

Abstract
The applicability of immunotherapy would be dramatically broadened to a greater number of recipients if direct “off-the-shelf” products could be engineered to engender functionally potent immune responses against true “self”-tumor antigens. This would obviate the need for ex vivo culture of dendritic cells or T cells on a patient-by-patient basis, for example. The carcinoembryonic antigen (CEA) is a glycoprotein expressed in normal gut epithelium that is up-regulated in the majority of colon cancers, non-small cell lung cancers, and half of all breast cancers. Such properties make CEA an excellent and important target for cancer immunotherapy. In this study, we show stabilization of 14-day established s.c. mGC4CEA tumors in human CEA (huCEA) transgenic mice following two direct low-dose injections of 0.15 × 106 transducing units of a lentiviral vector (LV) that directs expression of huCEA (LV-huCEA). This stabilization result was reproducible and detailed analyses including antibody assays, multiplex cytokine analyses on unstimulated splenocytes, lymph node cell characterizations, tetramer staining, and immunofluorescence staining of tumor sections showed that this outcome correlated with both a cellular and humoral immune response. Similar tumor outcomes were not seen when mice were vaccinated with a control LV that engineered expression of enGFP only. The long-term potency of this vaccination strategy was also studied and revealed the requirement for maintenance of tumor antigen-specific immunity for efficient tumor control. These data support the use of direct injections of low doses of LV-huCEA for enhancement of tumor immunotherapy directed against CEA.
Introduction
Immunotherapy against cancer aims to either broadly stimulate the immune system or generate a specific immune response against a unique tumor-associated antigen or both (1, 2). Dendritic cells prime resident T cells to become either activated cytotoxic T lymphocytes (CTLs) or T helper cells for inducing both cellular and humoral immunity. As they are the most potent antigen-presenting cells (APC) in the immune system, a promising immunotherapy strategy is the transduction of autologous dendritic cells by a virus engineered to generate expression of a tumor-associated antigen (1, 2). In this context, the use of lentiviral vectors (LV) as vectors has several advantages. Not only are LVs less immunogenic themselves and have improved safety coefficients compared with some other gene delivery vehicles, they are also capable of efficiently transducing dendritic cells ex vivo (see refs. 3–5 for example). Another potential target for LV transduction in this area is effector T cells (6, 7).
An alternative approach to ex vivo transduction of immune system cells that has recently shown potential is direct in vivo administration of LVs (8). A major advantage here is that the same vaccine may be administrated to all recipients, whereas autologous dendritic cell transduction, for example, requires cells to be isolated and prepared from each individual. This direct administration schema stems from the observation that straightforward LV delivery in mice allows for APC transduction (9). More specifically, following footpad injections of LV, transduced splenic and draining lymph node cells were found to be CD11c+ dendritic cells (10). In addition, an increased amplitude and longevity of the CTL response was obtained by footpad injections of LV compared with that obtained by transplantation of transduced dendritic cells (10).
Relatively low levels of vector transduction seem to be sufficient to generate immune responses against expressed antigens: effective antitumor immunity targeting the chicken OVA antigen has been shown by footpad injections of 107 LV transducing units (TU) despite the fact that possibly <0.1% of cells in the draining lymph nodes were functionally transduced (11). The antitumor efficiency of this approach has also been shown by targeting the Neu antigen in a mouse model of spontaneous breast tumor development (12). However, the need for production of highly concentrated purified virus to achieve the high doses of LV often used in these studies may be a limitation of this approach, especially when scaled-up appropriately for patients in the clinic. In the present study, we chose to test the outcome of using low doses of a LV vaccine in a human carcinoembryonic antigen (CEA)-expressing tumor model. CEA is an important target in the context of the development of effective immunotherapy strategies. Use of human CEA (huCEA) transgenic mice makes this a true self-antigen model; thus, results may be more informative than those obtained using heterologous antigens.
CEA expression is up-regulated in the majority of colon cancers, non-small lung cancers, and half of all breast cancers (13). Colon cancer is a good target for the development and implementation of effective immunotherapy approaches as, unfortunately, >20% of patients have metastatic disease at the time of diagnosis, and surgery can only be done in ∼20% of cases (14). In addition, some CEA vaccine strategies have already been employed in clinical trials (15). These trials have shown safety and the induction of anti-CEA immune responses, but clinical results have generally been limited thus far (15). Several virus-based anti-CEA-expressing cancer vaccine schemas have contributed to these clinical responses but have required boosting the immune system (15) either by using different viruses for prime-and-boost injections (16, 17) or by treating with costimulatory molecules (16, 18) and/or cytokines (17).
Our goal is to develop a broadly applicable vaccination system against CEA as a tumor-associated antigen on a scale that may be relevant for clinical trials. Toward this, we have employed in vivo administration of low doses of a LV that directs expression of huCEA in this study and a stringent huCEA transgenic (huCEA Tg) mouse model to examine true self-antigen responses. We have also performed numerous assays to better understand the immune response generated. We observed that we can safely break tolerance to huCEA and engineer an efficient antitumor response along with anti-huCEA antibody and CTL responses. This work thus provides a platform for the further development of this strategy toward full clinical realization against this important target molecule.
Materials and Methods
Lentiviruses
The huCEA cDNA was graciously provided by Dr. Jeffrey Schlom (NIH) and used as a template by Lentigen to construct LV-huCEA. LV expressing enhanced green fluorescent protein (LV-enGFP; Lentigen) was used as a control. Viral functional titers (TU/mL) were determined by infection of 293T cells and subsequent analysis of transgene expression by flow cytometry.
Mice
huCEA Tg mice (19) were housed in a pathogen-free environment in the animal facility at the University Health Network and studies were done under the Animal Care Committee approval. Eight- to 10-week-old huCEA Tg mice were used for antitumor immunity studies. For ethical reasons, mice were euthanized before the tumor diameter exceeded 15 mm.
mGC4CEA and mGC8 Tumor Cell Lines
The murine gastric carcinoma cell line expressing huCEA (mGC4CEA) was derived from CEA424/SV40Tag mice (20) and used to establish s.c. tumors. Similarly, mGC8 cells were derived from SV40Tag mice and used as specificity control in assays. These cells were cultured in RPMI 1640 supplemented with 10% heat-inactivated FCS (FCS “Gold”; PAA Laboratories), 2 mmol/L l-glutamine, 100 units/mL penicillin, 100 μg/mL streptomycin, nonessential amino acids, and 1 mmol/L sodium pyruvate (Life Technologies/Invitrogen).
In vivo Transduction Assessment by Microscopy
Mice were immunized in the footpad with 1 × 107 TU LV-enGFP control vector. Two days later, the popliteal lymph nodes were removed and frozen in Shandon Cryomatrix (Thermo Scientific). Tissues were cut into 5-μm-thick sections on glass slides. Cryosections were fixed with ice-cold methanol/acetone, washed, and incubated with blocking solution (PBS containing 0.25% Triton X-100 and 1% bovine serum albumin) for 30 min at room temperature. A mixture of primary antibodies [PE anti-mouse CD11c (eBioscience) and rabbit anti-GFP (Invitrogen) diluted in 1% bovine serum albumin, PBS-Triton X-100] were added to the sections and incubated overnight at 4°C. Sections were then washed and incubated with Alexa Fluor 488 anti-rabbit IgG (Molecular Probes) for 30 min at room temperature. After incubation with 4′,6-diamidino-2-phenylindole (DAPI), the slides were sealed with Fluoromount-G mounting medium (Southern Biotechnology Associates). Sections were analyzed with an Olympus BX-50 microscope using a Photometrics Cool-SnaPHQ2 camera. Five areas (×40 objective) per section were captured and the double staining (GFP + CD11c) was analyzed by Image J software.
In vivo Transduction Assessment by Flow Cytometry
Male C57BL/6 mice (12 weeks old) were obtained from The Jackson Laboratory. Mice were anesthetized with isoflurane. LV-enGFP (manufactured by Lentigen) was then injected into the right footpad (30 μL volume containing 6.4 × 105 infectious units); the LV-enGFP titer was 2.1 × 107 IU/mL (tested on 293T cells by serial dilution). On day 3 after LV administration, one ipsilateral and one contralateral inguinal lymph node was collected per mouse; a total of 14 mice were injected with LV, and two to three lymph nodes were pooled for each analysis. Five mice that were not injected with LV served as controls (bilateral lymph nodes were harvested). Mechanical disruption and collagenase treatment (30 min; Roche) were used to generate lymph node single-cell suspensions. Cells were filtered (70 μm filters; BD Biosciences) and stained with the following antibodies or appropriate isotype controls: anti-CD11c Pacific Blue (BioLegend), anti-CD45RA PE, anti-CD8α PE-Cy5, anti-CD4 PE-Cy7, anti-CD11b APC, and anti-Gr-1 APC-Cy7 (all from BD Biosciences). Fluorescence was measured by flow cytometry using a LSRII instrument (BD Biosciences) and analyzed using CellQuest (BD Biosciences).
Tumor Rejection Experiments
For therapeutic tumor treatment, mice were grafted s.c. at day 0 with 0.8 × 106 mGC4CEA tumor cells in the flank and subsequently immunized in the footpad on days 14 and 21 with PBS, 0.15 × 106 TU LV-enGFP control, or 0.15 × 106 TU LV-huCEA. Tumor growth was evaluated by caliper measurement until 28 days post-tumor challenge. Tumor volumes were calculated using the formula: L × W × H. In the second experiment, one additional group of LV-huCEA-vaccinated mice was randomly assigned at the beginning to receive one more dose of 0.1 × 106 TU LV-huCEA vaccine at day 28. This group was called LV-huCEA_Lg (for long-term), whereas the other huCEA group of this second experiment was designated LV-huCEA_Sh (for short-term). Tumor growth in the LV-huCEA_Lg group was assessed for >50 days post-tumor challenge.
Detection and Characterization of the Anti-huCEA Antibody Response
Serum samples were collected from huCEA Tg mice before and during the tumor rejection experiments. Based on a protocol by Cusi et al. (21), 96-well microtiter plates were coated with 1 μg/mL purified huCEA (Chemicon International) and incubated at 4°C overnight. Wells were washed with PBS-0.05% Tween 20 and blocked with 5% heat-inactivated FCS in PBS for 2 h at room temperature. Duplicate 100 μL aliquots of sample (sera diluted 1:40) were allowed to react in the wells for 1 h at 37°C. Mouse anti-huCEA monoclonal antibody, COL-1 (Zymed), was diluted 1:200 and used as a positive control. Following washes with PBS-0.05% Tween 20, 100 μL of a 1:30,000 dilution of goat horseradish peroxidase-labeled anti-mouse IgG (Bio-Rad) was added, and the plate was incubated for 1 h at 37°C. After washes with PBS-0.05% Tween 20, the substrate 3,3′,5,5′-tetramethylbenzidine (Sigma-Aldrich) was added to each well and allowed to react at room temperature for 30 min in the dark. The reaction was stopped with 100 μL of 1 mol/L H3PO4 and plates were analyzed by spectrophotometry at 450 nm. Induction rate of antibodies for individual mice were calculated at each time point relative to the absorbance measured for the pre-immune serum. Positive sera were considered to be those showing an absorbance at least 2.2 times the pre-immune value.
For the complement-dependent cytotoxicity assay, mGC4CEA cells (105) were plated in 96-well plates and incubated overnight at 37°C. The supernatant was removed and the cells were incubated with 50 μL mouse serum (diluted 1:20) for 30 min at 37°C. Rabbit complement (Sera Laboratories International; diluted 1:4, 50 μL total volume) was then added into wells. After 1 h at 37°C, 20 μL MTS reactive reagent (Promega) was added. Cells treated with 0.25% Triton X-100 were included as a 100% lysis control and cells treated with complement alone as a 0% lysis control. MGC8 cells were used to determine the specificity of the assay. Absorbance readings were obtained at 490 nm using a SpectraMax Plus 384 microplate reader (Molecular Devices) at 1, 2, and 3 h after incubation. The cytotoxicity activity of the antiserum was expressed as percentage of specific lysis (% total lysis - % nonspecific lysis).
Splenocyte Harvest and Culture
Splenic homogenates from huCEA Tg mice were filtered through a 40 μm cell strainer (BD Falcon) and collected by low-speed centrifugation. RBC were lysed in 1 mL RBC lysing buffer (Sigma). The remaining splenocytes were cultured at a concentration of 2 × 106 cells/mL in RPMI 1640 supplemented with 10% heat-inactivated bovine serum (PAA Laboratories), 2 mmol/L l-glutamine, 100 units/mL penicillin, 100 μg/mL streptomycin, 50 μmol/L β-mercaptoethanol, and 3.3 mmol/L N-acetylcysteine. This medium was supplemented every 48 h with 20 units/mL recombinant human interleukin (IL)-2 (Roche) and 20 ng/mL human IL-7 (Peprotech).
Multiple Cytokine Analysis
Splenocytes were prepared and cultured as above without any stimulation. Supernatants from cell cultures were harvested after 24 and 48 h and frozen for later analysis. IFN-γ, IL-2, tumor necrosis factor-α, IL-4, and IL-10 levels were measured from thawed supernatant samples by Luminexusing Bio-Plex multiplex sandwich immunoassays according to the manufacturer's protocol (Bio-Rad Laboratories). IL-4 and IFN-γ secretion by the cells were also assessed by single separate ELISA according to the manufacturer's instructions (BD Biosciences).
IFN-γ Secretion Assay
Splenocytes were pooled according to group and cocultured for 48 h with mGC4CEA cells at a maximum spleen cell-to-tumor cell ratio of 20:1. Each cell type was also cultured alone to determine constitutive cytokine secretion. Evaluation of IFN-γ secretion by the cells was done by ELISA according to the manufacturer's instructions (BD Biosciences).
Tetramer Staining
To detect huCEA-specific CTLs in huCEA Tg mice after vaccination, pooled splenocytes were stained with APC-conjugated anti-CD8 monoclonal antibody and PE-conjugated huCEA/H-2Db tetramer specific for the EAQNTTYL immunogenic peptide (iTag; Beckman Coulter). After blocking, immunofluorescence staining was done following the manufacturer's instructions. Immunofluorescence was compared with the appropriate isotype-matched controls and analyzed with CellQuest software using a FACSCalibur cytometer (BD Biosciences).
Immunofluorescence Staining of Tumor Sections
At the end of the second tumor rejection experiment, tumors from 3 mice per group were harvested and frozen. Sections (4 μm) were fixed for 15 min at room temperature in a 1:1 methanol/acetone solution. Dry sections were washed with cold PBS and incubated for 1 h with a blocking solution (PBS containing 1% bovine serum albumin and 0.2% gelatin). Sections were incubated for 45 min with anti-CD4 or anti-CD8 primary antibody (eBioscience) at a 1:50 dilution in PBS containing 0.5% bovine serum albumin and 0.2% gelatin. After washes with PBS, Alexa Fluor 488-conjugated secondary anti-IgG antibody (Molecular Probes) was added at a dilution of 1:200 for 45 min. Secondary antibody alone was used as a background control. DAPI solution was used for nuclei staining. Two to three pictures per section were analyzed using an Axioskop 2 microscope linked to an AxioCam MRc camera (Zeiss). For each area, the ratios between the positive area fractions obtained with Alexa Fluor 488 and DAPI was evaluated using ImageJ software and designated as A/D ratios. Average ratios were calculated per mouse and the background was deducted by subtracting the ratio obtained for the negative control. The result was designated as “absolute A/D ratios.”
Statistical Analyses
Student's t test analysis was done with P < 0.05 designated to be statistically significant.
Results
LV-huCEA Induces Therapeutic Immunity Leading to mGC4CEA Tumor Regression in huCEA Tg Mice
We generated a novel recombinant LV that engineered expression of huCEA. This LV employed a human codon-optimized cDNA sequence that also removed cryptic splice sites to maximize stability and transgene expression in transduced cells.
The potential efficacy of LV-huCEA as a vaccine against huCEA-expressing tumors was assessed in vivo employing nonconditioned huCEA Tg mice. We evaluated the antitumor effect in a relatively stringent model using 14-day established tumors. Mice were injected s.c. in the flank with mGC4CEA tumor cells. After the tumors became palpable, mice were vaccinated in the footpad with PBS or recombinant LVs that direct expression of huCEA or enGFP (negative control). Vaccinated mice received a boost injection 1 week later. Tumor growth was assessed until day 28 in short-term experiments. In duplicate experiments done according to this schedule, the kinetics of tumor growth in analogous groups was the same. Figure 1 shows results from the first experiment. We observed stabilization of average tumor sizes in the LV-huCEA-vaccinated mice beginning after the first vaccination on day 14. In contrast, tumors in PBS- or LV-enGFP-vaccinated mice continued to grow at the same rate until the end of the experiment (Fig. 1). Overall, in both experiments, tumors in the LV-huCEA-vaccinated mice were significantly smaller than those in the mice of the PBS and LV-enGFP groups (P < 0.05 at day 23 and P < 0.005 at day 28).
Figure 1.
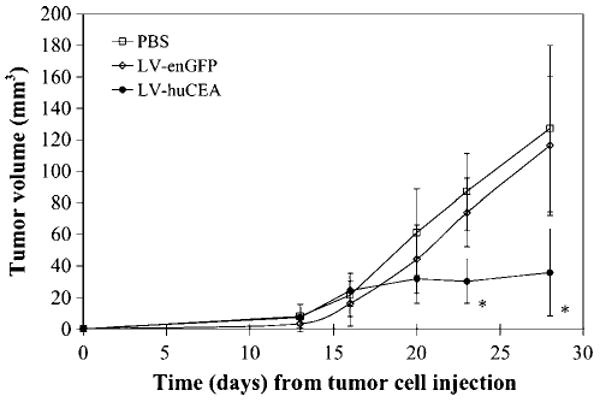
Direct immunization with LV-huCEA induces huCEA-expressing tumor stabilization. In this representative experiment, huCEA Tg mice were injected s.c. with mGC4CEA cells in the flank. Mice were then directly vaccinated with PBS (5 mice; empty squares), LV-huCEA (6 mice; filled circles), or LV-enGFP (5 mice; empty diamonds). Tumors were measured over time using a caliper. *, P < 0.005, compared with PBS and LV-enGFP controls.
Interestingly, although the collective average tumor size observed in the LV-huCEA-treated groups stabilized over the course of the experiments, differential growth rates of individual tumors were observed. In some mice, a regression of the tumor actually occurred, whereas tumors grew slower or at the normal rate in other mice. Yet, the latter effect was rare, with >65% of the LV-huCEA-vaccinated mice actually showing tumor regression in both first (4 of 6) and second (6 of 9) experiments. In most cases, this regression was seen a few days after the second LV injection, except for in a few mice for which tumor regression began before the second injection (1 of 4 and 2 of 6 mice, respectively). In each experiment, only one LV-huCEA-vaccinated mouse did not respond to the vaccine and showed unrestrained tumor growth (data not shown).
Vaccination with LV-huCEA Breaks Tolerance and Leads to Anti-huCEA Antibody Production in huCEA Tg Mice
Blood was collected from the huCEA Tg mice during the tumor stabilization experiments to check for anti-huCEA antibodies in sera by ELISA (Fig. 2A and B, respectively) to determine whether this vaccination strategy was able to break immune tolerance and induce a humoral response. The antibody levels detected were noticeably higher in the sera of LV-huCEA-vaccinated mice only after the boost injection. On average, the absorbance readings measured at day 28 for sera from mice of the LV-huCEA group were significantly higher than the absorbance measured before immunization (P < 0.05, Student's t test), whereas differences were not observed for the control groups. This significant induction showed the ability of this vaccination strategy to break self-tolerance to huCEA in this model. Looking at the results from individual animals, we did not detect any significant anti-huCEA antibody induction in any of the sera from the PBS groups. In each LV-enGFP group, one mouse showed some increase in anti-huCEA antibody production. On the other hand, induction of anti-huCEA antibody production was observed following the immunizations in the sera from 4 of 6 and 7 of 9 LV-huCEA-vaccinated mice from the first and second studies, respectively. In addition, this relevant anti-huCEA antibody production appeared to correlate with antitumor immunity, because there was no antibody induction in the sera from the 2 LV-huCEA-vaccinated mice that did not show any tumor regression.
Figure 2.
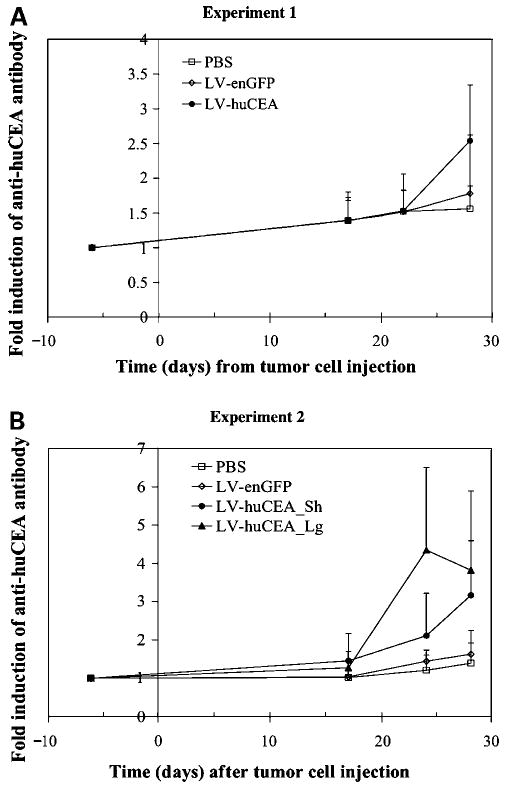
Direct immunization with LV-huCEA induces anti-huCEA antibody secretion in huCEA Tg mice sera. huCEA Tg mice bearing mGC4CEA tumors were vaccinated with PBS (5 and 4 mice in A and B, respectively; empty squares), LV-huCEA (6 mice in A and 5 + 4 mice in B; filled circles and triangles, respectively), or LV-enGFP (5 mice in both A and B experiments; empty diamonds). Anti-huCEA antibodies were titered by ELISA. Fold induction of antibody production was calculated relative to values obtained from the pre-immune serum. LV-huCEA_Sh stands for short-term and LV-huCEA_Lg stands for long-term (see text for details).
Vaccination with LV-huCEA Induces a Balanced Th1/Th2 Pattern of Cytokine Activation in huCEA-Tg Mice
To assess the constitutive activation status of the immune system in the vaccinated mice, splenocytes from individual mice were harvested and cultured without additional in vitro stimulation. Supernatants (24 and 48 h) were collected and subjected to multiple cytokine detection assays. Results revealed that LV-huCEA immunization generated cytokines from splenocytes characteristic of both humoral and cellular immune responses (Fig. 3). Indeed, a clear increase in the secretion of both IFN-γ, a Th1-type cytokine, and IL-4, a Th2-type cytokine, were detected at significant levels (Fig. 3A and B). The induction of IL-4 secretion following LV-huCEA vaccination was significant compared with the levels measured for splenocytes from control groups in both experiments (Fig. 3B; P < 0.05, Student's t test). Regarding IFN-γ secretion, the difference with control groups was significant in the first experiment only (P < 0.05, Student's t test). However, for the second experiment, the only mouse of the LV-huCEA group that had uninhibited tumor growth did not show any clear induction of IFN-γ secretion, suggesting a direct link between the Th1 response and the tumor regression. When data from this nonresponsive mouse were removed from analysis, IFN-γ secretion induction for this group reached statistical significance. Note that a significant induction of IL-2 and IL-10 secretion was also shown in the first experiment (P < 0.05, Student's t test; Fig. 3C and D). In the second experiment, we also detected an increase of IL-2 and IL-10 cytokine secretion that was significant compared with the PBS group levels (P < 0.05, Student's t test). Nevertheless, this induction was not statistically different from the one measured for the LV-enGFP group. Indeed, IL-2 and IL-10 secretion were also slightly stimulated by the LV-enGFP vaccine. This suggests that the transduction itself or enGFP might be involved in the induction of a full immune response. Levels of tumor necrosis factor-α observed were more variable and we only detected a trend toward a higher secretion of this cytokine after LV-huCEA vaccination in both experiments (data not shown).
Figure 3.
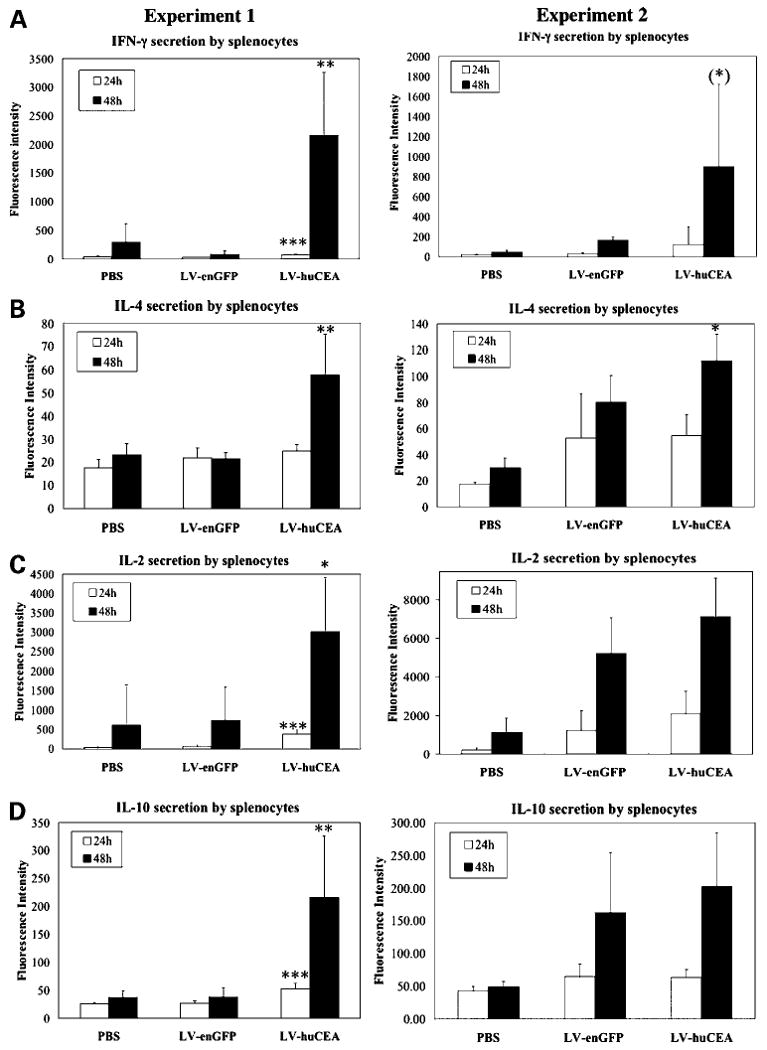
Direct immunization with LV-huCEA induces cytokine secretion by unstimulated splenocytes. Splenocytes from huCEA Tg mice sacrificed at day 28 were cultured and secreted cytokines were detected at 24 h (open columns) and 48 h (shaded columns). A to D, fluorescence intensity measured for IFN-γ, IL-4, IL-2, and IL-10, respectively. Left, first tumor stabilization experiment; right, second stabilization experiment. *, P < 0.05; **, P < 0.005; ***, P < 0.0005 (LV-huCEA versus LV-enGFP).
LV-huCEA Vaccinations Induced a huCEA-Specific T-Cell Response
In the second stabilization experiment, we wanted to determine whether huCEA-specific T cells had been induced by the LV-huCEA immunizations. Specific T-cell activity should be detectable in the presence of target cells expressing huCEA. To show this, we cultured pooled splenocytes from immunized mice alone or in presence of the target mGC4CEA cells for 48 h and then measured IFN-γ secretion by ELISA. The optimal effector-to-target cell ratio was found to be 20:1. Vaccinated mice had high constitutive IFN-γ secretion (Fig. 4A). Splenocytes from LV-huCEA-vaccinated mice subsequently secreted significantly more IFN-γ than splenocytes from the control group (P < 0.005, Student's t test; Fig. 4A), confirming the general activation status elucidated by quantitation of selected cytokines (as above). In addition, we detected significantly more IFN-γ secreted by the splenocytes from LV-huCEA-vaccinated mice when they were in the presence of mGC4CEA target cells (P < 0.05, Student's t test). This did not occur in the other groups. This result suggests the presence of active T cells specific for the huCEA antigen. Tumor specificity was confirmed by the absence of IFN-γ secretion when splenocytes were cultured in the presence of CEA-negative mGC8 cells (data not shown).
Figure 4.
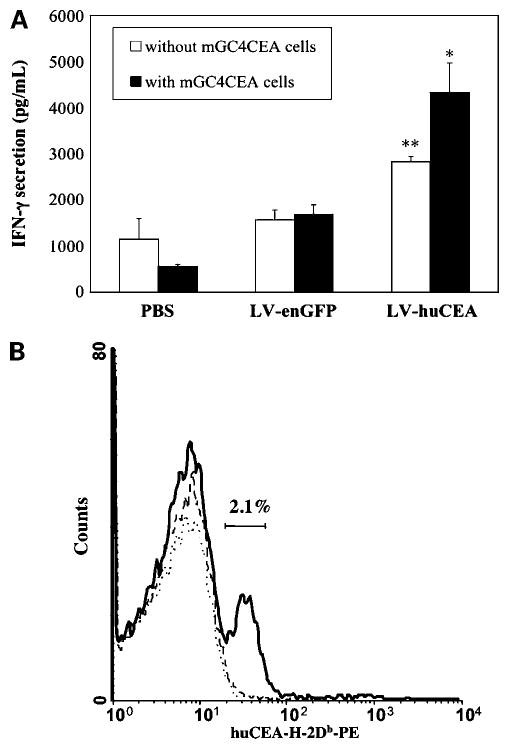
Direct immunization with LV-huCEA induces a huCEA-specific T-cell response. A, specific quantification of IFN-γ secretion in the presence of huCEA-expressing target cells. At the end of the second tumor stabilization experiment, pools of splenocytes from huCEA Tg mice were cultured alone (open columns) or with mGC4CEA cells (shaded columns). For the cocultures, cells were cultured with an effector-to-target ratio of 20:1 and IFN-γ secretion was measured by ELISA. **, P < 0.005 compared with PBS and LV-enGFP controls; *, P < 0.05 for LV-huCEA with mGC4CEA cells compared with splenocytes incubated without mGC4CEA cells. B, immunization with LV-huCEA induces the production of a CD8+ population specific for a CEA peptide. After the second tumor stabilization experiment, pooled splenocytes from vaccinated huCEA Tg mice were stained either with an APC-conjugated anti-CD8 antibody and a PE-conjugated CEA/H-2Db-tetramer (shown here) or with the matched isotype controls (flat curves; not shown). Number of cells relative to FL-2 fluorescence, gated on CD8+ cells. Continuous thick line, representative of splenocytes from LV-huCEA-vaccinated mice; thin dotted lines, staining of the control splenocytes.
To actually enumerate huCEA-specific CTLs that could be responsible for the antitumor activity, we double-stained splenocytes from the second vaccination experiment with an APC-conjugated anti-CD8 antibody and a PE-conjugated huCEA/H-2Db tetramer. It was found that ∼2.1% of CD8+ splenocytes were specific for the huCEA peptide (Fig. 4B). These data provide evidence for the induction of a huCEA-specific CTL response and opens the door to future gene therapy studies that may increase this percentage.
Long-term Persistence of the Antitumor Immune Response Likely Requires the Presence of Anti-huCEA Antibody and Antigen-Specific T Cells
In the second tumor vaccination experiment, we also wanted to address barriers to the long-term persistence of the antitumor immunity induced by LV-huCEA immunization. To this end, we included an additional recipient group to be followed for 2 months. This group (LV-huCEA_Lg) received a third viral vaccination 1 week after the second administration but with a 33% lower dose of 0.1 × 106 TU. The sizes of all tumors in this cohort remained stable until at least 8 days after the last low-dose LV-huCEA injection (corresponding to day 36 after the tumor cell inoculation). Then, tumors resumed growth in 3 of 4 mice between days 36 and 43; one mouse had a very small stable tumor (<3 mm3) for the duration of the experiment (Fig. 5A). Concomitant with accelerated tumor growth, we measured a rapid decline in the anti-huCEA antibody levels in the sera (Fig. 5B). This correlated with a decrease in IL-4 secretion by splenocytes as measured by the Luminex method (data not shown). Further on this topic, the only mouse that maintained appreciable anti-huCEA antibody levels was the one harboring the small stable tumor (Fig. 5A and B). Lastly, we used the complement-dependent cytotoxicity assay to determine if the antibodies produced in this cohort of animals were able to specifically lyse target cells mediated by the complement pathway. Serum samples from 5 mice were analyzed at 36, 43, 51, and 56 days after tumor cell inoculation. At 43 days, serum from 4 of 5 animals showed significant complement-dependent cytotoxicity activity with levels up to 20% specific lysis (data not shown). This percentage declined, however, by the next assay point wherein only 2 animals showed significant complement-dependent cytotoxicity activity (data not shown). Indeed, this reduction correlated with increased tumor growth, especially for animal M276 (Fig. 5A).
Figure 5.
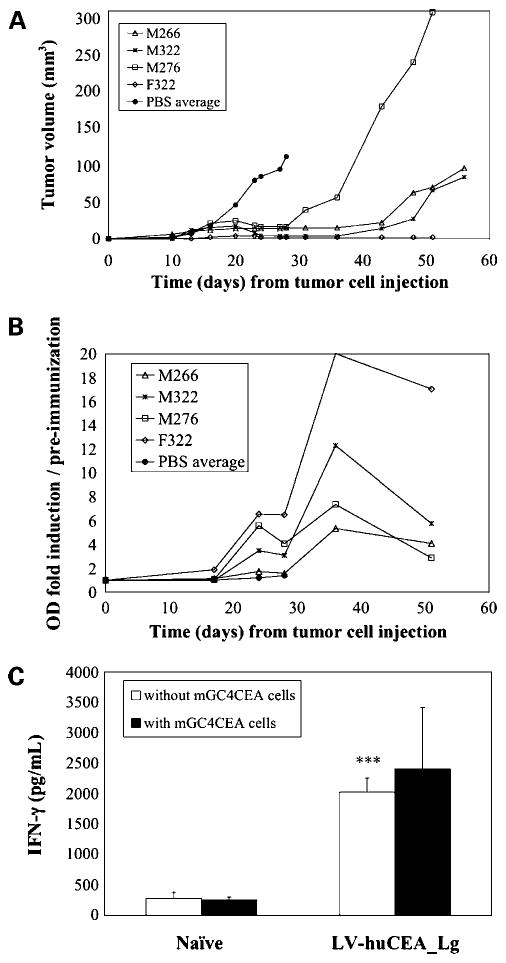
Characterization of the long-term antitumor response in LV-huCEA-vaccinated mice. In the second tumor stabilization experiment, one group of LV-huCEA-vaccinated mice received a third vaccination at day 28 with a lower dose of virus (0.1 × 106 TU) and was followed for 51 days (LV-huCEA_Lg). A, tumor measurement data. B, calculated antibody response data from 4 mice sacrificed at day 57. Naive mice were used as controls. C, IFN-γ secretion from pooled splenocytes of sacrificed mice done in triplicate. ***, P < 0.005 for LV-huCEA compared with naive mice.
The long-term immune response was also assessed by assaying splenocytes from 2 mice injected three times with the LV-huCEA vaccine. At day 57, no huCEA-specific CTLs were detected by tetramer staining of the splenocytes (data not shown). When assayed, IFN-γ secretion by splenocytes from LV-huCEA-vaccinated mice in the absence of tumor cells was found to be maintained at the same level (Fig. 5C) as seen for spleen cells from short-term vaccinated mice and was still significantly different (P < 0.0005, Student's t test) from that obtained with splenocytes from naive mice. However, the specific increase of IFN-γ secretion by the presence of the target cells was no longer detectable (P > 0.05, Student's t test). Taken together, these data suggest that broad antitumor immunity is likely less effective in the long run than the antigen-specific form.
Enumeration of Productively Transduced Dendritic Cells in Murine Lymph Nodes following Direct LV Injection
Toward ascertainment and counting of the possible immune effector cells that may be involved in generation of this potent antitumor response in vivo, lymph nodes were collected from a separate cohort of mice directly injected in the footpad with LV-enGFP. After lymph node harvest, cells were disaggregated and analyzed by flow cytometry for enGFP expression and cell surface antigen expression following staining with a panel of antibodies (Table 1). Large numbers of cell events were analyzed (up to 8.7 × 105 per group). Interestingly, dendritic cells positive for enGFP expression were found in both the ipsilateral and contralateral lymph nodes collected from injected mice. Further, it was found that a greater percentage of enGFP-positive dendritic cells were of myeloid origin compared with plasmacytoid or lymphoid lineages, although even then these transduced cells comprised <1.3% of the total dendritic cells per lymph node (Table 1). Analogous results were found by immunohistochemical analyses of lymph node sections for CD11c and enGFP coexpression (data not shown).
Table 1. In vivo LV injection: effect on lymph node dendritic cell subset numbers and enGFP expression.
| Dendritic cell subset* | Cohort† | Total dendritic cell number per lymph node‡ |
enGFP+ dendritic cell number per lymph node |
||
|---|---|---|---|---|---|
| n | Statistics | n | Statistics | ||
| Myeloid | [A] LV, ipsilateral | 2.3 × 103 ± 0.4 | A vs C, P = 0.008 | 11 ± 3 | A vs C, P = 0.01 |
| [B] LV, contralateral | 1.5 × 103 ± 0.6 | B vs C, P = 0.08 | 19 ± 3 | B vs C, P = 0.20 | |
| [C] control | 0.4 × 103 ± 0.1 | 1 ± 1 | |||
| Plasmacytoid | [A] LV, ipsilateral | 2.0 × 103 ± 0.6 | A vs C, P = 0.05 | 3 ± 1 | A vs C, P = 0.04 |
| [B] LV, contralateral | 1.2 × 103 ± 0.3 | B vs C, P = 0.20 | 4 ± 1 | B vs C, P = 0.02 | |
| [C] control | 0.6 × 103 ± 0.1 | 1 ± 1 | |||
| Lymphoid | [A] LV, ipsilateral | 8.3 × 102 ± 3.0 | A vs C, P = 0.10 | BDL | |
| [B] LV, contralateral | 6.2 × 102 ± 2.3 | B vs C, P = 0.20 | BDL | ||
| [C] control | 3.2 × 102 ± 0.4 | BDL | |||
On day 3 after footpad injection of LV-enGFP, one ipsilateral and one contralateral inguinal lymph node was harvested from each mouse and controls. Total dendritic cells (DCs) and enGFP+ dendritic cells per lymph node were enumerated by flow cytometry. The following panels were used to identify DC subsets: myeloid, CD11c+, CD11b-B220-; plasmacytoid, CD11c+, B220+, Gr-1+; and lymphoid, CD11c+, CD11b-, B220-, CD8+.
Fourteen mice were injected with LV-enGFP; lymph nodes in each group were pooled from n = 2-3 mice, thereby giving overall n =5. n = 5 controls were also evaluated. Cell events analyzed ranged from 5.1 × 105 to 8.7 × 105 ipsilaterally, from 1.6 × 105 to 6.2 × 105 contralaterally, and 1 × 105 in controls.
Absolute numbers of cells per lymph node were calculated from flow and absolute count data. Mean ± SE. P values were determined by two-sided Student's t test. BDL, below detection limit (no events recorded).
LV-huCEA Induces Immune Cell Infiltration into huCEA Tumors
Lastly, in addition to looking at the immune response generated by splenocytes following vaccinations, we analyzed the infiltration of immune cells into the tumor (Fig. 6). Tumor sections were made from 3 mice per group and the presence of CD4+ and CD8+ cells was ascertained by immunofluorescence staining (Fig. 6A and B). Absolute ratios of Alexa Fluor 488/DAPI fluorescent fraction areas (absolute A/D ratios) were calculated as described above and reported per group (Fig. 6C). CD4+ and CD8+ cells were found in all tumors, suggesting an effective infiltration of these cells. There is also a clear trend toward an increase in CD4+ and CD8+ cells following LV-huCEA vaccination (Fig. 6C); however, significant differences could not be shown due to individual variability. That said, no CD8+ cell response was detected in the one mouse of the LV-huCEA_Sh group that showed a normal tumor growth rate (data not shown). Removing the data from this mouse lead to a significant difference between the absolute A/D ratios of the LV-huCEA_Sh group compared with the LV-enGFP or PBS groups (P < 0.05, Student's t test). The 2 other mice of the LV-huCEA_Sh group that had shown tumor regression were found to have a high absolute A/D ratio of CD8+ cells (0.22 and 0.43, respectively). Such ratios were always <0.1 for both anti-CD4 and anti-CD8 analyses of the tumors of the control groups, except for one PBS-injected mouse that showed a 0.15 absolute A/D ratio with anti-CD4 analysis. These observations suggest a crucial role for CD8+ cells in tumor regression. The absolute A/D ratio for the CD4+ labeling was also higher than 0.2 for 2 of the 3 analyzed tumors of the LV-huCEA_Sh group. Immunofluorescence staining of tumor sections for the LV-huCEA_Lg group showed that the absolute A/D ratios were intermediate between values obtained for control and LV-huCEA_Sh groups (0.11-0.19) but still elevated.
Figure 6.

Direct immunization with LV-huCEA induces CD8+ and CD4+ cell infiltration into tumors. huCEA Tg mice tumors were harvested from animals in the second tumor stabilization experiment. Sections were made and stained with DAPI as well as either an anti-CD4 antibody or an anti-CD8 antibody. For each area, the ratios between the positively staining area fractions obtained with Alexa Fluor 488 and DAPI were evaluated and designated “absolute A/D ratios” after subtraction of the ratio obtained from the negative control. A, differential patterns of DAPI and Alexa Fluor 488 overlays obtained from one PBS-vaccinated mouse after control, anti-CD4, and anti-CD8 stainings. B, images obtained from one LV-huCEA-vaccinated mouse. C, “absolute A/D ratios” obtained from the different groups.
Discussion
CEA is an important target for the refinement of immunotherapy approaches. LVs are efficient at stably transferring cDNA sequences into a variety of cells. Following the recent successful completion of the first human trial using recombinant LVs (22), several clinical protocols using this delivery system have been undertaken. In this study, we wanted to assess the potential of direct injections of low doses of LV as a vaccine directed against the tumor-associated antigen CEA and to characterize subsequent T- and B-cell responses. Our study thus expands results from other laboratories using this delivery system toward this important tumor-associated antigen. For example, in 2004, Gyobu et al. transduced nonspecifically activated T cells with a LV allowing for expression of a chimeric immunoglobulin T-cell receptor composed of single-chain variable fragments derived from a CEA-specific antibody. Such T bodies showed promising antitumor activities against CEA-positive human lung cancer cells (6). More recently, Compte et al. transduced peripheral blood lymphocytes so that they secreted bispecific anti-CEA and anti-CD3 diabodies, leading to in vivo tumor inhibition (7).
We showed that direct footpad administration of low doses of LV-huCEA (∼0.15 × 106 TU) were able to break immune tolerance against huCEA (a true self-antigen in this instance) and induce efficient immunity against CEA-positive tumors in huCEA Tg mice. In experiments using direct injection of low doses of LV-enGFP, we found that gene transfer using this approach predictably occurred at an apparently low frequency, with only ∼1% of myeloid dendritic cells expressing the transgene in vivo; further experiments will be required to better characterize the cellular events that account for the gene therapeutic effect. As well, note that no vaccinated mouse showed any signs of gross pathologic consequences resulting from the vaccination procedure. In addition to actual regression of the s.c. tumors observed in most of the vaccinated mice, both cellular and humoral immune responses were induced. Moreover, the tumor regression appeared to correlate with both the antibody and the CTL responses and was further linked to CD8+ cell infiltration into the tumors. This finding is in accordance with the observation that CD8+ T cells within cancer cell nests were significantly associated with a better survival of patients with colorectal cancer (23).
Both the measurable anti-huCEA antibody response and tumor regression occurred after the second virus injection. This is expected as a strong secondary immune response may be required to actually reduce tumor size. In future studies, we will evaluate whether multiple LV injections (or coinjection of LVs that engineer expression of other immune modulating factors) transform the current transient therapeutic effect into a stable curative modality. As well, we appear to have generated a strong Th2 response by this in vivo infection protocol. It is possible that LV-mediated induction of Th2 cytokines may contribute to antitumor effects through the promotion of tumor-specific antibodies; on the other hand, it is possible that induction of Th2 cytokines may be cross-regulatory to a protective Th1-type response, thereby yielding a net reduction in the antitumor effect. In future studies, methods to tip this Th1/Th2 balance differentially, such as addition of anti-IL-4 antibodies or the adoptive transfer of host-type Th1 cells, will be tested.
In our study, we observed tumor growth resuming after day 36, a week after the last LV boost. In the study by Kim et al. in which LVs were also used, s.c. tumor growth was not followed beyond day 32 (12). Therefore, no information exists whether long-lasting tumor immunity was induced in their experiments. In the Dullaers et al. study, the in vivo CTL activity was shown to be maintained until day 30; however, it was not assessed after this time and mouse survival decreased around day 35 (11). This phenomenon may also be due to the lack of induction of a persistent antitumor immune response as observed in our experiments.
CD8+ T cells are a critical component of the immune response against cancer. Indeed, CTLs play a major role in tumor rejection (24–26). To induce a full memory response, it is also crucial that some of these CD8+ T cells survive long-term. Using our strategy, we observed that tumor regrowth was correlated with disappearance of the specific CTL population that had been detected by huCEA peptide/MHC tetramer staining at day 28. Immunofluorescence staining of tumor sections also suggested a decrease in the CD8+ cell infiltration in the tumors of LV-huCEA-vaccinated mice in the long-term. We hypothesize that maintenance of anti-CEA CTLs should provide for long-term antitumor immunity. Different approaches have been used to achieve this goal and could be tested in combination with our strategy, such as the use of low doses of IL-15 to stimulate the proliferation and maintenance of memory CD8+ T cells or the administration of 4-11B agonists that can rescue CD8+ T cells from activation-induced cell death (27,28).
Another factor that could explain our difficulty in generating a persistent immune response is tolerance induced by the tumor environment. T regulatory cells constitute a major obstacle to immunotherapy (29). IL-10 can promote T regulatory cell development; we observed high levels of this cytokine produced from splenocytes following vaccination. Myeloid-derived suppressor cells represent another tolerance-inducing population. In cancer, these cells accumulate and persist ultimately leading to suppression of T-cell responses and development of T regulatory cells (30). Myeloid-derived suppressor cell accumulation has recently been linked to inflammation (31). In our study, the high induction of IFN-γ secretion might have induced myeloid-derived suppressor cell accumulation, leading to the long-term disappearance of the immune response. Several ways of dealing with myeloid-derived suppressor cells are being explored, such as promoting myeloid-derived suppressor cell differentiation by administration of all-trans retinoic acid (30, 32) or blockade of tumor-derived stem cell factor, which prevents tumor-specific T-cell anergy, T regulatory cell development, and tumor angiogenesis (33).
Overall, our work showed the potency of direct vaccinations with low doses of LV-huCEA to induce efficient immunity against huCEA-expressing tumors in huCEA Tg mice. In addition, because it has been shown that this mode of injection leads to priming of T cells in lymph nodes (34), these data could be extended to clinical applications using intranodal injections, an administration route that is showing promise for anticancer immunotherapy (35).
Acknowledgments
We thank R. Head, X. Fan, and Dr. J. Liu (University Health Network) for technical assistance and discussions.
Grant support: Colorectal Cancer Campaign in Canada.
Footnotes
Disclosure of Potential Conflicts of Interest: R. Keefe and B. Dropulic, employees of Lentigen Corp. No other potential conflicts of interest were disclosed.
References
- 1.Mossoba ME, Medin JA. Cancer immunotherapy using virally transduced dendritic cells: animal studies and human clinical trials. Expert Rev Vaccines. 2006;5:717–32. doi: 10.1586/14760584.5.5.717. [DOI] [PubMed] [Google Scholar]
- 2.Loisel-Meyer S, Foley R, Medin JA. Immuno-gene therapy approaches for cancer: from in vitro studies to clinical trials. Front Biosci. 2008;13:3202–14. doi: 10.2741/2921. [DOI] [PubMed] [Google Scholar]
- 3.He Y, Zhang J, Mi Z, Robbins P, Falo LD., Jr Immunization with lentiviral vector-transduced dendritic cells induces strong and long-lasting T cell responses and therapeutic immunity. J Immunol. 2005;174:3808–17. doi: 10.4049/jimmunol.174.6.3808. [DOI] [PubMed] [Google Scholar]
- 4.Lopes L, Fletcher K, Ikeda Y, Collins M. Lentiviral vector expression of tumour antigens in dendritic cells as an immunotherapy strategy. Cancer Immunol Immunother. 2006;55:1011–6. doi: 10.1007/s00262-005-0095-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mossoba ME, Walia JS, Rasaiah VI, et al. Tumor protection following vaccination with low doses of lentivirally transduced DCs expressing the self-antigen erbB2. Mol Ther. 2008;16:607–17. doi: 10.1038/sj.mt.6300390. [DOI] [PubMed] [Google Scholar]
- 6.Gyobu H, Tsuji T, Suzuki Y, et al. Generation and targeting of human tumor-specific Tc1 and Th1 cells transduced with a lentivirus containing a chimeric immunoglobulin T-cell receptor. Cancer Res. 2004;64:1490–5. doi: 10.1158/0008-5472.can-03-2780. [DOI] [PubMed] [Google Scholar]
- 7.Compte M, Banco B, Serrano F, et al. Inhibition of tumor growth in vivo by in situ secretion of bispecific anti-CEA × anti-CD3 diabodies from lentivirally transduced human lymphocytes. Cancer Gene Ther. 2007;14:380–8. doi: 10.1038/sj.cgt.7701021. [DOI] [PubMed] [Google Scholar]
- 8.Breckpot K, Aerts JL, Thielemans K. Lentiviral vectors for cancer immunotherapy: transforming infectious particles into therapeutics. Gene Ther. 2007;14:847–62. doi: 10.1038/sj.gt.3302947. [DOI] [PubMed] [Google Scholar]
- 9.VandenDriessche T, Thorrez L, Naldini L, et al. Lentiviral vectors containing the human immunodeficiency virus type-1 central polypurine tract can efficiently transduce nondividing hepatocytes and antigen-presenting cells in vivo. Blood. 2002;100:813–22. doi: 10.1182/blood.v100.3.813. [DOI] [PubMed] [Google Scholar]
- 10.Esslinger C, Chapatte L, Finke D, et al. In vivo administration of a lentiviral vaccine targets DCs and induces efficient CD8(+) T cell responses. J Clin Invest. 2003;111:1673–81. doi: 10.1172/JCI17098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dullaers M, Van Meirvenne S, Heirman C, et al. Induction of effective therapeutic antitumor immunity by direct in vivo administration of lentiviral vectors. Gene Ther. 2006;13:630–40. doi: 10.1038/sj.gt.3302697. [DOI] [PubMed] [Google Scholar]
- 12.Kim JH, Majumder N, Lin H, Watkins S, Falo LD, Jr, You Z. Induction of therapeutic antitumor immunity by in vivo administration of a lentiviral vaccine. Hum Gene Ther. 2005;16:1255–66. doi: 10.1089/hum.2005.16.1255. [DOI] [PubMed] [Google Scholar]
- 13.Berinstein NL. Carcinoembryonic antigen as a target for therapeutic anticancer vaccines: a review. J Clin Oncol. 2002;20:2197–207. doi: 10.1200/JCO.2002.08.017. [DOI] [PubMed] [Google Scholar]
- 14.Lorenz M, Staib-Sebler E, Hochmuth K, et al. Surgical resection of liver metastases of colorectal carcinoma: short and long-term results. Semin Oncol. 2000;27:112–9. [PubMed] [Google Scholar]
- 15.Nagorsen D, Thiel E. Clinical and immunologic responses to active specific cancer vaccines in human colorectal cancer. Clin Cancer Res. 2006;12:3064–9. doi: 10.1158/1078-0432.CCR-05-2788. [DOI] [PubMed] [Google Scholar]
- 16.Marshall JL, Gulley JL, Arlen PM, et al. Phase I study of sequential vaccinations with fowlpox-CEA(6D)-TRICOM alone and sequentially with vaccinia-CEA(6D)-TRICOM, with and without granulocyte-macrophage colony-stimulating factor, in patients with carcinoembryonic antigen-expressing carcinomas. J Clin Oncol. 2005;23:720–31. doi: 10.1200/JCO.2005.10.206. [DOI] [PubMed] [Google Scholar]
- 17.Marshall JL, Hoyer RJ, Toomey MA, et al. Phase I study in advanced cancer patients of a diversified prime-and-boost vaccination protocol using recombinant vaccinia virus and recombinant nonreplicating avipox virus to elicit anti-carcinoembryonic antigen immune responses. J Clin Oncol. 2000;18:3964–73. doi: 10.1200/JCO.2000.18.23.3964. [DOI] [PubMed] [Google Scholar]
- 18.Morse MA, Clay TM, Hobeika AC, et al. Phase I study of immunization with dendritic cells modified with fowlpox encoding carcinoembryonic antigen and costimulatory molecules. Clin Cancer Res. 2005;11:3017–24. doi: 10.1158/1078-0432.CCR-04-2172. [DOI] [PubMed] [Google Scholar]
- 19.Eades-Perner AM, van der Putten H, Hirth A, et al. Mice transgenic for the human carcinoembryonic antigen gene maintain its spatiotemporal expression pattern. Cancer Res. 1994;54:4169–76. [PubMed] [Google Scholar]
- 20.Nockel J, van den Engel NK, Winter H, Hatz RA, Zimmermann W, Kammerer R. Characterization of gastric adenocarcinoma cell lines established from CEA424/SV40 T antigen-transgenic mice with or without a human CEA transgene. BMC Cancer. 2006;6:57. doi: 10.1186/1471-2407-6-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cusi MG, Del Vecchio MT, Terrosi C, et al. Immune-reconstituted influenza virosome containing CD40L gene enhances the immunological and protective activity of a carcinoembryonic antigen anticancer vaccine. J Immunol. 2005;174:7210–6. doi: 10.4049/jimmunol.174.11.7210. [DOI] [PubMed] [Google Scholar]
- 22.Levine BL, Humeau LM, Boyer J, et al. Gene transfer in humans using a conditionally replicating lentiviral vector. Proc Natl Acad Sci U S A. 2006;103:17372–7. doi: 10.1073/pnas.0608138103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Naito Y, Saito K, Shiiba K, et al. CD8+ T cells infiltrated within cancer cell nests as a prognostic factor in human colorectal cancer. Cancer Res. 1998;58:3491–4. [PubMed] [Google Scholar]
- 24.Hung CF, Cheng WF, He L, et al. Enhancing major histocompatibility complex class I antigen presentation by targeting antigen to centrosomes. Cancer Res. 2003;63:2393–8. [PubMed] [Google Scholar]
- 25.Pilon SA, Piechocki MP, Wei WZ. Vaccination with cytoplasmic ErbB-2 DNA protects mice from mammary tumor growth without anti-ErbB-2 antibody. J Immunol. 2001;167:3201–6. doi: 10.4049/jimmunol.167.6.3201. [DOI] [PubMed] [Google Scholar]
- 26.Xu D, Gu P, Pan PY, Li Q, Sato AI, Chen SH. NK and CD8+ T cell-mediated eradication of poorly immunogenic B16-10 melanoma by the combined action of IL-12 gene therapy and 4-1BB costimulation. Int J Cancer. 2004;109:499–506. doi: 10.1002/ijc.11696. [DOI] [PubMed] [Google Scholar]
- 27.Takahashi C, Mittler RS, Vella AT. Cutting edge: 4-1BB is a bona fide CD8 T cell survival signal. J Immunol. 1999;162:5037–40. [PubMed] [Google Scholar]
- 28.Li B, Lin J, Vanroey M, Jure-Kunkel M, Jooss K. Established B16 tumors are rejected following treatment with GM-CSF-secreting tumor cell immunotherapy in combination with anti-4-1BB mAb. Clin Immunol. 2007;125:76–87. doi: 10.1016/j.clim.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 29.Wilczynski JR, Kalinka J, Radwan M. The role of T-regulatory cells in pregnancy and cancer. Front Biosci. 2008;13:2275–89. doi: 10.2741/2841. [DOI] [PubMed] [Google Scholar]
- 30.Marx J. Cancer immunology Cancer's bulwark against immune attack: MDS cells. Science. 2008;319:154–6. doi: 10.1126/science.319.5860.154. [DOI] [PubMed] [Google Scholar]
- 31.Bunt SK, Yang L, Sinha P, Clements VK, Leips J, Ostrand-Rosenberg S. Reduced inflammation in the tumor microenvironment delays the accumulation of myeloid-derived suppressor cells and limits tumor progression. Cancer Res. 2007;67:10019–26. doi: 10.1158/0008-5472.CAN-07-2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nefedova Y, Fishman M, Sherman S, Wang X, Beg AA, Gabrilovich DI. Mechanism of all-trans retinoic acid effect on tumor-associated myeloid-derived suppressor cells. Cancer Res. 2007;67:11021–8. doi: 10.1158/0008-5472.CAN-07-2593. [DOI] [PubMed] [Google Scholar]
- 33.Pan PY, Wang GX, Yin B, et al. Reversion of immune tolerance in advanced malignancy: modulation of myeloid-derived suppressor cell development by blockade of stem-cell factor function. Blood. 2008;111:219–28. doi: 10.1182/blood-2007-04-086835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.He Y, Zhang J, Donahue C, Falo LD., Jr Skin-derived dendritic cells induce potent CD8(+) T cell immunity in recombinant lentivector-mediated genetic immunization. Immunity. 2006;24:643–56. doi: 10.1016/j.immuni.2006.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Spaner DE, Astsaturov I, Vogel T, et al. Enhanced viral and tumor immunity with intranodal injection of canary pox viruses expressing the melanoma antigen, gp100. Cancer. 2006;106:890–9. doi: 10.1002/cncr.21669. [DOI] [PubMed] [Google Scholar]