

Abstract
Synaptic vesicle fusion in brain synapses occurs in phases that are either tightly coupled to action potentials (synchronous), immediately following action potentials (asynchronous) or as stochastic events in the absence of action potentials (spontaneous). Synaptotagmin-1, -2 and -9 are vesicle-associated Ca2+-sensors for synchronous release. Here we found that Double C2 domain (Doc2) proteins act as Ca2+-sensors to trigger spontaneous release. Although Doc2 proteins are cytosolic, they function analogously to synaptotagmin-1 but with a higher Ca2+-sensitivity and superior in vitro fusion-efficiency. Doc2 proteins bound to SNARE-complexes in competition with synaptotagmin-1. Thus, different classes of multiple C2 domain-containing molecules trigger synchronous versus spontaneous fusion, which suggests a general mechanism for synaptic vesicle fusion triggered by the combined actions of SNAREs and multiple C2 domain-containing proteins.
Neurotransmitter release is triggered by a rise in intracellular Ca2+, which activates sensors that subsequently trigger vesicle fusion. Synchronous release, the fastest mode of neurotransmission, involves the Ca2+ sensors synaptotagmin-1, -2 or -9 which are anchored in the vesicle membrane and contain two cytoplasmic C2 domains that bind phospholipids in a Ca2+-dependent manner and interact with the soluble N-ethylmaleimide-sensitive factor attachment receptor (SNARE) complex (1-5). Synaptotagmin-1-deficient neurons lack synchronous release but display an increase in spontaneous release (6-9) except in autapses (1, 10), suggesting a distinct mechanism for spontaneous release. Spontaneous release occurs in the absence of action potentials and is largely Ca2+-dependent (12-16), although truly Ca2+-independent fusion may also exist (11).
Doc2a and Doc2b are soluble proteins that contain C2 domains with high similarity to synaptotagmins (17). They are expressed in nerve terminals and interact with the secretory molecules Munc18, Munc13 and the SNARE proteins syntaxin-1 and SNAP25 (18, 19). Overexpression of Doc2b enhances exocytosis in chromaffin cells (18), pancreatic beta cells (20, 21) and adipocytes (22) but its function in neurons is elusive.
Role of Doc2b and Ca2+ in spontaneous synaptic release
We generated Doc2b−/− mice by deleting the promoter and exon 1 of the Doc2b gene (fig. S1) (23). Doc2b−/− mice did not express the remaining exons and lacked Doc2b immunoreactivity. Doc2b−/− mice were viable and fertile without gross abnormalities. Other proteins implicated in neurotransmitter secretion were expressed at normal levels (fig. S1D). Compensatory ectopic expression of Doc2a was not detected in Doc2b-deficient brains by in situ hybridization and the Doc2a protein level was unchanged (fig. S1). Doc2a−/− Doc2b−/− double knock-out (DKO) mice were also viable, fertile and indistinguishable with regard to gross anatomy. To study neurotransmission and synaptic plasticity in Doc2 mutants, hippocampal neurons were cultured on glial microislands to promote self-innervation (autapses). Because Doc2a and Doc2b are both expressed in the hippocampus (17), DKO mice were compared to wild-type controls. All aspects of evoked release were normal, with excitatory postsynaptic currents (EPSC) of normal amplitude and shape (Fig. 1A), a normal synaptic depression during prolonged stimulation at 5 or 40 Hz (Fig. 1B and fig. S2A,B), a normal contribution to the total postsynaptic charge transfer (24) of asynchronous release (Fig. 1B) and a normal size of the readily releasable pool (RRP, fig. S2D).
Fig. 1.

Impaired spontaneous neurotransmission in Doc2a/b-deficient mice. Hippocampal neurons were cultured on microislands to promote self-innervation. (A) Averaged evoked excitatory postsynaptic currents (EPSCs) in control and DKO neurons. (B) Evoked EPSC charge (mean ± sem) during repeated stimulation at 5 or 40 Hz. The synchronous and asynchronous component were estimated as previously reported (24). (C) Representative traces of spontaneous EPSCs in wild-type or DKO cells. To rescue the phenotype, GFP or Doc2b were acutely expressed in DKO cells. (D) Average spontaneous release frequency in control and DKO cells and rescue by Doc2b overexpression. Cell numbers are indicated between brackets. **, P<0.01 (E) Average mEPSC frequency before and after intracellular loading of the Ca2+ chelator BAPTA. The average spontaneous release rate varied between experiments, but our experimental design prevents confounding effects thereof (fig. S2E) (F) Example trace representative for the data in (E), taken from a DKO cell before and after BAPTA loading. (G) Typical recordings before and after repetitive stimulation at 5 Hz. (H) Individual release events were binned in 1 s time intervals to monitor their average frequency immediately after repetitive stimulation. Squares were calculated as 50% of the frequency in control cells.
However, DKO mice exhibited a twofold reduction in the spontaneous release frequency (6.2 Hz in wild-type to 3.3 Hz in DKO; Fig. 1C,D). Acute expression of Doc2b in DKO neurons restored this phenotype. Doc2b immunoreactivity was confirmed in all cellular compartments including synapses, before the start of recordings (fig. S3). As a control, acute expression of green fluorescent protein (GFP) in DKO cells did not affect the spontaneous release frequency (3.1 Hz, Table S1). A normal organization of the postsynaptic apparatus was indicated by a normal mEPSC amplitude, rise time and decay time in DKO neurons (fig. S2F) or after overexpression (fig. S3B).
Because Doc2 proteins require Ca2+ for their phospholipid-binding and secretion enhancing activity (18, 25), we investigated if spontaneous release also depended on Ca2+. The spontaneous release frequency was measured in the same cells before and after loading them with the Ca2+-chelator BAPTA. This significantly reduced the frequency to 2.0 Hz (p<0.0001) in wild-type cells and to 1.4 Hz in DKO cells (p<0.0001, Fig. 1E,F). The frequency did not decrease further upon longer incubation with BAPTA-AM (fig. S2H). Thus, as expected (13-16), spontaneous events are often triggered by short-lived Ca2+ transients in resting cells.
Consistent with the role of Ca2+ in spontaneous release, the frequency of individual release events markedly increased after intense activity (5 Hz for 20 s; Fig. 1G,H) and decayed after the last stimulus with an exponential halftime of 11 s. Similar observations have been made in the neuromuscular junction and calyx of Held (26, 27). In DKO cells after intense stimulation the number of observed events was reduced to 50% of wildtype, except for the first time point that was sampled 200-1200 ms after the last depolarizing stimulus, where asynchronous release may still contribute (Fig. 1H). The Doc2-independent events remaining in DKO cells were also affected by BAPTA and repetitive firing, suggesting that additional Ca2+-dependent mechanisms exist besides Doc2a/b-dependent events. Thus Doc2 proteins responding to intracellular Ca2+ are required for approximately half of the spontaneous release events in hippocampal neurons.
Disinhibition in Purkinje cells lacking Doc2b
Doc2b mRNA was abundant in Purkinje cells (PCs) of the cerebellum (Fig. 2A) while Doc2a was not detectable. Doc2b expression was exclusive to the PC layer with no detectable mRNA in other cerebellar cells including interneurons. PCs synapse onto neighboring PCs via recurrent axon collaterals (28). We performed whole cell voltage-clamp recordings at postnatal days 7-8 because at this time, recurrent synapses are the predominant source of GABAergic input while stellate and basket cells are still functionally immature (29). In the presence of 6,7-dinitroquinoxaline-2,3-dione (DNQX) to block AMPA receptors and tetrodotoxin (TTX) to block sodium currents, the PCs had a stable resting potential without NMDA receptor currents or Ca2+-spikes. Under these conditions, any remaining inhibitory postsynaptic currents (IPSCs) can be interpreted as spontaneous release events.
Fig. 2.
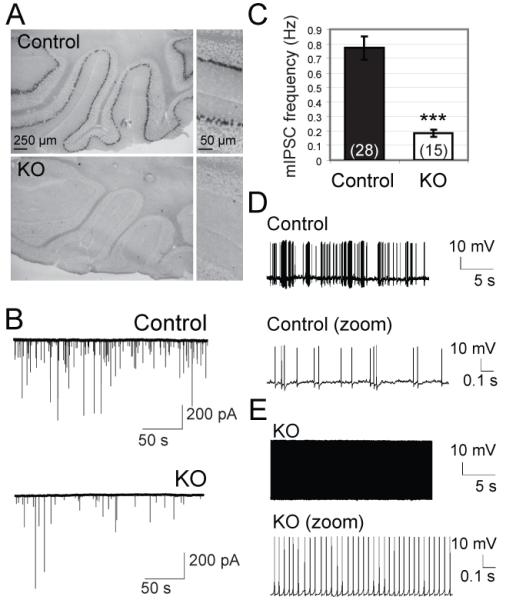
Reduced frequency of spontaneous events in Purkinje cells lacking Doc2b. (A) Doc2b mRNA was detected by in situ hybridization in cerebellar Purkinje cells from wildtype, but not Doc2b−/− (KO) mice. (B) Typical Voltage-clamp recordings in acute slices (C) Mean frequency of spontaneous inhibitory postsynaptic currents (mIPSCs) in KO mice and age-matched control littermates (n=15 and 28 cells; N=2 and 3 mice respectively). ***, p<0.0001. (D-E) Current clamp recordings of Purkinje cell firing patterns in controls (D, 8 cells from 3 mice) and KO cells (E, 7 cells from 2 mice).
The frequency of spontaneous IPSCs was reduced fourfold in Doc2b−/− mice compared to control littermates (Fig. 2B,C). Postsynaptic parameters were normal (i.e. amplitude, rise time and decay). In young rodents, GABAergic input inhibits PC firing (29) and recurrent PC-PC synapses are the major GABA source (30). We thus tested if the disinhibition in Doc2b−/− PC-PC synapses affected PC spiking. Whole cell current clamp recordings were performed at postnatal day P17 in the absence of DNQX or TTX. We observed various firing patterns in PCs from wild-type or heterozygous mice: irregular trains of simple spikes in most cells (Fig. 2D) or trimodal firing patterns comprised of tonic, burst and silent periods, as expected (30, 31). In contrast, PCs from Doc2b−/− mice showed continuous spiking without interruptions (Fig. 2E). This pattern occurred with a frequency of 20-25 Hz and was observed in all Doc2b−/− cells tested, but never in control cells.
We also investigated neurotransmission in the calyx of Held synapse, a giant glutamatergic synapse in the auditory brainstem where the Doc2-Munc13 interaction is suggested to contribute to presynaptic plasticity (32). DKO mice exhibited normal evoked responses with a 2-fold lower spontaneous release frequency (1.9 Hz compared to 4.8 Hz in control littermates), but this difference did not reach statistical significance (fig. S4).
Phospholipid- and SNARE interactions
Doc2b is structurally similar to synaptotagmin-1 but lacks a transmembrane domain and its C2 domains have a relatively high Ca2+ affinity (25). It is thus conceivable that Doc2b mediates spontaneous release events similar to synaptotagmin-dependent synchronous release, only with slower kinetics and a higher Ca2+-sensitivity. We thus tested if Doc2b is biochemically similar to synaptotagmin-1.
Doc2b C2A domain bound in a Ca2+-dependent manner to phosphatidylserine-containing membranes (Fig. 3A) (33, 34). In addition, the C2B domain showed weak Ca2+-dependent phospholipid binding (Fig. 3A). The C2AB fragment of Doc2b showed stronger liposome binding than the isolated C2A and C2B domains (Fig. 3A). Ca2+-dependent membrane binding by synaptotagmin-1 was enhanced in the presence of phosphatidyinositol(4,5)bisphosphate (PIP2) (35). For the C2A and C2B domains of Doc2b the inclusion of PIP2 enhanced both Ca2+-dependent and -independent binding (Fig. 3B). This Ca2+-independent binding was localized to two polybasic patches on the C2A and C2B domains, respectively (figs. S5 and S6). The use of brain-derived Folch lipids (36) generally increased binding, including the Ca2+-independent binding by the C2A domain (Fig. 3C, figs. S5 and S6). The C2AB domains of Doc2a and synaptotagmin-1 displayed similar behavior (Fig. 3D).
Fig. 3.

Membrane-binding and curvature induction by Doc2b. (A-D) Liposome cosedimentation assays to characterize the membrane binding properties of the Doc2b C2A, C2B, C2AB, Doc2a C2AB and synaptotagmin-1 (Syt1) C2AB domains. Liposomes composed of the indicated lipids were incubated with the Doc2b C2A, C2B or C2AB domains. Liposome-bound protein was co-sedimented by ultracentrifugation. 8% of the supernatant and pellet fraction were run on 4-12% gradient gels. Proteins were visualized by Coomassie staining. S indicates unbound protein in the supernatant while P indicates liposome-bound and thus co-pelleted protein. No or very little protein sedimented in the absence of liposomes. (E) Ca2+- dependent tubulation of liposomes by the Doc2b C2AB domain. Folch liposomes were incubated in the absence or presence of Doc2b C2AB and/or Ca2+ and processed for electron microscopy by negative stain. The arrows indicate bundles of closely aligned tubules. Scale bar 100 nm. All data shown are representatives of at least 3 independent experiments. (PS: phosphatidylserine, PC: phosphatidylcholine, PIP2: phosphatidylinositol(4,5)bisphosphate)
The C2AB domain of synaptotagmin-1, -3 and -5 induce a high degree of positive membrane curvature in a Ca2+-dependent manner (3), due to the insertion of the tips of both C2 domains into the hydrophobic phase of the membrane. Shallow insertions into a monolayer are known to induce bending of the membrane towards the insertion (37-39). In order to examine if Doc2b also induces membrane curvature, we incubated liposomes derived from Folch lipids with 10 μM Doc2b C2AB domain in the absence (EDTA) or presence of Ca2+ (Fig. 3E). Indeed, the Doc2b C2AB domain efficiently induced tubulation of otherwise spherical liposomes in a strictly Ca2+-dependent manner (Fig. 3E). We frequently observed closely aligned tubules indicating that the Doc2b C2AB domain could cluster liposomes in addition to their tubulation. Tubulation was still apparent at a Doc2b concentration of 5 μM but was barely detectable below 1 μM.
Synaptotagmin-1 requires SNARE complex binding to promote fusion in vitro and in vivo (4, 5). We thus tested if Doc2b also bound SNARE complexes. In one set of experiments we assembled the SNARE core complex on glutathione beads using the synaptobrevin SNARE domain fused to GST (fig. S7). The Doc2b and synaptotagmin-1 C2AB domains were subsequently added in the presence or absence of Ca2+. In another set of experiments we used the GST-Doc2b C2AB domain to pull down the soluble SNARE complex (Fig. 4A). As expected (40), we found efficient Ca2+-dependent binding of the synaptotagmin-1 C2AB domain to SNARE complexes (fig. S7). The Doc2b C2AB domain also bound to SNARE complexes (Fig. 4A and fig. S7). In the C2B domain of synaptotagmin-1 the same poly-lysine motif that binds PIP2 mediates SNARE complex binding (35). We thus tested SNARE complex binding by the mutant C2ABK237,319E domain of Doc2b. As expected, it showed a profound loss of SNARE complex binding (Fig. 4A and fig. S7). Next we tested if the putative membrane inserting hydrophobic amino acids in the tips of the Ca2+-binding loops (Fig. 4A, fig. S7) contributed to SNARE complex binding. To this end we mutated H158, F222 and I360A. The first Ca2+-binding loop of the Doc2b C2B domain contains an alanine and so was not mutated. We refer to this mutant as the 4A mutant. It showed no defect in SNARE complex binding indicating that, as for synaptotagmin-1, SNARE complex and membrane binding can occur at the same time (Fig. 4A and fig. S7). In contrast, Ca2+-dependent liposome binding was only affected by the 4A but not the K237, 319E mutation (Fig. 4C). The decreased binding of the 4A mutant was less effective if the liposomes were made with higher percentages of phosphatidylserine.
Fig. 4.

SNARE complex binding and promotion of SNARE-dependent membrane fusion by Doc2b. (A) Coomassie stained gels showing a pull down experiment in the presence or absence of Ca2+ using the indicated Doc2b fusion proteins as bait and the purified SNARE complex as prey. The co-pelleted SNARE complex is indicated by an arrow. (B) Coomassie stained gels showing a pull down in the presence of Ca2+ using the GST-SNARE complex as bait and the Doc2b and synaptotagmin-1 C2AB domains as prey. GST served as control for unspecific binding by the C2AB domains of Doc2B and synaptotagmin-1, respectively. The co-pelleted Doc2B and synaptotagmin-1 C2AB domains are indicated by arrows. (C) Liposome co-sedimentation assay in the presence or absence of Ca2+ using the indicated proteins and liposomes composed of 20% PS, 70% PC and 10% cholesterol. (D) In vitro membrane fusion assay using reconstituted full length SNAREs in the absence or presence of Ca2+ and/or 7.5 μM Doc2b C2AB. (E) In vitro membrane fusion assay as in (D) but in the presence or absence of the soluble SNARE domain of synaptobrevin. Ca2+ was added at 500 s. (F) Ca2+ dose-dependence curve showing the change of fluorescence at 120 s in the reconstituted fusion assay in the presence of 7.5 μM Doc2b. Doc2b efficiently promotes fusion at sub-μM Ca2+ concentrations. The graph summarizes data from 3 independent experiments. (G) Comparison of the fusion promoting activities of the Doc2b, Doc2a and synaptotagmin-1 C2AB domains. (H) Fusion experiment using the indicated Doc2b C2AB proteins reveals that Ca2+-dependent membrane and SNARE-binding are required for efficient fusion promotion. All data shown are representatives of at least 3 independent experiments.
The high structural and biochemical similarities between Doc2b and synaptotagmin-1 suggest that the binding site on the SNARE complex may be identical. We thus tested if the C2AB domain of synaptotagmin-1 competes with the C2AB domain of Doc2b for SNARE complex binding (Fig. 4B). Indeed, increasing amounts of the synaptotagmin-1 C2AB domain competed off the Doc2b C2AB domain suggesting that the binding sites for synaptotagmin-1 and DOC2B are overlapping.
Ca2+-dependent promotion of membrane fusion by Doc2b
The C2AB domains of several members of the synaptotagmin family promote fusion of liposomes containing reconstituted SNAREs in a Ca2+-dependent manner (41). Similarly, the C2AB domain of Doc2b promoted fusion of SNARE-containing liposomes and this activity was strictly Ca2+-dependent (Fig. 4D-H). When Ca2+ was introduced at a later time point the fusion promoting effect was even more apparent (Fig. 4E). The fusogenic activity of Doc2b was SNARE-dependent (Fig. 4E). If Doc2b acts as a Ca2+ sensor for spontaneous synaptic vesicle fusion it is likely to be activated by intracellular Ca2+ in the sub-μM range (15). Consistent with this, Doc2b bound to membranes with a sub-μM affinity (fig. S8) and at sub-μM Ca2+ Doc2b promoted fusion (Fig. 4F). The in vitro fusion-promoting effect of Doc2b far exceeded that of synaptotagmin-1 and Doc2a (Fig. 4G). Next we tested if Ca2+-dependent membrane binding and SNARE binding were required for the strong fusion promoting effect of Doc2b. Indeed, both mutant Doc2b4A impaired in Ca2+-dependent phospholipid binding (fig. S8) and mutant Doc2bK237, 319E impaired in SNARE binding (Fig. 4A) were less effective (Fig. 4H).
Ca2+-dependence of Doc2b-driven spontaneous release
If Doc2b generates spontaneous events by acting as a Ca2+ sensor, then mutagenesis of its Ca2+-binding site should affect the Ca2+-dependence of spontaneous release. We thus expressed wildtype or mutant Doc2b in DKO neurons in network cultures (10, 12). The Ca2+-dependence of spontaneous release was assessed by superfusing neurons with increasing extracellular Ca2+ concentrations (from 0.2 mM to 10 mM; Fig. 5A). As expected, the spontaneous release rate increased with increasing Ca2+ concentrations (Fig. 5B) (12). Mutant Doc2bD218,220N, which mimics a dominant active Ca2+-bound state of the C2A domain (18), caused a massive increase in spontaneous release rate, especially at low Ca2+-concentrations (Fig. 5B-C). The Ca2+- dependence of spontaneous release was almost completely lost in Doc2bD218,220N expressing cells. This correlated well with the Ca2+-independent binding of the isolated C2AD218,220N domain to liposomes (Fig. 5E and fig. S8). DKO neurons expressing wildtype Doc2b had a higher spontaneous release rate at each Ca2+ concentration than DKO neurons expressing GFP (Fig. 5B). Even in cells lacking Doc2 we observed more spontaneous release events at higher Ca2+ concentrations, albeit less pronounced than in wildtype or rescued cells. Thus, as seen before (Fig 1), additional high affinity Ca2+-sensors for spontaneous release may exist. At high extracellular Ca2+-concentrations (5-10 mM) the spontaneous release rate increased in all groups, a release which may be supported by synaptotagmins (12). We also expressed Doc2bWT and Doc2bD218,220N in neurons from wildtype mice. Consistent with the data from DKO neurons, expression of each construct increased the spontaneous release rate. This was most pronounced in Doc2bD218,220N-expressing cells (Fig. 5C). Conversely, overexpression of a loss of function mutant, Doc2bK237,319E, caused significantly lower mEPSC frequencies at all Ca2+ concentrations compared to Doc2bWT in DKO neurons (Fig. 5D). Because PIP2 binds to the same region as SNAREs (17, 20), we cannot exclude a contribution of PIP2 binding in this experiment. However our in vitro fusion assay was performed in absence of PIP2 so here, the profound loss of Doc2bK237,319E function was almost certainly due to its loss of Ca2+-dependent SNARE-binding (Fig. 4). Thus, manipulation of the Ca2+-dependence of Doc2b affected the Ca2+-dependence of spontaneous release, implicating Doc2b as a Ca2+ sensor.
Fig. 5.

The Ca2+-dependence of Doc2b determines that of spontaneous release in rescued DKO neurons. (A) Typical mEPSC recordings in DKO cells rescued with GFP (control), Doc2bWT or Doc2bD218,220N. Spontaneous mEPSCs were recorded in network cultures of hippocampal neurons in the presence of TTX and gabazine. Increasing concentrations of extracellular Ca2+ were administered to the same cell. (B) Mean mEPSC frequencies ± sem in DKO cells expressing Doc2bWT (n=10), Doc2bD218,220N (n=26) or GFP as a control (n=21) (C) Mean mEPSC frequencies of the same constructs expressed in wildtype cells. The average mEPSC frequency varied between experiments, but our experimental design prevents confounding effects thereof (see Fig S2E) (D) Mean mEPSC frequencies in DKO cells expressing Doc2bWT (n=35) or mutant Doc2bK237,319E (n=31) (E) Ca2+-dependent liposome-binding by the isolated C2A domain of Doc2bWT and Doc2bD218,220N (n=6). *** (p<0.005); ** (p<0.01), * (p<0.05).
Conclusions
Here we have found that Doc2b is a high affinity Ca2+-sensor for half (hippocampus) or most (cerebellum) of the spontaneous neurotransmitter release events in CNS synapses. Doc2b acts as a functional synaptotagmin-1 analogue operating at smaller Ca2+ increases and with higher in vitro fusion efficiency. In contrast to synaptotagmin-1 that has the appropriate affinity to respond to high Ca2+ peaks occurring at release sites, Doc2b requires less than 1 μM Ca2+ to enhance fusion. Doc2b is not associated with the synaptic vesicle via a transmembrane domain like synaptotagmins and translocates to membranes in response to Ca2+ (25), a quality that would explain why evoked release was unaffected in our study. Nevertheless, it is likely that Doc2 proteins are located not far from release sites due to its interactions with SNARE-complexes (Fig 4A), Munc13 (42) and Munc18 (17). Given the sensitivity of Doc2-dependent spontaneous release to BAPTA-chelation (Fig 1E) the protein must be activated close to a source of Ca2+, whether this be a Ca2+-channel or an intracellular Ca2+-store (13-16).
Spontaneous release events appear to be mechanistically heterogeneous. In the synapses studied here, most spontaneous events are Ca2+-dependent and require Doc2. Because the spontaneous release in the absence of Doc2 remained sensitive to BAPTA and prolonged neuronal firing at 5 Hz, one or more additional high-affinity Ca2+- sensors probably exist. Finally, BAPTA did not block all spontaneous release events and thus, some events are probably truly Ca2+-independent, which may be supported by factors such as synaptotagmin-12 (11).
Because multiple C2 domain-containing proteins are co-expressed in synapses and at least Doc2b and synaptotagmin-1 compete for SNARE complex binding, a certain degree of redundancy probably exists among these protein families. The concentrations of various C2-domain proteins and their Ca2+-affinity will determine the Ca2+-sensitivity of release and the release probability of vesicles. Differences in this balance may explain the large differences in spontaneous release frequencies observed among neurons. Along the same lines, it is conceivable that the increased frequency of spontaneous release in synaptotagmin-1 deficient mice is triggered by Doc2 or other C2 domain-containing proteins (40, 43, 44) that replace synaptotagmin- 1 in its absence, where effectively synaptotagmin would appear to clamp fusion (12). Finally, while spontaneous vesicle fusion could conceivably have been dependent on SNARE assembly alone, we now show that many of these events require the action of curvature inducing multiple C2 domain proteins.
One-sentence summary
Doc2 proteins are functional analogs of synaptotagmins and act by membrane curvature induction to trigger spontaneous neurotransmitter release upon small Ca2+ increases.
Supplementary Material
Acknowledgments
We thank R.Zalm, D. Schut, I. Saarloos, B. de Vries, C. van der Meer, and M. Schindler for technical assistance. H. Mansvelder and H. Lodder are acknowledged for expert advice/help in neurophysiology. This work was supported by the EU (EUSynapse project 019055 to A.G. and M.V.), the Medical Research Council, U.K. to H.M.M. and S.M., an EMBO Long-Term Fellowship to S.M. and the NeuroBsik Mouse Phenomics Consortium BSIK03053.
Footnotes
The authors declare no conflicting interests.
REFERENCES AND NOTES
- 1.Geppert M, et al. Cell. 1994;79:717. doi: 10.1016/0092-8674(94)90556-8. [DOI] [PubMed] [Google Scholar]
- 2.Fernandez-Chacon R, et al. Nature. 2001;410:41. doi: 10.1038/35065004. [DOI] [PubMed] [Google Scholar]
- 3.Martens S, Kozlov MM, McMahon HT. Science. 2007;316:1205. doi: 10.1126/science.1142614. [DOI] [PubMed] [Google Scholar]
- 4.Lynch KL, et al. Mol Biol Cell. 2008 [Google Scholar]
- 5.Pang ZP, Shin OH, Meyer AC, Rosenmund C, Sudhof TC. J Neurosci. 2006;26:12556. doi: 10.1523/JNEUROSCI.3804-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Littleton JT, Stern M, Schulze K, Perin M, Bellen HJ. Cell. 1993;74:1125. doi: 10.1016/0092-8674(93)90733-7. [DOI] [PubMed] [Google Scholar]
- 7.DiAntonio A, Schwarz TL. Neuron. 1994;12:909. doi: 10.1016/0896-6273(94)90342-5. [DOI] [PubMed] [Google Scholar]
- 8.Sun J, et al. Nature. 2007;450:676. doi: 10.1038/nature06308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu J, Mashimo T, Sudhof TC. Neuron. 2007;54:567. doi: 10.1016/j.neuron.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 10.Liu H, Dean C, Arthur CP, Dong M, Chapman ER. J Neurosci. 2009;29:7395. doi: 10.1523/JNEUROSCI.1341-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maximov A, Shin OH, Liu X, Sudhof TC. J Cell Biol. 2007;176:113. doi: 10.1083/jcb.200607021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu J, Pang ZP, Shin OH, Sudhof TC. Nat Neurosci. 2009 [Google Scholar]
- 13.Collin T, Marty A, Llano I. Curr Opin Neurobiol. 2005;15:275. doi: 10.1016/j.conb.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 14.Emptage NJ, Reid CA, Fine A. Neuron. 2001;29:197. doi: 10.1016/s0896-6273(01)00190-8. [DOI] [PubMed] [Google Scholar]
- 15.Llano I, et al. Nat Neurosci. 2000;3:1256. doi: 10.1038/81781. [DOI] [PubMed] [Google Scholar]
- 16.De Crescenzo V, et al. J Neurosci. 2004;24:1226. doi: 10.1523/JNEUROSCI.4286-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Verhage M, et al. Neuron. 1997;18:453. doi: 10.1016/s0896-6273(00)81245-3. [DOI] [PubMed] [Google Scholar]
- 18.Friedrich R, et al. J Neurosci. 2008;28:6794. doi: 10.1523/JNEUROSCI.0538-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Orita S, et al. J Biol Chem. 1997;272:16081. doi: 10.1074/jbc.272.26.16081. [DOI] [PubMed] [Google Scholar]
- 20.Ke B, Oh E, Thurmond DC. J Biol Chem. 2007;282:21786. doi: 10.1074/jbc.M701661200. [DOI] [PubMed] [Google Scholar]
- 21.Miyazaki M, et al. Biochem Biophys Res Commun. 2009;384:461. doi: 10.1016/j.bbrc.2009.04.133. [DOI] [PubMed] [Google Scholar]
- 22.Fukuda N, et al. Diabetes. 2009;58:377. doi: 10.2337/db08-0303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Materials and methods are available as supporting material on Science Online.
- 24.Otsu Y, et al. J Neurosci. 2004;24:420. doi: 10.1523/JNEUROSCI.4452-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Groffen AJ, Friedrich R, Brian EC, Ashery U, Verhage M. J Neurochem. 2006;97:818. doi: 10.1111/j.1471-4159.2006.03755.x. [DOI] [PubMed] [Google Scholar]
- 26.Zengel JE, Sosa MA. J Physiol. 1994;477(Pt 2):267. doi: 10.1113/jphysiol.1994.sp020189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Korogod N, Lou X, Schneggenburger R. J Neurosci. 2005;25:5127. doi: 10.1523/JNEUROSCI.1295-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chan-Palay V. Z Anat Entwicklungsgesch. 1971;134:200. doi: 10.1007/BF00519300. [DOI] [PubMed] [Google Scholar]
- 29.Bernard C, Axelrad H. Brain Res. 1993;626:234. doi: 10.1016/0006-8993(93)90584-a. [DOI] [PubMed] [Google Scholar]
- 30.McKay BE, Turner RW. Journal of Physiology-London. 2005;567:829. doi: 10.1113/jphysiol.2005.089383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Womack M, Khodakhah K. J Neurosci. 2002;22:10603. doi: 10.1523/JNEUROSCI.22-24-10603.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hori T, Takai Y, Takahashi T. J Neurosci. 1999;19:7262. doi: 10.1523/JNEUROSCI.19-17-07262.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Groffen AJ, et al. J Biol Chem. 2004;279:23740. doi: 10.1074/jbc.M400731200. [DOI] [PubMed] [Google Scholar]
- 34.Kojima T, Fukuda M, Aruga J, Mikoshiba K. J Biochem (Tokyo) 1996;120:671. doi: 10.1093/oxfordjournals.jbchem.a021464. [DOI] [PubMed] [Google Scholar]
- 35.Bai J, Tucker WC, Chapman ER. Nat Struct Mol Biol. 2004;11:36. doi: 10.1038/nsmb709. [DOI] [PubMed] [Google Scholar]
- 36.Folch J, Lees M, Sloane Stanley GH. J Biol Chem. 1957;226:497. [PubMed] [Google Scholar]
- 37.Campelo F, McMahon HT, Kozlov MM. Biophys J. 2008;95:2325. doi: 10.1529/biophysj.108.133173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ford MG, et al. Nature. 2002;419:361. doi: 10.1038/nature01020. [DOI] [PubMed] [Google Scholar]
- 39.Martens S, McMahon HT. Nat Rev Mol Cell Biol. 2008;9:543. doi: 10.1038/nrm2417. [DOI] [PubMed] [Google Scholar]
- 40.Tang J, et al. Cell. 2006;126:1175. doi: 10.1016/j.cell.2006.08.030. [DOI] [PubMed] [Google Scholar]
- 41.Tucker WC, Weber T, Chapman ER. Science. 2004;304:435. doi: 10.1126/science.1097196. [DOI] [PubMed] [Google Scholar]
- 42.Duncan RR, Betz A, Shipston MJ, Brose N, Chow RH. J Biol Chem. 1999;274:27347. doi: 10.1074/jbc.274.39.27347. [DOI] [PubMed] [Google Scholar]
- 43.Pang ZP, Sun J, Rizo J, Maximov A, Sudhof TC. Embo J. 2006;25:2039. doi: 10.1038/sj.emboj.7601103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chicka MC, Hui E, Liu H, Chapman ER. Nat Struct Mol Biol. 2008;15:827. doi: 10.1038/nsmb.1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.