

Abstract
Objectives
Self-reported progression was evaluated as a predictor of survival in patients with metastatic gastrointestinal stromal tumor (GIST).
Methods
This is a follow-up of an open cohort study of Life Raft Group (LRG) members with a diagnosis of KIT-positive metastatic GIST receiving imatinib from May 2000–December 2007 reporting their subjective response to therapy by completion of an internet-based questionnaire. Subjects received ≥ 1 year of imatinib and reported an initial positive response. Members reporting stable disease or progression were excluded. Self-reported progression-free survival (srPFS) was compared with overall survival (OS) and analyzed by starting and last reported dose.
Results
One hundred sixty-nine subjects reported a mean starting dose of 527.8±177.9 mg/d at a mean age of 53.8±11.6 years at initial diagnosis. Of those reporting progression, 66% died versus 11% of those not reporting progression (P < 0.0001). When analyzed by last reported dose, a median srPFS benefit of 27.3 months was observed for the >400 mg/d group (P = 0.0017). Sixty-two percent of subjects who initiated therapy at >400 mg/d reported a dose reduction. When analyzed by last reported dose, a significant benefit in OS (P = 0.0229) and srPFS (P = 0.0069) was observed for subjects taking 600 over 400 mg/d.
Conclusions
srPFS strongly correlated with OS. Significant advantages were observed when last reported dose was considered, as was higher daily dose. These observations suggest that careful escalation to intermediate daily doses should be investigated further for its potential to reduce the incidence and severity of adverse events, but also as a strategy against developing secondary resistance to imatinib.
Keywords: gastrointestinal stromal tumor, GIST, imatinib, self-reported progression-free survival, overall survival, open cohort study
Introduction
Gastrointestinal stromal tumor (GIST) is a soft tissue sarcoma that can arise anywhere along the gastrointestinal tract, most commonly in the stomach and small intestine. The incidence of GIST is believed to be between six to 15 cases per million per year [1–3]. The understanding of the biology of GIST was revolutionized by the discovery of the roles of mutated growth factor receptor tyrosine kinases KIT (c-KIT, CD117) [4, 5] and PDGFRα [6] in pathogenesis. These mutations result in the constitutive activation of these receptors and drive the proliferation of GIST tumor cells. Activating mutations in the KIT receptor are observed in about 80% of GISTs and PDGFRα mutations in a further 5–10% [7]. Primary GIST tumors most frequently metastasize to the peritoneum and liver [8, 9]. Cytotoxic chemotherapy is ineffective in GIST, and until the introduction of targeted therapies, the prognosis was poor, with median survival time less than 2 years in the case of metastatic disease [10].
Imatinib (also known as STI-571, Glivec®, or Gleevec®) is an oral, small-molecule tyrosine kinase inhibitor (TKI) originally developed as an inhibitor of the BCR–ABL fusion protein for the treatment of chronic myelogenous leukemia [11]. Imatinib also inhibits KIT activity [12, 13], proved to be highly effective against GIST [14], and is now the first-line treatment for advanced and metastatic disease [9, 15–17]. Despite the success of imatinib for GIST, its effectiveness is limited by several considerations. First, patients experience a wide variety of dose-related side effects, including nausea, edema, cytopenias, rash, bleeding, and severe hepatotoxicity [18]. These side effects can necessitate dose reductions or discontinuation. Second, GIST tumors are highly heterogeneous with respect to the mutations they harbor [7, 19], and receptors bearing different mutations may have very different sensitivities to inhibition by imatinib [20, 21]. GIST tumors may also not respond well initially if they harbor mutations that are resistant to imatinib therapy (primary resistance), and even tumors that initially respond well may subsequently become drug-resistant (secondary resistance), leading to tumor progression and treatment failure [22]. This resistance may result from development/selection of additional KIT mutations or other mechanisms [23]. Sunitinib, a newer TKI, has recently been approved in many countries for imatinib-resistant GIST; however, more effective therapies for imatinib- and sunitinib-resistant GIST are needed. A number of new tyrosine kinase-targeted therapies are now in clinical trials for GIST.
In August 2000, the European Organization for Research and Treatment of Cancer (EORTC) Soft Tissue and Bone Sarcoma Group initiated a multi-national, phase I, dose-escalation study of imatinib for patients with advanced soft tissue sarcoma, including patients with GIST. A daily dose of 800 mg/d (400 mg b.i.d.) was determined to be the maximum tolerated dose [24]. In the phase II B2222 trial, a total of 147 patients were randomized to receive either 400 (n = 73) or 600 mg/d (n = 74). Twenty-eight percent of patients remained on study with a follow-up of up to 71 months [25]. A slightly longer progression-free survival was reported in the 600 mg/d dose group than in the 400 mg/d dose group, although the result was not statistically significant (24 versus 20 months, median time to progression, P = 0.371, log-rank test). Data on dose reductions occurring during this trial have not been reported.
Two large randomized phase III trials have been conducted that compared 400 and 800 mg/d imatinib in advanced or metastatic GIST patients. The United States/Canadian Intergroup S0033 trial by the National Cancer Institute accrued 746 patients between December 2000 and September 2001 [26]. An intent-to-treat analysis of this study observed no significant between-group differences in either progression-free survival (median PFS 18 and 20 months, respectively, P = 0.13) or overall survival (median overall survival 55 months and 51 months, respectively, P = 0.83). The larger phase III trial (EORTC 62005) was conducted by the EORTC, the Soft Tissue and Bone Sarcoma Group, the Italian Sarcoma Group, and the Australasian Gastro-Intestinal Tumors Group [27]. This pivotal trial accrued 946 patients between February 2001 and February 2002. At a median follow-up of 760 days, 56% of patients on 400 mg/d had progressed compared with 50% on 800 mg/d (P = 0.026, HR = 0.82). In the study reported by Verweij et al., 60% of the patients assigned to 800 mg imatinib daily were subsequently reduced in dose owing to side effects, versus 16% on 400 mg daily (P < 0.0001) [27]; similar numbers were reported by Blanke et al., in the other phase III pivotal trial [26]. Patients may also choose to take lower (or perhaps even higher) doses than prescribed, or may alter or interrupt their dosing schedule, all issues collectively relating to treatment compliance [28].
Preliminary results of MetaGIST, an initial meta-analysis of the two pivotal trials, reported in 2007, a small but statistically significant progression-free survival benefit for the higher dose arm (median benefit was 4 months, P = 0.04); however, there was no difference between dose groups in overall survival, with a median overall survival of 49 months for both (P = 0.97, HR = 1.00). Approximately 60% of patients survived for 3 years [29]. A more recent meta-analysis of these trials confirmed 400 mg/d as the recommended starting dose of imatinib for metastatic and advanced GIST patients; however, for patients who either progressed at this standard dose or harbor an exon 9 KIT mutation, the higher daily dose should be considered [30]. Some members of the JNCCN task force concur with 800 mg/d of imatinib for patients with documented KIT exon 9 mutations; this is a category 2B recommendation [31].
The analysis of clinical trial outcomes and end points is usually performed on the basis of intent-to-treat. Patients in these trials are analyzed as randomized, and therefore, by their initial daily dosage groups regardless of any subsequent changes in treatment. Intent-to-treat analysis therefore avoids possible confounding effects due to non-random changes, thus preserving the randomized nature of the trial. This simplification, however, would systematically discard any available information regarding reported dose histories, e.g., last observation carried forward and exposure estimates, information which can potentially provide insight into the agent’s effectiveness.
The Life Raft Group is an international, internet-based, non-profit organization providing support, through education and innovative research, to GIST patients and their families. To monitor and evaluate information about treatment effectiveness and side effects, the Life Raft Group has created and maintained its own patient-driven database.
Objectives
The objectives of this study were to evaluate whether self-reported progression was a robust predictor of survival and how rates of progression-free and overall survival reported in this manner correlated to imatinib dose, in patients with metastatic GIST.
Methods
Study Design
This is an open cohort study of the effect of imatinib dosage upon overall and self-reported progression-free survival of metastatic gastrointestinal stromal tumor (GIST) patients within the Life Raft Group’s member database from May 2000–December 2007. This report is a continuation of a study of 169 subjects presented at the 2004 Connective Tissue Oncology Society Meeting [32] in Montreal, Canada, with a follow-up analysis from December 2007.
Setting
The Life Raft Group database contains information provided by its members worldwide since May 8, 2000. Members were recruited by referrals from attending physicians or other GIST patients; however, the majority of members were acquired through patient-initiated contacts, following internet searches or other requests for information and assistance. All members had a reported diagnosis of KIT (CD117)-positive GIST. There were no other criteria for membership, and no one was excluded owing to age, confirmation of diagnosis, disease status, or prior or current treatment regimen or response. Agreeing to supply the Life Raft Group (LRG) with data was part of the initial membership agreement; however, there was no change in membership or status upon withdrawal of participation. All data were collected using an electronic questionnaire periodically forwarded as reminders to members, a relative, or a caregiver. Data fields collected included dates of diagnosis, initiation of imatinib treatment and last update, initial and last reported daily dosages, and response to treatment. Demographic data included sex, date of birth, country, and primary tumor site. Outcomes were based upon subjective status in response to imatinib therapy: no evident disease (NED), shrinkage, progression, stable disease, or deceased. Responses were reviewed, and the data were logged and curated by LRG staff who confirmed all end points, e.g., death or progression.
Selection and Description of Study Participants
All subjects included within this study had a diagnosis of KIT-positive metastatic GIST and had initiated imatinib therapy at least 1 year prior to inclusion.
In order to select for subjects who might receive the greatest benefit, only members who reported initial tumor response (“shrinkage”) to imatinib therapy and remained progression-free for at least 1 year were included. In addition, members who reported only stable disease (“no shrinkage”) throughout this first year were excluded. These criteria were intended to select for subjects who were most likely to benefit from imatinib by excluding those who might harbor a mutation associated with primary resistance to the drug.
Treatment interruptions and decreases in dosage were permitted within the first year to manage adverse events, and dose increases were also permitted for reasons other than disease progression. There were no additional patient- or disease-specific exclusion criteria, nor any restrictions on comorbid conditions or polypharmacy.
All treatment decisions were at the discretion of the treating physician; however, 47 of the included subjects reported being enrolled in a clinical trial of imatinib for the treatment of metastatic or advanced GIST (phase II, B2222 [25]; phase III, EORTC 62005 [27], S0033 [26]). The starting imatinib dosages for these subjects would have been initially assigned at random and subsequent treatment decisions made according to their respective trial protocols.
Outcomes and End Points
Owing to the open, observational, and exploratory nature of this study, overall survival was selected as the primary outcome, with self-reported progression-free survival as a secondary parameter. Demographic, disease, and treatment details were obtained at membership registration, and updates were anticipated on a periodic but unscheduled basis. Updates were expected to coincide most often with specific, unambiguous events including periodic CAT scans, the onset/resolution of adverse events, and the consequent physician-directed interruptions, or other treatment regimen adjustments, as well as end points. End points included tumor progression, termination of imatinib therapy for reasons other than progression, and death. Subjects were followed from registration until September 2004, in the initial analysis and again in December 2007, in this follow-up analysis.
Statistical Analyses
Survival analysis using a log-rank (Mantel–Cox) test was performed using GraphPad Prism (version 5.01, for Windows, GraphPad Software, San Diego, CA, USA, www.graphpad.com). Two-sided P value < 0.05 was considered significant. Hazard ratios were computed by the Mantel–Haenszel method also using GraphPad Prism. All other calculations were carried out using Microsoft Excel 2004 for Mac (version 11.5.4).
Results
Participant and Disease Characteristics
The study analyzed 169 subjects with metastatic GIST who had been treated with imatinib for an average of 4.7 (1.6 SD) years. The population contained 92 males and 77 females from 17 countries at a mean age of 53.8 (11.6 SD) years at initial diagnosis (Table 1).
Table 1.
Selection of study subjects and data acquired from LRG members
| Total LRG database | |||
|---|---|---|---|
| (2000–2004) | |||
| N = 482 | |||
| (325 with metastatic disease in 2004) | |||
| ⇓ | |||
| Metastatic patients on imatinib for ≥1year | |||
| N = 252 | |||
| ⟹ Excluded owing to: | Males | Females | |
| ⇓ | Progression in first year | 7 | 3 |
| (83 members excluded) | Stable disease | 14 | 11 |
| Insufficient data | 23 | 25 | |
| Patients at 1 year with reported shrinkage and NED only | Males | Females | |
| ⇓ | n ≤ 400 mg/d | 52 | 48 |
| N = 169 ⟹ |
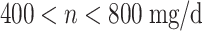
|
13 | 13 |
| n ≥ 800 mg/d | 27 | 16 | |
Location of primary tumor was provided as free text and sites were grouped post hoc as: upper/mid-GI (esophagus, stomach, abdomen, omentum), bowel (small intestine, colon, rectum), other (liver, bladder, pancreas), or unknown (Table 2). Half (51%) of all reported primary tumors were located within the upper- to mid-gastrointestinal tract; however, the majority were situated in either the stomach (n = 60) or abdomen (n = 20), and there were no reported cases of an esophageal site. “Bowel” was a composite of mostly small intestinal tumors (n = 53) with few colon (n = 6) or rectal (n = 2) tumors. There were four reported liver tumors, six cases of unspecified “other” location, and nine reported primary tumors of unknown location. Other subject and disease characteristics are reported in Table 3.
Table 2.
Patient and disease characteristics: included subjects and remaining members
| Study population (N = 169) | Remaining members (N = 313) | |
|---|---|---|
| Median age (range) at imatinib start in years | 54 (21–86) | 53 (8–85) |
| Males | 92 (54%) | 163 (52%) |
| Continent | ||
| The Americasa | 145 (86%) | 256 (82%) |
| US | 133 (79%) | 240 (77%) |
| Europe | 16 (9%) | 29 (9%) |
| Asia | 4 (2%) | 9 (3%) |
| Africa | 1 (0.6%) | 1 (0.3%) |
| Oceania | 1 (0.6%) | 4 (1%) |
| Unknown | 2 (1%) | 14 (4%) |
| Primary tumor siteb | ||
| Upper-/mid-GI | 86 (51%) | 157 (50%) |
| Bowel | 64 (38%) | 111 (35%) |
| Other | 10 (6%) | 18 (6%) |
| Unknown | 9 (5%) | 27 (9%) |
| Clinical trial participants | 47 (28%) | 21 (7%) |
| Starting dose (mg/d) | ||
| Median (range) | 400 (0–800) | 400 (100–800) |
| Mean (SD) | 527.8 (177.9) | 424.8 (205.7) |
aMembers located in the Americas were almost exclusively in N. America. There was a single member in Costa Rica that was not included in the study and a single study subject in Mexico; no members were in S. America
bPrimary tumor sites were grouped post hoc as: upper-/mid-GI (esophagus, stomach, abdomen, omentum), bowel (small intestine, colon, rectum), other (liver, bladder, pancreas), and unknown
Table 3.
Subject and disease characteristics by starting imatinib dosage group
| Number of subjects | n ≤ 400 mg/day |
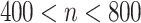
|
≥ 800 mg/day |
|---|---|---|---|
| Sex | |||
| Males | 52 | 13 | 27 |
| Females | 48 | 13 | 16 |
| Continent | |||
| N. America | 83 | 25 | 37 |
| Europe | 10 | 0 | 6 |
| Asia | 4 | 0 | 0 |
| Africa | 1 | 0 | 0 |
| Oceania | 1 | 0 | 0 |
| Unknown | 1 | 1 | 0 |
| Primary tumor sitea | |||
| Upper-/mid-GI | 50 | 11 | 25 |
| Bowels | 36 | 11 | 17 |
| Other | 8 | 1 | 1 |
| Unknown | 6 | 3 | 0 |
| Clinical trial participants | 21 | 9 | 17 |
| Mean age at diagnosis in years (SD) | 55.8 (12.2) | 50.3 (10.1) | 51.1 (10.3) |
| Minimum to maximum | 28.2–86.4 | 21.0–71.9 | 31.0–74.9 |
| Mean age at study start in years (SD) | 59.4 (12.2) | 54.9 (10.4) | 55.7 (10.4) |
| Minimum to maximum | 32.9–90.1 | 23.0–76.0 | 37.3–78.3 |
| Mean imatinib exposure in years (SD) | 4.6 (1.6) | 4.9 (1.7) | 4.9 (1.6) |
| Minimum to maximum | 0.9–7.1 | 1.8–7.3 | 1.1–6.8 |
aPrimary tumor sites were grouped post hoc as: upper-/mid-GI (esophagus, stomach, abdomen, omentum), bowel (small intestine, colon, rectum), other (liver, bladder, pancreas), and unknown.
Mutational data were provided for only 15% (26 of 169) of the subjects, too low for meaningful analysis. Of the 26 patients with mutational data, one was wild-type GIST, four had KIT exon 9 mutations, 20 had KIT exon 11 mutations, and one had a PDGFRα mutation within an unspecified exon.
Subjects reported taking an average initial daily dosage of 572.8 mg (177.9 SD) of imatinib. At the time of follow-up analysis, subjects last reported taking an average daily dosage of 455.0 mg/d (161.0 SD) of imatinib with a mean exposure of 30.2 (8.9 SD) months.
Self-Reported Progression as a Surrogate for Survival in Metastatic GIST
Since the validity of patient-reported progression had not previously been established, members who reported no progression (n = 91) by October 2004 were compared with those who reported progression (n = 44). Of the 78 subjects who had reported progression by October 2004, 29 had died by December 2007 and 15 were still alive. The 34 subjects who had died by 2004 were excluded. Of those reporting progression in 2004, 29 of 44 (66%) died before the second analysis (Dec 2007) compared with ten of 91 (11%) of those who had not reported progression (P < 0.0001, HR = 17.46; Fig. 1). As of December 2007, a total of 81% (63 of 78 subjects) of those who reported tumor progression by Oct 2004 died versus 11% (ten of 91 subjects) for those who had not reported progression by 2004.
Fig. 1.

Overall survival comparison of those reporting progression by 2004 versus those who did not
Imatinib Dose Changes
Dosage changes and last reported status are summarized in Table 4. Of the 69 subjects who began treatment in the high-dose group (> 400 mg/d), 43% had dose reductions that moved them into the low-dose (≤ 400 mg/d) group, and 19% had dose reductions but remained in the range of the high-dose group when last reported dose was considered; 38% had no dose reductions (Table 5). Of those subjects who initiated treatment in the low-dose group, 14 (14%) had dose increases that moved them into the high-dose group prior to progression using last reported dose. Nine percent of the subjects that started on ≤ 400 mg/d required a dose reduction.
Table 4.
Schedule of dosage changes and last reported status at end point
| Dose changes (mg/day) | ||||
|---|---|---|---|---|
| Starting dosage | End point dosage | Progression | No progression | Total |
| 100 | 400 | 1 | 1 | |
| 200 | 600 | 1 | 1 | |
| 300 | 250 | 1 | 1 | |
| 300 | 1 | 1 | ||
| 400 | 1 | 1 | ||
| 400 | 0 | 2 | 2 | 4 |
| 300 | 4 | 1 | 5 | |
| 400 | 57 | 16 | 73 | |
| 600 | 5 | 6 | 11 | |
| 800 | 1 | 1 | 2 | |
| ≤ 400 subtotal | 72 | 28 | 100 | |
| 600 | 300 | 2 | 1 | 3 |
| 400 | 4 | 2 | 6 | |
| 500 | 1 | 1 | ||
| 600 | 7 | 7 | 14 | |
| 800 | 2 | 2 | ||
| 600 subtotal | 13 | 13 | 26 | |
| 800 | 0 | 2 | 2 | |
| 100 | 1 | 1 | ||
| 400 | 13 | 5 | 18 | |
| 450 | 1 | 1 | ||
| 500 | 1 | 1 | ||
| 600 | 6 | 4 | 10 | |
| 800 | 6 | 4 | 10 | |
| 800 subtotal | 30 | 13 | 43 | |
| Grand total | 115 | 54 | 169 | |
Progression includes subjects who reported either tumor progression or death at last follow-up. No progression includes a last report of no evident disease, tumor shrinkage, and stable disease
Table 5.
Dose reductions in the starting high-dose group (n = 69 at >400 mg/d)
| Dose reduced to | n (%) |
|---|---|
| 400 mg/d | 30 (43) |
| 450–600 mg/d | 13 (19) |
| Total dose reductions | 43 (62) |
| No dose reduction | 26 (38) |
Males were better able to tolerate higher imatinib doses than females (Table 6). Nineteen percent of females who started at 800 mg/d remained at this highest daily dose. However, despite their relatively greater tolerance, only 26% of males who started on 800 mg/d remained at this dose. Eighty-five percent of males and 38% of females who began at 600 mg/d remained at that level.
Table 6.
Imatinib dose reductions by gender and starting dose
| Gender/starting dose | Start (n) | End point, n (% remaining) |
|---|---|---|
| Female | ||
| 400 mg/d | 44 | 38 (86) |
| 600 mg/d | 13 | 5 (38) |
| 800 mg/d | 16 | 3 (19) |
| Male | ||
| 400 mg/d | 51 | 48 (94) |
| 600 mg/d | 13 | 11 (85) |
| 800 mg/d | 27 | 7 (26) |
Evaluation of Overall Survival, High- Versus Low-Dose Imatinib
In an analysis of overall survival by starting imatinib dose, a small benefit of 5.7 months was observed for the high- over low-dose group and did not reach statistical significance (77 versus 71.3 months, P = 0.336, HR = 1.25; Fig. 2). When the analysis was repeated using last reported dose, a statistically significant benefit in overall survival was observed for the higher dose group. The median survival was 70.8 months for the low-dose group and had not yet been reached for the high-dose group (P = 0.037, HR = 1.66; Fig. 3).
Fig. 2.

Overall survival using reported initial daily imatinib dose: high versus low dose
Fig. 3.

Overall survival using last reported imatinib daily dose: high versus low dose
Evaluation of Self-Reported Progression-Free Survival, High- Versus Low-Dose Imatinib
In analyzing the progression-free survival of subjects categorized by starting daily dose of imatinib, a 7.7 month median benefit in srPFS was observed for the high-dose group that did not reach significance (P = 0.1029, Fig. 4). In contrast, using last reported dose, the median srPFS was 27.3 months longer in the high-dose group (58.8 versus 31.5 months, P = 0.0017, HR = 1.826; Fig. 5).
Fig. 4.

Self-reported progression-free survival using starting daily imatinib dose: high versus low dose
Fig. 5.

Self-reported progression-free survival using last reported daily imatinib dose: high versus low dose
Effectiveness of Imatinib, 600 mg/day
Because the 800 mg/d dose was not tolerated well by persons of either sex, the effectiveness of 600 mg/d, as well as the difference in effectiveness between 600 and 800 mg/d was evaluated by last reported dose. We compared subjects taking 400 mg/d (n = 98) to those taking 600 mg/d (n = 36). In addition, we compared subjects on 600 mg/d directly to those on 800 mg/d (n = 15). However, the comparison of 600 to 800 mg/d should be viewed with caution due to the small size of the 800 mg/d group.
Overall Survival, 600 mg/day
Subjects who last reported taking 600 mg/d showed a statistically significant survival advantage over those last reported taking 400 mg/d (P = 0.0229, HR = 1.877; Fig. 6). For comparison, the hazard ratio for overall survival comparing low (≤ 400 mg/d) to high (> 400 mg/d) doses was 1.663 (P = 0.037; Fig. 3). The small advantage for the 600 mg/d versus the entire high-dose group, including subjects on 800 mg/d, was not statistically significant. The median overall survival was 70.8 months for the entire low-dose group (≤ 400 mg/d) and 67.8 months for patients taking 400 mg/d only (excluding those taking < 400 mg/d). The median overall survival had not been reached for either the entire high-dose group or subjects taking 600 mg/d (Figs. 3 and 6). No significant difference in overall survival was observed between subjects last reporting a daily dosage of 600 (n = 36) or 800 mg/d (n = 15) of imatinib (P = 0.75, HR = 0.8466).
Fig. 6.

Overall survival using last reported dose, 400 mg/d versus 600 mg/d
Self-Reported Progression-Free Survival, 600 mg/day
In an analysis of last reported dose, a significantly longer srPFS was observed for 600 over 400 mg/d imatinib. Subjects last reportedly taking 400 mg/d (n = 98) had a median srPFS of 30.2 months compared with 58.6 months srPFS for subjects taking 600 mg/d imatinib (n = 36), a benefit of 28.4 months for the latter group (P = 0.0069, HR = 1.811; Fig. 7). In addition, the median srPFS for patients taking 600 mg/d was almost identical to that for the entire high-dose group (> 400 mg/d), 58.6 months and 58.8 months, respectively. There was no significant difference in srPFS between patients taking 800 (n = 15) and 600 mg/d of imatinib (n = 36; data not shown).
Fig. 7.

Self-reported progression-free survival using last reported imatinib dose, 400 versus 600 mg/d
Survival by Gender
In contrast to the entire sample of patients, overall survival by gender did not show a statistically significant benefit. Median overall survival for females in the low-dose group was 70.8 months and had not yet been reached in the high-dose group at the time of follow-up analysis. The hazard ratio was 1.88. The median overall survival times for males in the low- and high-dose group were 74.9 and 77.0 months, respectively (P = 0.087, HR = 1.70). In a 400 versus 600 mg/d comparison of last reported dose, the median overall survival for females on 400 mg/d was 68.9 months and had not yet been reached in the 600 mg/d group (P = 0.079, HR = 2.31). For males with a last reported daily dose of 400 mg/d, the median overall survival was 60.2 months and had also yet to be reached at end point for those taking 600 mg/d (P = 0.096, HR = 1.783).
When last reported dose was used to evaluate srPFS, both males and females showed a benefit from reportedly higher doses of imatinib. The median srPFS for males was 29 months in the low-dose group and 49.3 months in the high-dose group, a benefit for of 20.3 months (P = 0.017, HR = 1.83). The median srPFS for females was 37.5 months in the low-dose group and had not been reached in the high-dose group (P = 0.023, HR = 2.04). In a 400 versus 600 mg/d comparison, median srPFS time for females on a last reported dose of 400 mg/d were 36.7 months and had also not been reached in the high-dose group (P = 0.0357, HR = 2.187). The median srPFS times for males on a last reported dose of 400 versus 600 mg/d were 25.9 and 49.3 months, respectively, a difference that was near the borderline of statistical significance (P = 0.0505, HR = 1.722).
Discussion
The utility of patient-reported tumor progression at last reported dose as a surrogate for survival in metastatic GIST patients was investigated, based on analysis of an internet-driven open cohort study of members of the Life Raft Group who completed a self-reported subjective assessment of progression-free survival compared against overall survival. Excluding members who had died by the initial analysis, subjects who had reported stability by 2004 were far more likely to be alive in 2007 than those who reported progression at that time (P < 0.0001, HR = 17.46).
LRG members demonstrated a clear advantage at higher daily doses of imatinib in terms of both overall and progression-free survival, when last reported dose was considered. These advantages no longer reached significance when analyzed by prescribed initiating dose. Recognizing that many of the potential side effects from imatinib therapy improve over time [27], careful dose-escalation strategies may be considered to increase imatinib tolerability, and hence treatment compliance, and reduce the risk of developing secondary resistance to the drug. This analysis of member-supplied data also suggests that 600 mg/d may be as effective as 800 mg/d of imatinib. However, due to the small number of subjects initially prescribed the highest daily dose, only a qualitative comparison should be made.
The data presented here suggest that higher daily doses of imatinib reduce rates of progression. In spite of this fact, higher doses may not be appropriate for all patients with metastatic GIST. For patients taking the same dose of imatinib, plasma levels can vary by 4-fold or more [33, 34]. Side effects from imatinib also vary significantly, from very mild to life threatening and even fatal [26]. Most side effects are dose-dependent and thus both more likely and more severe at higher doses [18]. It is also important to note that there were some long-term responders in the low-dose group, including some patients taking as little as 300 mg/d. Fewer females than males were able to tolerate the higher doses of imatinib, which suggests that females may benefit from more careful dose-escalation strategies. Since side effects can improve over time, the utilization of supportive medical interventions to directly manage these events may prove invaluable in the ongoing development of treatment strategies.
Subjects who developed imatinib resistance over the course of treatment had much higher mortality rates than those that remained stable, despite crossover to higher doses or other treatments. This population was initially analyzed in October 2004, at which time 91 reported stable disease on imatinib therapy but 78 had reported progression, of whom 34 had died by November 1, 2004. Those who progressed moved to various other treatment regimes, often through several treatment strategies. Sixty percent of those on a lower initial dose of imatinib crossed over to a higher dose; 65% were given sunitinib (SU11248, Sutent®); 41% underwent surgery, and 12% received nilotinib (AMN107, Tasigna®).
Despite these subsequent treatments, the mortality rate among those who had progressed by October 2004 was 81% by December 2007, compared with a mortality rate of 11% among those who reported stable disease in October 2004. Seventy-two percent of these deaths occurred within 2 years from the point of relapse.
A recent meta-analysis of the two pivotal phase III trials of imatinib for unresectable or metastatic GIST confirmed a typical initial starting daily dose of 400 mg/d. A dose of 800 mg/d was found to benefit those patients with exon 9 KIT mutations and those who progressed at the standard, starting dose of 400 mg/d [30]. In addition, preliminary data based upon imatinib plasma levels from 73 of the 147 patients enrolled in the phase II B2222 trial suggested that at least 25% of these GIST patients were under-dosed [35]. In the EORTC pharmacokinetic (PK) study, clearance of imatinib increased by 33% in GIST patients in the 12-month period studied. This study found a 42% decrease in area under the curve over the 12 months. While the EORTC PK study was not conclusive, it adds to the concern that patients treated with lower doses of imatinib might be at higher risk of developing resistance.
At least three different theories have been proposed to explain this phenomenon: (1) Imatinib clearance increases over time, possibly related to improved liver function [36]. (2) Multi-drug resistance transport proteins are induced over time, decreasing the net transport of imatinib across the intestinal membrane [37]. (3) Patient adherence to the drug falls off over time [28].
The results of this analysis show a strong correlation between imatinib treatment failure and lower survival and suggest that the avoidance of developing resistance may yield better outcomes than overcoming secondary resistance. However, only a clinical trial will be able to resolve the question of whether dose-escalation or other strategies (possibly including measuring imatinib plasma levels) would produce superior results to the currently recommended dose of 400 mg/d.
This analysis of patient-reported progression at last reported dose offers a real-world complementary perspective to that seen in investigator-initiated randomized trials. While there is no substitute for hypothesis-driven clinical research, given the relative ease, flexibility, and expense of collecting observational data, such investigations should be encouraged as possible hypothesis-generating resources towards improvements in care.
Limitations
Due to its relatively small size and non-randomized nature, care must be taken when interpreting the results of this study. In terms of study design and the potential for selection bias, the LRG members captured in this analysis may not be representative of the entire population of GIST patients. In order to address this concern, every attempt has been made to compare the characteristics of both the included and excluded LRG member populations, with the underlying assumption that the event of seeking LRG membership initially was itself effectively at random.
Excluding members who progressed during the first year or who did not show initial shrinkage effectively eliminated those with primary resistance. These exclusion criteria may have also eliminated a higher proportion of KIT exon 9 primary mutations, biasing the results towards increased median survival rates compared with other studies that include this population. One such study indicated that exon 9 patients receive a greater benefit from higher doses of imatinib [38]. This study indicates that the same may be true for other GIST genotypes as well. These criteria may have also selected for healthier patients who are more likely to be able to tolerate higher doses of imatinib and may have fared better regardless of dose, initial or last reported. In addition, as all members reported their own daily dosages, this study was not controlled in any fashion, and known dosages, both low and high, could have influenced the subjective reporting of disease progression.
Acknowledgments
Jerry Call and Norman Scherzer are employees of The Life Raft Group, which receives financial support from several pharmaceutical companies including Novartis Pharmaceuticals, the manufacturer of imatinib (Gleevec™). P. David Josephy has received travel expense reimbursement support from Novartis to attend several GIST patient meetings. He is the president of the Canadian affiliate of the Life Raft Group, which has received financial support from Novartis. Christopher Walentas is an independent consultant for the Life Raft Group.
An abstract of the initial analysis was presented at the Annual Meeting of Connective Tissue Oncology Society in Montreal, Canada, in November 2004 [32]. Preliminary data were presented in the LRG Newsletter (www.liferaftgroup.org/docs/newsletters/March2008nwsltr.pdf). We thank Jim Hughes for helpful data analysis, Steve Rigg for database design and support, and Pamela Barckett for data management. We also thank the LRG members who participated in the study, and dedicate this work to the memory of those members who did not live to see its results reported.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Abbreviations
- CAT
computer-aided tomography
- GI
gastrointestinal
- GIST
gastrointestinal stromal tumor
- HR
hazard ratio
- LRG
Life Raft Group
- NED
no evident disease
- NS
not significant
- OS
overall survival
- srPFS
self-reported progression-free survival
- TKI
tyrosine kinase inhibitor
- JNCCN
Journal of the National Comprehensive Cancer Network
Contributor Information
Jerry Call, Phone: +1-973-8379092, FAX: +1-973-8379095, Email: jcall@liferaftgroup.org.
Norman J. Scherzer, Phone: +1-973-8379092, FAX: +1-973-8379095, Email: nscherzer@liferaftgroup.org
P. David Josephy, Phone: +1-519-8244100, FAX: +1-519-7661499, Email: djosephy@UOGUELPH.CA.
Christopher Walentas, Phone: +1-212-8107361, Email: cwalentas@gmail.com.
References
- 1.Mucciarini C, Rossi G, Bertolini F, Valli R, Cirilli C, Rashid I, Marcheselli L, Luppi G, Federico M. Incidence and clinicopathologic features of gastrointestinal stromal tumors. A population-based study. BMC Cancer. 2007;7:230. doi: 10.1186/1471-2407-7-230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goettsch WG, Bos SD, Breekveldt-Postma N, Casparie M, Herings RM, Hogendoorn PC. Incidence of gastrointestinal stromal tumours is underestimated: results of a nation-wide study. Eur J Cancer. 2005;41:2868–2872. doi: 10.1016/j.ejca.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 3.Reddy P, Boci K, Charbonneau C. The epidemiologic, health-related quality of life, and economic burden of gastrointestinal stromal tumours. J Clin Pharm Ther. 2007;32:557–565. doi: 10.1111/j.1365-2710.2007.00852.x. [DOI] [PubMed] [Google Scholar]
- 4.Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T, Ishiguro S, Kawano K, Hanada M, Kurata A, Takeda M, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998;279:577–580. doi: 10.1126/science.279.5350.577. [DOI] [PubMed] [Google Scholar]
- 5.Kitamura Y, Hirota S, Nishida T. Gastrointestinal stromal tumors (GIST): a model for molecule-based diagnosis and treatment of solid tumors. Cancer Sci. 2003;94:315–320. doi: 10.1111/j.1349-7006.2003.tb01439.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Heinrich MC, Corless CL, Duensing A, McGreevey L, Chen CJ, Joseph N, Singer S, Griffith DJ, Haley A, Town A, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 2003;299:708–710. doi: 10.1126/science.1079666. [DOI] [PubMed] [Google Scholar]
- 7.Fletcher JA, Rubin BP. KIT mutations in GIST. Curr Opin Genet Dev. 2007;17:3–7. doi: 10.1016/j.gde.2006.12.010. [DOI] [PubMed] [Google Scholar]
- 8.Burkill GJ, Badran M, Al-Muderis O, Meirion Thomas J, Judson IR, Fisher C, Moskovic EC. Malignant gastrointestinal stromal tumor: distribution, imaging features, and pattern of metastatic spread. Radiology. 2003;226:527–532. doi: 10.1148/radiol.2262011880. [DOI] [PubMed] [Google Scholar]
- 9.Dematteo RP, Heinrich MC, El-Rifai WM, Demetri G. Clinical management of gastrointestinal stromal tumors: before and after STI-571. Hum Pathol. 2002;33:466–477. doi: 10.1053/hupa.2002.124122. [DOI] [PubMed] [Google Scholar]
- 10.Gold JS, van der Zwan SM, Gonen M, Maki RG, Singer S, Brennan MF, Antonescu CR, De Matteo RP. Outcome of metastatic GIST in the era before tyrosine kinase inhibitors. Ann Surg Oncol. 2007;14:134–142. doi: 10.1245/s10434-006-9177-7. [DOI] [PubMed] [Google Scholar]
- 11.Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S, Zimmermann J, Lydon NB. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med. 1996;2:561–566. doi: 10.1038/nm0596-561. [DOI] [PubMed] [Google Scholar]
- 12.Buchdunger E, Cioffi CL, Law N, Stover D, Ohno-Jones S, Druker BJ, Lydon NB. Abl protein-tyrosine kinase inhibitor STI571 inhibits in vitro signal transduction mediated by c-kit and platelet-derived growth factor receptors. J Pharmacol Exp Ther. 2000;295:139–145. [PubMed] [Google Scholar]
- 13.Mol CD, Dougan DR, Schneider TR, Skene RJ, Kraus ML, Scheibe DN, Snell GP, Zou H, Sang BC, Wilson KP. Structural basis for the autoinhibition and STI-571 inhibition of c-Kit tyrosine kinase. J Biol Chem. 2004;279:31655–31663. doi: 10.1074/jbc.M403319200. [DOI] [PubMed] [Google Scholar]
- 14.Joensuu H, Roberts PJ, Sarlomo-Rikala M, Andersson LC, Tervahartiala P, Tuveson D, Silberman S, Capdeville R, Dimitrijevic S, Druker B, Demetri GD. Effect of the tyrosine kinase inhibitor STI571 in a patient with a metastatic gastrointestinal stromal tumor. N Engl J Med. 2001;344:1052–1056. doi: 10.1056/NEJM200104053441404. [DOI] [PubMed] [Google Scholar]
- 15.Cassier PA, Dufresne A, Arifi S, El Sayadi H, Labidi I, Ray-Coquard I, Tabone S, Meeus P, Ranchere D, Sunyach MP, et al. Imatinib mesilate for the treatment of gastrointestinal stromal tumour. Expert Opin Pharmacother. 2008;9:1211–1222. doi: 10.1517/14656566.9.7.1211. [DOI] [PubMed] [Google Scholar]
- 16.Schnadig ID, Blanke CD. Gastrointestinal stromal tumors: imatinib and beyond. Curr Treat Options in Oncol. 2006;7:427–437. doi: 10.1007/s11864-006-0018-5. [DOI] [PubMed] [Google Scholar]
- 17.Siehl J, Thiel E. C-kit, GIST, and imatinib. Recent Results Cancer Res. 2007;176:145–151. doi: 10.1007/978-3-540-46091-6_12. [DOI] [PubMed] [Google Scholar]
- 18.Guilhot F. Indications for imatinib mesylate therapy and clinical management. Oncologist. 2004;9:271–281. doi: 10.1634/theoncologist.9-3-271. [DOI] [PubMed] [Google Scholar]
- 19.Hoeben A, Schoffski P, Debiec-Rychter M. Clinical implications of mutational analysis in gastrointestinal stromal tumours. Br J Cancer. 2008;98:684–688. doi: 10.1038/sj.bjc.6604217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carter TA, Wodicka LM, Shah NP, Velasco AM, Fabian MA, Treiber DK, Milanov ZV, Atteridge CE, Biggs WH, 3rd, Edeen PT, et al. Inhibition of drug-resistant mutants of ABL, KIT, and EGF receptor kinases. Proc Natl Acad Sci U S A. 2005;102:11011–11016. doi: 10.1073/pnas.0504952102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heinrich MC, Corless CL, Demetri GD, Blanke CD, von Mehren M, Joensuu H, McGreevey LS, Chen CJ, Van den Abbeele AD, Druker BJ, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol. 2003;21:4342–4349. doi: 10.1200/JCO.2003.04.190. [DOI] [PubMed] [Google Scholar]
- 22.Van Glabbeke M, Verweij J, Casali PG, Le Cesne A, Hohenberger P, Ray-Coquard I, Schlemmer M, van Oosterom AT, Goldstein D, Sciot R, et al. Initial and late resistance to imatinib in advanced gastrointestinal stromal tumors are predicted by different prognostic factors: a European organisation for research and treatment of cancer–Italian sarcoma group–Australasian gastrointestinal trials group study. J Clin Oncol. 2005;23:5795–5804. doi: 10.1200/JCO.2005.11.601. [DOI] [PubMed] [Google Scholar]
- 23.Desai J, Shankar S, Heinrich MC, Fletcher JA, Fletcher CD, Manola J, Morgan JA, Corless CL, George S, Tuncali K, et al. Clonal evolution of resistance to imatinib in patients with metastatic gastrointestinal stromal tumors. Clin Cancer Res. 2007;13:5398–5405. doi: 10.1158/1078-0432.CCR-06-0858. [DOI] [PubMed] [Google Scholar]
- 24.van Oosterom AT, Judson I, Verweij J, Stroobants S, Donato di Paola E, Dimitrijevic S, Martens M, Webb A, Sciot R, Van Glabbeke M, et al. Safety and efficacy of imatinib (STI571) in metastatic gastrointestinal stromal tumours: a phase I study. Lancet. 2001;358:1421–1423. doi: 10.1016/S0140-6736(01)06535-7. [DOI] [PubMed] [Google Scholar]
- 25.Blanke CD, Demetri GD, von Mehren M, Heinrich MC, Eisenberg B, Fletcher JA, Corless CL, Fletcher CD, Roberts PJ, Heinz D, et al. Long-term results from a randomized phase II trial of standard- versus higher-dose imatinib mesylate for patients with unresectable or metastatic gastrointestinal stromal tumors expressing KIT. J Clin Oncol. 2008;26:620–625. doi: 10.1200/JCO.2007.13.4403. [DOI] [PubMed] [Google Scholar]
- 26.Blanke CD, Rankin C, Demetri GD, Ryan CW, von Mehren M, Benjamin RS, Raymond AK, Bramwell VH, Baker LH, Maki RG, et al. Phase III randomized, intergroup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumors expressing the kit receptor tyrosine kinase: S0033. J Clin Oncol. 2008;26:626–632. doi: 10.1200/JCO.2007.13.4452. [DOI] [PubMed] [Google Scholar]
- 27.Verweij J, Casali PG, Zalcberg J, LeCesne A, Reichardt P, Blay JY, Issels R, van Oosterom A, Hogendoorn PC, Van Glabbeke M, et al. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomised trial. Lancet. 2004;364:1127–1134. doi: 10.1016/S0140-6736(04)17098-0. [DOI] [PubMed] [Google Scholar]
- 28.Feng, W., Henk, H., Thomas, S., Baladi, J., Hatfield, A., Goldberg, G.A., Cortes J. (2006). Compliance and persistency with imatinib. Journal of Clinical Oncology (ASCO Annual Meeting), 24:6038.
- 29.Van Glabbeke MM, Owzar K, Rankin C, Simes J, Crowley J, GIST Meta-analysis Group (MetaGIST). Comparison of two doses of imatinib for the treatment of unresectable or metastatic gastrointestinal stromal tumors (GIST): a meta-analysis based on 1,640 patients (pts). Journal of Clinical Oncology (2007 ASCO Annual Meeting ), 2007. p. 10004.
- 30.Patel S, Zalcberg JR. Optimizing the dose of imatinib for treatment of gastrointestinal stromal tumours: lessons from the phase 3 trials. Eur J Cancer. 2008;44:501–509. doi: 10.1016/j.ejca.2007.11.021. [DOI] [PubMed] [Google Scholar]
- 31.Demetri GD, Benjamin RS, Blanke CD, Blay JY, Casali P, Choi H, Corless CL, Debiec-Rychter M, DeMatteo RP, Ettinger DS, et al. NCCN Task Force report: management of patients with gastrointestinal stromal tumor (GIST)—update of the NCCN clinical practice guidelines. J Natl Compr Canc Netw. 2007;5(Suppl 2):S1–29. [PubMed] [Google Scholar]
- 32.Barckett P, Call J, Josephy PD, Scherzer NJ, Singleton R. Disease progression in patients with metastatic gastrointestinal stromal tumor (GIST) receiving Gleevec (imatinib): the effect of drug dosage. In: Connective Tissue Oncology Society 10th Annual Meeting, 2004. p. 227.
- 33.Delbaldo C, Chatelut E, Re M, Deroussent A, Seronie-Vivien S, Jambu A, Berthaud P, Le Cesne A, Blay JY, Vassal G. Pharmacokinetic–pharmacodynamic relationships of imatinib and its main metabolite in patients with advanced gastrointestinal stromal tumors. Clin Cancer Res. 2006;12:6073–6078. doi: 10.1158/1078-0432.CCR-05-2596. [DOI] [PubMed] [Google Scholar]
- 34.Widmer N, Decosterd LA, Csajka C, Leyvraz S, Duchosal MA, Rosselet A, Rochat B, Eap CB, Henry H, Biollaz J, Buclin T. Population pharmacokinetics of imatinib and the role of alpha-acid glycoprotein. Br J Clin Pharmacol. 2006;62:97–112. doi: 10.1111/j.1365-2125.2006.02719.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Demetri GD, Wang Y, Wehrle E, Blanke C, Joensuu H, von Mehren M. Correlation of imatinib plasma levels with clinical benefit in patients (Pts) with unresectable/metastatic gastrointestinal stromal tumors (GIST). In: Gastrointestinal Cancers Symposium, 2008. p. 3.
- 36.Judson I, Ma P, Peng B, Verweij J, Racine A, di Paola ED, van Glabbeke M, Dimitrijevic S, Scurr M, Dumez H, van Oosterom A. Imatinib pharmacokinetics in patients with gastrointestinal stromal tumour: a retrospective population pharmacokinetic study over time. EORTC Soft Tissue and Bone Sarcoma Group. Cancer Chemother Pharmacol. 2005;55:379–386. doi: 10.1007/s00280-004-0876-0. [DOI] [PubMed] [Google Scholar]
- 37.Burger H, Nooter K. Pharmacokinetic resistance to imatinib mesylate: role of the ABC drug pumps ABCG2 (BCRP) and ABCB1 (MDR1) in the oral bioavailability of imatinib. Cell Cycle. 2004;3:1502–1505. doi: 10.4161/cc.3.12.1331. [DOI] [PubMed] [Google Scholar]
- 38.Debiec-Rychter M, Sciot R, Le Cesne A, Schlemmer M, Hohenberger P, van Oosterom AT, Blay JY, Leyvraz S, Stul M, Casali PG, et al. KIT mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumours. Eur J Cancer. 2006;42:1093–1103. doi: 10.1016/j.ejca.2006.01.030. [DOI] [PubMed] [Google Scholar]