science. 2003;302:890–893. doi: 10.1126/science.1090842. [DOI] [PubMed] [Google Scholar]
- Miller FD, Kaplan DR. Signaling mechanisms underlying dendrite formation. Curr Opin Neurobiol. 2003;13:391–398. doi: 10.1016/s0959-4388(03)00072-2. [DOI] [PubMed] [Google Scholar]
- Molyneaux BJ, Arlotta P, Macklis JD. Molecular development of corticospinal motor neuron circuitry. Novartis Found Symp. 2007;288:3–15. doi: 10.1016/j.expneurol.2006.02.074. discussion 15-20, 96-18. [DOI] [PubMed] [Google Scholar]
- Molyneaux BJ, Arlotta P, Hirata T, Hibi M, Macklis JD. Fezl is required for the birth and specification of corticospinal motor neurons. Neuron. 2005;47:817–831. doi: 10.1016/j.neuron.2005.08.030. [DOI] [PubMed] [Google Scholar]
- Moretti P, Zoghbi HY. MeCP2 dysfunction in Rett syndrome and related disorders. Curr Opin Genet Dev. 2006;16:276–281. doi: 10.1016/j.gde.2006.04.009. [DOI] [PubMed] [Google Scholar]
- Nelson ED, Kavalali ET, Monteggia LM. MeCP2-dependent transcriptional repression regulates excitatory neurotransmission. Curr Biol. 2006;16:710–716. doi: 10.1016/j.cub.2006.02.062. [DOI] [PubMed] [Google Scholar]
- Okabe M, Ikawa M, Kominami K, Nakanishi T, Nishimune Y. ‘Green mice’ as a source of ubiquitous green cells. FEBS Lett. 1997;407:313–319. doi: 10.1016/s0014-5793(97)00313-x. [DOI] [PubMed] [Google Scholar]
- Ozdinler PH, Macklis JD. IGF-I specifically enhances axon outgrowth of corticospinal motor neurons. Nat Neurosci. 2006;9:1371–1381. doi: 10.1038/nn1789. [DOI] [PubMed] [Google Scholar]
- Pelka GJ, Watson CM, Radziewic T, Hayward M, Lahooti H, Christodoulou J, Tam PP. Mecp2 deficiency is associated with learning and cognitive deficits and altered gene activity in the hippocampal region of mice. Brain. 2006;129:887–898. doi: 10.1093/brain/awl022. [DOI] [PubMed] [Google Scholar]
- Rett A. Uber ein zerebral-atrophisches Syndrome bei Hyperammonemie. Wien Med Wochenschr. 1966;116:723–726. [PubMed] [Google Scholar]
- Schule B, Armstrong DD, Vogel H, Oviedo A, Francke U. Severe congenital encephalopathy caused by MECP2 null mutations in males: central hypoxia and reduced neuronal dendritic structure. Clin Genet. 2008;74:116–126. doi: 10.1111/j.1399-0004.2008.01005.x. [DOI] [PubMed] [Google Scholar]
- Shahbazian M, Young J, Yuva-Paylor L, Spencer C, Antalffy B, Noebels J, Armstrong D, Paylor R, Zoghbi H. Mice with truncated MeCP2 recapitulate many Rett syndrome features and display hyperacetylation of histone H3. Neuron. 2002;35:243–254. doi: 10.1016/s0896-6273(02)00768-7. [DOI] [PubMed] [Google Scholar]
- Sholl DA. Dendritic organization in the neurons of the visual and motor cortices of the cat. J Anat. 1953;87:387–406. [PMC free article] [PubMed] [Google Scholar]
- Smrt RD, Eaves-Egenes J, Barkho BZ, Santistevan NJ, Zhao C, Aimone JB, Gage FH, Zhao X. Mecp2 deficiency leads to delayed maturation and altered gene expression in hippocampal neurons. Neurobiol Dis. 2007;27:77–89. doi: 10.1016/j.nbd.2007.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spruston N. Pyramidal neurons: dendritic structure and synaptic integration. Nat Rev Neurosci. 2008;9:206–221. doi: 10.1038/nrn2286. [DOI] [PubMed] [Google Scholar]
- Young JI, Zoghbi HY. X-chromosome inactivation patterns are unbalanced and affect the phenotypic outcome in a mouse model of rett syndrome. Am J Hum Genet. 2004;74:511–520. doi: 10.1086/382228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Hong EJ, Cohen S, Zhao WN, Ho HY, Schmidt L, Chen WG, Lin Y, Savner E, Griffith EC, Hu L, Steen JA, Weitz CJ, Greenberg ME. Brain-Specific Phosphorylation of MeCP2 Regulates Activity-Dependent Bdnf Transcription, Dendritic Growth, and Spine Maturation. Neuron. 2006;52:255–269. doi: 10.1016/j.neuron.2006.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]