

Abstract
Background. Chediak-Higashi syndrome (CHS) is a rare, autosomal recessive disorder characterized by oculocutaneous albinism, immunodeficiency, coagulopathy and late-onset, progressive neurological dysfunction. It also has an “accelerated phase” characterized by hemophagocytic lymphohistiocytosis (HLH). The disease is caused by mutations in the CHS1/LYST gene located on chromosome 1, which affects lysosome morphology and function. We report the case of an African-American child with CHS in Case. This 16-month old African-American girl presented with fever and lethargy. The proband had pale skin compared to her parents, with light brown eyes, silvery hair and massive hepatosplenomegaly. Her laboratory evaluation was remarkable for pancytopenia, high serum ferritin and an elevated LDH. Bone marrow aspirate revealed large inclusions in granulocytes and erythrophagocytosis consistent with HLH. Genetic evaluation revealed two novel nonsense mutations in the CHS1 gene: c.3622C > T (p.Q1208X) and c.11002G > T (p.E3668X). Conclusions. Our patient is one of the few cases of CHS reported in the African American population. We identified 2 nonsense mutations in the CHS1 gene, the first mutation analysis published of an African-American child with Chediak-Higashi Syndrome. These two mutations predict a severe phenotype and thus identification of these mutations has an important clinical significance in CHS.
1. Introduction
Chediak-Higashi Syndrome (CHS) (MIM: 214500) is a rare autosomal recessive immune disease characterized by oculocutaneous albinism, a predisposition for infections, coagulopathies, neurological dysfunction, and large granules in many cell types [1–3]. CHS is the result of a genetic defect in the human CHS1/LYST gene (chromosome 1q421-q42.2), coding for the lysosomal trafficking regulator gene CHS1, resulting in defective formation of secretory vesicles and lysosomes [3–7]. Patients with CHS contain giant cytoplasmic inclusion bodies, large lysosomes, and lysosome-related organelles such as melanosomes. Patients have an increased risk of infection due to defects in natural killer cell activity, T-cell cytotoxicity, chemotaxis, and bactericidal killing by granulocytes and monocytes. A complication of CHS is hemophagocytic lymphohistiocytosis (HLH), which is also known as the “accelerated phase” of the disorder [5]. This phase is characterized by fever, jaundice, hepatosplenomegaly, lymphadenopathy, pancytopenia, and bleeding [8]. Ten to fifteen percent of CHS patients exhibit a milder clinical phenotype, with fewer or no severe infections and without an “accelerated phase” of the disease. Though these patients typically survive to adulthood, they are prone to developing progressive neurological dysfunction with age [6, 9].
The exact role of the CHS1 protein is unknown, but has been better defined through recent studies. CHS1 is thought to play a role in regulating vesicular trafficking and lysosomal and lysosome-related organelle size [3, 10, 11]. The milder phenotype of this disease occurs in patients who carry at least one missense mutation causing a partially functioning CHD1. Individuals with the more severe CHS phenotype usually have null mutant CHS1 alleles, predicting the absence of the CHD1 protein. The greater the impairment of the lysosomal function, the more susceptible an individual is to infections [3, 5, 6].
The “accelerated phase” is the most life threatening clinical feature of CHS, due to the high risk of infection and hemorrhage [5, 8, 12]. Treatments with etoposide and corticosteroids have resulted in transient remissions, but relapses are frequent and bone marrow transplants have been the only effective cure [8, 12]. The determining factor for remission after bone marrow transplantation is disease status at transplantation, and transplantation is most successful when a patient with CHS is not in the “accelerated phase” [12].
The majority of patients with CHS are generally of Caucasian or Japanese descent [5]. So far, not more than 5 cases of Chediak-Higashi syndrome in African-American patients have been described, and to our knowledge, no mutation analysis in these patients was reported [13–18]. Here we present a detailed clinical presentation of an African-American child with CHS, born to nonconsanginous parents. In addition, we describe the CHS1 mutation analysis identifying two novel nonsense mutations in her CHS1 gene.
2. Case Presentation
A sixteen-month-old African-American girl, presented to the Children's Hospital at Montefiore emergency room with a history of fever, decreased activity, poor feeding, increased sleepiness and irritability. She had no nausea, vomiting, diarrhea, urinary symptoms, cough or runny nose. The child was previously healthy, born full term without complications to a 24-year-old G2P1001 mother. On arrival to the emergency room she was febrile, tachypneic, tachycardic, and clinically in respiratory distress.
On examination she was noted to have silvery hair, blue eyes, and pale skin. Her eyelids were edematous, but she otherwise had a normal ear, nose, and throat exam. The heart and lung examination showed sinus tachycardia and tachypnea, but were otherwise within normal limits. The patient had a protuberant abdomen with massive hepatosplenomegaly. She had meningismus, but otherwise had a grossly normal neurologic exam and normal CSF studies and culture.
Initial laboratory results revealed pancytopenia, coagulopathy, metabolic acidosis, elevated LDH and normal uric acid. Our patient received a full sepsis, workup and was started on broad spectrum antibiotics for febrile neutropenia. The diagnosis of Chediak-Higashi syndrome was promptly determined after review of the peripheral blood smear and bone marrow biopsy (Figures 1 and 2). Bone marrow aspirate revealed large inclusions in myelocytes and erythrophagocytosis consistent with CHS and HLH. The bone marrow biopsy was diagnostic with sheets of histocytes with pale foamy cytoplasm, and pink granules in neutrophils both consistent with HLH in the accelerated phase. Electron microscopy revealed large inclusions in the granulocytes diagnostic of CHS (Figure 3). Interestingly this patient had platelets that were hypogranular to agranular as seen by electron microscopy, an unusual characteristic in CHS. Dermatology and genetics specialists assisted in confirmation of the diagnosis based on their clinical findings and the patient's hair analysis (Figure 4). The patient's hair analysis was consistent with other Chediak-Higashi hair samples [7, 19]. Further questioning of the parents revealed three maternal uncles had silvery hair, fair skin and frequent infections as children without any neurologic dysfunction.
Figure 1.
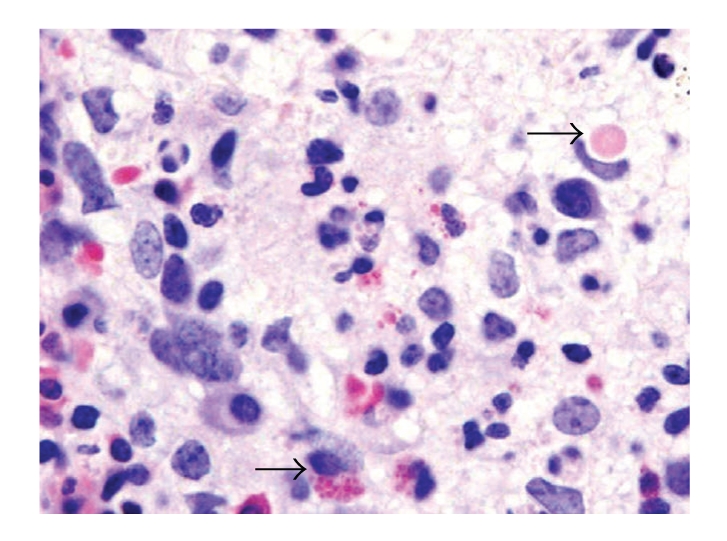
Wright stained bone marrow aspirate smear of the patient with Chediak-Higashi. The arrows in the upper right-hand side point to histiocytes engulfing intact red blood cells. The arrows in the lower left indicate fragmented red blood cells (hemophagocytosis).
Figure 2.
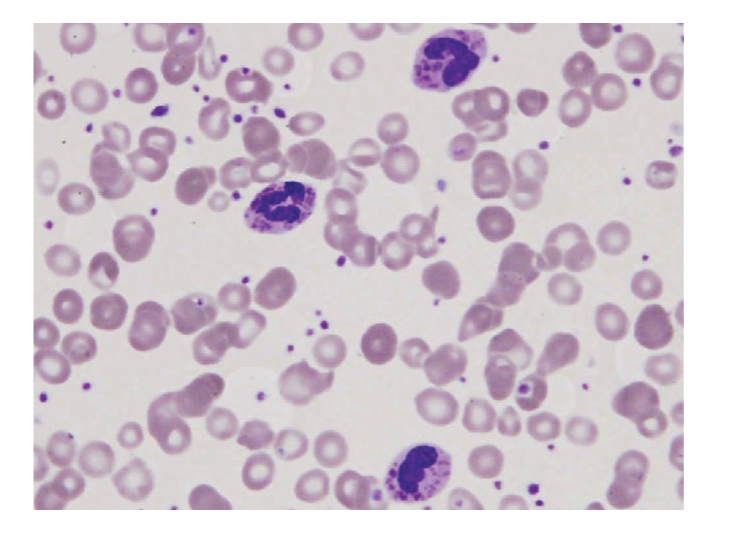
Peripheral blood smear of our patient with Chediak-Higashi depicting three polymorphonucelar leucocytes with inclusion bodies. These findings are typical in Chediak-Higashi syndrome.
Figure 3.
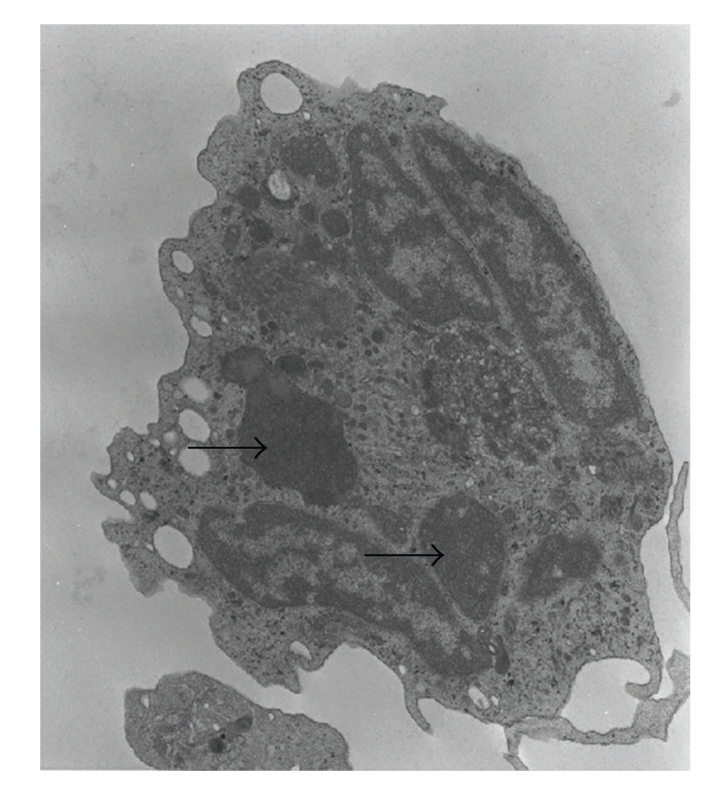
Electron microscopy of a neutrophil granulocyte of our patient. The arrows show abnormal large cytoplasmic inclusions.
Figure 4.

Hair shaft samples under high powered light microscopy. (a) African-American control hair sample, demonstrating evenly distributed pigment in the hairshaft. (b) Hair sample of a previously published severely affected Chediak-Higashi syndrome patient [19], demonstrating an atypical granular distribution of “pigmented clumps” in the hair shaft. (c) Hair sample of our African-American patient with Chediak-Higashi syndrome, demonstrating a similar atypical granular pigmentation pattern as the CHS patient in (b). All images were taken at the same settings using a 40X/1.3 oil objective on an Axiovert200M light microscope.
The patient received multiple transfusions including platelets, packed red blood cells, fresh frozen plasma and cryoprecipitate for her anemia, thrombocytopenia and coagulopathy. Within twenty-four hours of presentation she was given chemotherapy with etoposide, dexamethasone and cyclosporine as per protocol [20]. She had an excellent response after chemotherapy and a repeat bone marrow showed resolution of the histiocytic infiltration. Shortly after the patient was discharged she had to be readmitted for hypotensive shock and C difficile colitis. She recovered with broad spectrum antibiotics with fluid resuscitation and then was referred to another facility, but unfortunately, the patient succumbed to her disease before being able to receive a bone marrow transplant.
3. Molecular Analysis
After written consent was obtained, our patient's blood was sent for genetic testing to be done at the NIH. Genomic DNA was investigated for mutations in the CHD1 gene (GenBank NM_000081). Each of the 53 exons and their surrounding intronic regions were PCR amplified, sequenced and analyzed, using standard methods. Two heterozygous nonsense mutations in CHD1 were identified (Figure 5). Exon 5 displayed a c.3622C > T mutation, resulting in a change of a glutamine at codon 1208 to a termination codon (CAG > T A G; p.Q1208X). And exon 50 contained a heterozygous c.11002G > T mutation, resulting in a change of a glutamic acid at codon 3668 to a termination codon (GAA > T A A; p.E3668X). These mutations are predicted to result in nonsense mediated mRNA decay, and absence of a translated CHS1 protein.
Figure 5.

Two heterozygous nonsense mutations in the CHD1 gene in our patient; p.Q1208X in exon 8 and p.E3668X in exon 50.
4. Discussion
Chediak-Higashi syndrome is a rare autosomal recessive disorder characterized by varying degrees of oculocutaneous albinism, easy brusability and increased infections [3]. There is abnormal natural killer cell function with impaired chemotaxis and bactericidal activity. The underlying defects involve impaired lysosome-related organelle biogenesis [2, 6, 7, 21, 22].
We describe a case of CHS in the black population. Her clinical picture, laboratory studies, bone marrow, and genetic evaluation confirmed CHS. She presented with a severe phenotype in the accelerated phase of CHS and had certain unusual phenotypic features such as agranular platelets. We identified 2 novel nonsense mutations in the CHS1 gene (p.Q1208X and p.E3668X), which predict the absence of a translated CHD1 protein, along with a severe phenotype as seen in our patient. Hence identification of these mutations has an important clinical significance in predicting the phenotypic severity of patients with CHS. Furthermore, these mutations are the first CHD1 defects described in the black population, and may serve as a source for future mutation analysis in this population.
Acknowledgments
The authors thank Heidi Dorward for skillful analysis and imaging of hair samples. This work was partially supported by the Intramural Research programs of the National Human Genome Research Institute, National Institutes of Health, Bethesda, Md, USA.
References
- 1.White JG. The Chediak-Higashi syndrome: a possible lysosomal disease. Blood. 1966;28(2):143–156. [PubMed] [Google Scholar]
- 2.Introne W, Boissy RE, Gahl WA. Clinical, molecular, and cell biological aspects of Chediak-Higashi syndrome. Molecular Genetics and Metabolism. 1999;68(2):283–303. doi: 10.1006/mgme.1999.2927. [DOI] [PubMed] [Google Scholar]
- 3.Kaplan J, De Domenico I, Ward DM. Chediak-Higashi syndrome. Current Opinion in Hematology. 2008;15(1):22–29. doi: 10.1097/MOH.0b013e3282f2bcce. [DOI] [PubMed] [Google Scholar]
- 4.Nagle DL, Karim MA, Woolf EA, et al. Identification and mutation analysis of the complete gene for Chediak-Higashi syndrome. Nature Genetics. 1996;14(3):307–311. doi: 10.1038/ng1196-307. [DOI] [PubMed] [Google Scholar]
- 5.Karim M, Suzuki K, Fukai K, et al. Apparent genotype-phenotype correlation in childhood, adolescent, and adult Chediak-Higashi syndrome. American Journal of Medical Genetics. 2002;108(1):16–22. [PubMed] [Google Scholar]
- 6.Westbroek W, Adams D, Huizing M, et al. Cellular defects in Chediak-Higashi syndrome correlate with the molecular genotype and clinical phenotype. Journal of Investigative Dermatology. 2007;127(11):2674–2677. doi: 10.1038/sj.jid.5700899. [DOI] [PubMed] [Google Scholar]
- 7.Huizing M, Helip-Wooley A, Westbroek W, Gunay-Aygun M, Gahl WA. Disorders of lysosome-related organelle biogenesis: clinical and molecular genetics. Annual Review of Genomics and Human Genetics. 2008;9:359–386. doi: 10.1146/annurev.genom.9.081307.164303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Janka GE. Familial and acquired hemophagocytic lymphohistiocytosis. European Journal of Pediatrics. 2007;166(2):95–109. doi: 10.1007/s00431-006-0258-1. [DOI] [PubMed] [Google Scholar]
- 9.Tardieu M, Lacroix C, Neven B, et al. Progressive neurologic dysfunction 20 years after allogenic BMT for Chediak- Higashi Syndrome. Blood. 2005;106:40–42. doi: 10.1182/blood-2005-01-0319. [DOI] [PubMed] [Google Scholar]
- 10.Tchernev VT, Mansfield TA, Giot L, et al. The Chediak-Higashi protein interacts with SNARE complex and signal transduction proteins. Molecular Medicine. 2002;8(1):56–64. [PMC free article] [PubMed] [Google Scholar]
- 11.Ward DM, Shiflett SL, Huynh D, Vaughn MB, Prestwich G, Kaplan J. Use of expression constructs to dissect the functional domains of the CHS/beige protein: identification of multiple phenotypes. Traffic. 2003;4(6):403–415. doi: 10.1034/j.1600-0854.2003.00093.x. [DOI] [PubMed] [Google Scholar]
- 12.Eapen M, DeLaat CA, Baker KS, et al. Hematopoietic cell transplantation for Chediak-Higashi syndrome. Bone Marrow Transplantation. 2007;39(7):411–415. doi: 10.1038/sj.bmt.1705600. [DOI] [PubMed] [Google Scholar]
- 13.Valenzuela R, Aikawa M, O’Regan S, Makker S. Chediak-Higashi syndrome in a black infant. A light and electron microscopic study with special emphasis on erythrophagocytosis. American Journal of Clinical Pathology. 1976;65(4):483–494. doi: 10.1093/ajcp/65.4.483. [DOI] [PubMed] [Google Scholar]
- 14.O’Regan S, Newman AJ, Valenzuela R, Makker SP, Graham RC. The Chediak Higashi syndrome in a Negro infant. Journal of Pediatrics. 1976;88(3):522–523. doi: 10.1016/s0022-3476(76)80289-2. [DOI] [PubMed] [Google Scholar]
- 15.De Beer HA, Anderson R, Findlay GH. Chediak-Higashi syndrome in a “black” child. Clinical features, immunological studies, and optics of the hair and skin. South African Medical Journal. 1981;60(3):108–112. [PubMed] [Google Scholar]
- 16.Leal I, Merino F, Soto H, et al. Chediak-Higashi syndrome in a Venezuelan black child. Journal of the American Academy of Dermatology. 1985;13(2, supplement):337–342. doi: 10.1016/s0190-9622(85)70171-5. [DOI] [PubMed] [Google Scholar]
- 17.Anderson LL, Paller AS, Malpass D, Schmidt ML, Berger TG. Chediak-Higashi syndrome in a black child. Pediatric Dermatology. 1992;9(1):31–36. doi: 10.1111/j.1525-1470.1992.tb00322.x. [DOI] [PubMed] [Google Scholar]
- 18.Maari CH, Eichenfield LF. Congenital generalized hypomelanosis and immunodeficiency in a black child. Pediatric Dermatology. 2007;24(2):182–185. doi: 10.1111/j.1525-1470.2007.00371.x. [DOI] [PubMed] [Google Scholar]
- 19.Zarzour W, Kleta R, Frangoul H, et al. Two novel CHS1 (LYST) mutations: clinical correlations in an infant with Chediak-Higashi syndrome. Molecular Genetics and Metabolism. 2005;85(2):125–132. doi: 10.1016/j.ymgme.2005.02.011. [DOI] [PubMed] [Google Scholar]
- 20.Henter JI, Horne A, Aricó M, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatric Blood and Cancer. 2007;48(2):124–131. doi: 10.1002/pbc.21039. [DOI] [PubMed] [Google Scholar]
- 21.Spritz RA. Genetic defects in Chediak-Higashi syndrome and the beige mouse. Journal of Clinical Immunology. 1998;18(2):97–105. doi: 10.1023/a:1023247215374. [DOI] [PubMed] [Google Scholar]
- 22.Ward DM, Shiflett SL, Kaplan J. Chediak-Higashi Syndrome: a clinical and molecular view of a rare liposomal storage disorder. Current Molecular Medicine. 2002;2:469–477. doi: 10.2174/1566524023362339. [DOI] [PubMed] [Google Scholar]