

Abstract

The conventional notion that peptides are poor candidates for orally available drugs because of protease-sensitive peptide bonds, intrinsic hydrophilicity, and ionic charges contrasts with the diversity of antibiotic natural products with peptide-based frameworks that are synthesized and utilized by Nature. Several of these antibiotics, including penicillin and vancomycin, are employed to treat bacterial infections in humans and have been best-selling therapeutics for decades. Others might provide new platforms for the design of novel therapeutics to combat emerging antibiotic-resistant bacterial pathogens.
Keywords: antibiotics, biosynthesis, natural products, nonribosomal peptides, ribosomal peptides
1. Introduction
Peptide-based antibiotic natural products are produced ubiquitously and provide host organisms with frontline defense mechanisms to wage war against invading microbes. Some peptide scaffolds that give rise to antibiotics are generated on the ribosome (Figure 1). The defensins,[1-5] including fungal plectasin,[6] are one class of ribosomal peptide antibiotics that are synthesized as inactive precursor peptides and undergo regioselective proteolysis, which affords their biologically active forms. In other instances, ribosomal peptides undergo a series of enzymatic post-translational modifications, which confer hydrolytic stability or create conformational constraints that are essential for physiological function. Examples of such tailored ribosomal peptides include the microcins,[7-10] lantibiotics,[11-13] patellamides,[14] and streptolysin S (SLS).[15] Peptide-based antibiotics are also products of nonribosomal peptide synthetase (NRPS) assembly lines (Figure 2).[16] The nascent nonribosomal peptides (NRPs) synthesized on the assembly lines by thioester templating often undergo further enzymatic tailoring,[17] which results in remarkable structural modifications and provides potent antibiotic activity. Penicillin/cephalosporin,[18] vancomycin,[19] and daptomycin[20] are examples of NRP antibiotics that are employed to treat bacterial infections in humans. These peptide-based antibiotics exhibit diverse mechanisms of action, which include disruptions of membrane integrity, cell wall biosynthesis, protein synthesis, DNA replication, RNA transcription, and fatty acid biosynthesis.[21]
Figure 1.

Examples of ribosomally synthesized peptides that undergo post-translational modification. Lacticin 481 contains two lanthionines, one methyllanthionine, and one 2,3-didehydrobutyrine, all of which result from post-translational tailoring. Patellamide A and C are octapeptides that become macro- and heterocyclized during maturation. Microcin C7 exhibits an unusual C-terminal appendage that is comprised of an aminopropyl-modified AMP moiety that is linked to the peptide by a N—P bond.
Figure 2.

Examples of nonribosomally synthesized antibiotic peptides. Crosslinking of the vancomycin heptapeptide provides a dome-shaped architecture. Tailoring of the ACV tripeptide creates the rigidified β-lactam moiety of isopenicillin N. Daptomycin undergoes macrocyclization during release from its NRPS.
The purpose of the current review is to compare and contrast ribosomal and nonribosomal peptide antibiotic assembly. We will begin by describing several canonical and/or timely examples of ribosomal (Section 2) and NRP (Section 3) antibiotics by focusing on their unique structural attributes and the enzymatic machinery required for their biosynthesis and maturation. In Section 4, we will consider the advantages and limitations in building block utilization, scaffold construction, and enzymatic tailoring strategies of the ribosomal and nonribosomal peptide synthesis routes. Despite varied logic and mechanisms, these processes create potent antibacterial agents that are directed against specific physiological targets through remarkable chemical modifications to common peptide bond scaffolds.
2. Ribosomal Peptide Antibiotics
Compared to NRP antibiotics, the ribosomally derived peptide antibiotics are less celebrated as potential small-molecule medicines for humans. Nevertheless, the generation of antibacterial peptide scaffolds by the maturation of ribosomal protein precursors is a powerful strategy that is used by prokaryotes and eukaryotes in the fight against their competitors. In this section, we highlight several ribosomal peptide antibiotics, including the defensins, lantibiotics, cyanobactins, and microcins, which exhibit unique connectivity or post-translational tailoring. Other antibiotic ribosomal peptides, such as the cathelicidins,[22] cecropins,[23] and melittin,[24] are outside the scope of this review.
2.1. Defensins
The defensins constitute a large class of cysteine-rich ribosomally derived peptide antibiotics that are synthesized by eukaryotes.[1-5] Vertebrate defensins contain six cysteines that are regiospecifically oxidized to afford three disulfide bonds, which stabilize the peptide fold and provide protease resistance.[25] Some invertebrate defensins, including mussel defensin-1 (MGD-1)[26,27] and mytillin,[28] contain eight cysteines that are oxidized to four disulfide linkages. The nascent defensins are synthesized as ~60–100 aa precursor proteins, and the active forms, which are generally ~30–40 aa in length, arise from regiospecific post-translational proteolytic cleavage. Defensins have no other modifications to the peptide scaffold, N, or C terminus. Although the length and amino acid sequences vary, the vast majority of vertebrate defensins exhibit β-sheet tertiary structure and are cationic and amphipathic. They generally demonstrate broad-range antibacterial activity, and are potent against Gram-negative and Gram-positive bacteria. Some defensins are also active against viruses, fungi, and protozoa. Their bactericidal action is thought to result from association with and insertion into cell membranes, which disrupts the membrane integrity and induces cell death by efflux of metabolites and influx of ions. Detailed structure–function analysis of the murine defensin, cryptdin-4, indicates that specific arginine residues in the peptide backbone are necessary for membrane disruption.[29]
Three classes of defensins, α, β, and θ, are defined by disulfide connectivity. α-Defensins, which are produced by mammals, exhibit linkages between cysteines 1–6, 2–4 and 3–5. Disulfide bonds between cysteines 1–5, 2–4 and 3–6 characterize the β-defensins, which are synthesized by a variety of organisms that include crustaceans, birds, rodents, primates, and humans. Nonhuman primates are the only known producers of θ-defensins. θ-Defensins are comprised of two nine residue hemipeptides that are linked through three disulfide bonds. The two nine residue hemipeptides form head-to-tail dimers and thus afford 18-membered cyclic peptides. Higher plants and insects produce “defensin-like” peptides, which generally exhibit three disulfide linkages and α-helix–β-sheet structures.[5] Of particular relevance is the fungal defensin plectasin, which was isolated from the saprophytic ascomycete Pseudoplectania nigrella.[6] Nascent plectasin is a 95 aa peptide that contains a N-terminal signal sequence (residues 1–23), a propiece (residues 24–55), and a C-terminal domain (residues 56–95). Proteolytic cleavage affords the 40 aa mature toxin, which contains one α-helix and two β-strands stabilized by three disulfide bonds linking Cys4-Cys30, Cys15-Cys37, and Cys19-Cys39 (Figure 3). Plectasin exhibits excellent stability in serum, low toxicity in mice, and an efficacy comparable to that of vancomycin and penicillin in mouse model studies of Streptococcus pneumoniae infection; taken together, these features suggest a high therapeutic potential.
Figure 3.

NMR spectroscopic solution-phase structures of plectasin (PDB ID: 1ZFU). The disulfide linkages are shown in red. The imaged is colored from blue to red going from the N to C terminus.
Human cells make and secrete defensins to act locally against microbes (Figure 4).[2] α-Defensins are constitutively expressed in the alimentary canal and are thought to help control the microbial flora in the small intestine. β-Defensins are expressed in a variety of locations, including neutrophils, and their expression is generally induced as a result of infection or inflammation. Some defensins, and related cationic peptides, also have immunomodulatory activity in animals.[30,31]
Figure 4.

Structures of the human neutrophil defensin-3 (HNP3, left, PDB ID: 1DFN) and human beta defensin-2 (HBD2, right, PDB ID: 1KJ6). HNP3 is an α-defensin. The disulfide linkages are in black.
Significant work has gone into optimizing the antibiotic properties of such simple peptides, but these efforts have not resulted in successful commercialization.[32-36] From the stand-point of therapeutic development, defensins exhibit a number of advantageous features that include broad-spectrum activity, rapid killing, and anti-inflammatory properties. Nevertheless, obstacles are numerous and could include toxicity, pH and salt-dependent activity, poor tissue penetration, and high production costs. The stability and low toxicity of plectasin, and the fact that it is amenable to production in a fungal expression system that is currently employed for industrial-scale productions, bode well for its potential application as a therapeutic. The next sections address ribosomal peptides that undergo additional post-translational modifications that build hydrolytic and/or conformational stability into the nascent peptide backbones.
2.2. Lantibiotics
Lantibiotics (class I bacteriocins) are low-molecular-weight (<5 kDa) lanthionine-containing cyclic peptides of ribosomal origin that are produced by Gram-positive bacteria.[11-13] Lanthionine consists of two alanine moieties connected at the β-carbon atoms by a thioether linkage (Figure 5). Generally, lantibiotics also exhibit a methyl-derivatized lanthionine, (2S,3S,6R)-3-methyllanthionine, and the unsaturated amino acids 2,3-didehydroalanine (Dha) and (Z)-2,3-didehydrobutyrine (Dhb). Nisin (Figure 5), which is produced by Lactococcus lactis and has been widely used as a food preservative for more than 50 years, provides a paradigm for studies of lantibiotic structure, biosynthesis, and mechanism of antibiotic action.
Figure 5.

Structures of lanthionine (Lan), (2S,3S,3R)-3-methyllanthionine (MeLan), 2,3-didehydroalanine (Dha), (Z)-2,3-didehydrobutyrine (Dhb), and nisin.
Genes for lantibiotic synthesis, export, and immunity are clustered and located on transposable elements, chromosomes, or plasmids.[12,37] The nisin gene cluster contains eleven genes (nisABTCIPRKFEG), spans 14 kbp, and is located on a transposable element.[38] Like other ribosomally derived antibiotic scaffolds, the lantibiotics are initially synthesized as biologically inactive precursor peptides; the N-terminal leader sequences of 23–59 amino acids are cleaved by proteases to yield active toxins. For instance, nisA encodes the 57-residue nisin precursor peptide NisA, which has a 23-residue leader sequence. After post-translational tailoring by NisBC, the leader peptide is removed by a protease to afford the active form. The leader peptide is an essential recognition element for the post-translational tailoring enzymes and is also necessary for immunity and export signaling.
Post-translational modification of NisA involves dehydration and cyclization of residues in the peptide backbone. NisB is a dehydratase and catalyzes the dehydration of Ser and Thr to Dha and Dhb, respectively.[39,40] In this step, the side-chain hydroxyl moieties of specific Ser and Thr residues are phosphorylated, which allows the cleavage of the Cβ—O bonds by elimination of phosphate (Figure 6). Subsequently, the nascent eneamino acid side chains (dehydroalanine, dehydrobutyrine) are intramolecularly and regioselectively captured by the thiolate side chains of Cys residues; this creates the lanthionine and methyllanthionine moieties (Figure 6). These reactions are catalyzed by NisC, a cyclase that contains a catalytic ZnII site.[41] The enzyme-mediated post-translational Michael additions create macrocycles in the maturing peptide scaffolds and build in the conformational constraints that determine the final architecture of the peptide. The thioether linkages are stable to hydrolysis, account for the remarkable chemical stability of lantibiotics, and are also critical for physiological function.
Figure 6.

Formation of lanthionine. Dehydration of a serine residue in the peptide backbone affords Dha. A cysteine thiolate captures the eneamino acid side chain of Dha to form the thioether.
For many years, lantibiotics were thought to be nonselective, channel-forming bacterial membrane disruptors, but careful NMR spectroscopic analysis of SDS micelles (as a membrane model) revealed that nisin has very high affinity for lipid II, a key intermediate in assembly of the peptidoglycan layer of bacterial cell walls.[42-44] In particular, the N terminus of nisin interacts with lipid II, and its C terminus inserts into the membrane, which results in pore formation. Each pore contains four lipid II and eight nisin molecules.[45]
The transannular and intramolecular Michael chemistry that is displayed by NisBC and other lantibiotic-tailoring enzymes is a powerful reminder that Nature can turn acyclic, floppy peptides with hydrolytic lability into long-lived and conformationally restricted frameworks. Given the advances in the bacterial genomics of novel lantibiotic gene clusters and insights into the mechanisms of post-translational maturation that have been derived from enzymology, it is likely that engineered and optimized lantibiotic variants will be generated.[11,12,37] The extent to which the pharmacokinetics and pharmacodynamics will be readily adapted for human use as front-line antibiotics remains to be determined.
2.3. Aromatic heterocyclic peptide scaffolds
A second method of converting ribosomal peptides into conformationally constrained, hydrolytically stable, and protease-resistant scaffolds is exemplified by a large class of bacterial peptides that contain five-membered heterocycles derived from Cys, Ser, and Thr residues.[46] Some of these peptides have been named “thiopeptides,” as in thiostrepton[47] and its congeners, but the most distinguishing characteristic of this class is the high content of thiazoline/oxazoline/methyloxazoline, or the two-electron-oxidized thiazole/oxazole/methyloxazole rings in the peptide backbones (Figure 7). Examples of heterocyclic antibiotic peptides that arise from ribosomal precursors include microcin B17,[46] the cyanobactins,[48] and SLS.[15]
Figure 7.

Examples of antibiotic peptides that contain five-membered heterocycles derived from cysteine or serine/threonine. The first twelve residues of MccB17 (VGIGGGGGGGGG) are omitted.
2.3.1 Microcin B17
The first insight into the chemical logic behind and enzymatic machinery required for the formation of heterocyclic peptide scaffolds came from investigations of the Escherichia coli metabolite microcin B17 (MccB17).[49] Microcins are low molecular weight (< 10 kDa) ribosomal peptide antibiotics produced by some species of enterobacteria that exhibit diverse structural features and mechanisms of action.[8] MccB17 is a 43-residue peptide that contains four thiazole and four oxazole moieties (Figure 7).[50,51] It is produced by E. coli strains that harbor the pMccB17 plasmid, which contains genes for microcin production, post-translational modification, export, and host immunity. Uptake of mature MccB17 by neighboring enterobacteria lacking immunity results in cell death. This process is a natural way for E. coli strains to battle against their neighbors in microenvironments such as the vertebrate gastrointestinal tract.
The MccB17 operon contains seven genes (mcbABCDEFG) that are required for the ribosomal synthesis of McbA (mcbA), post-translational modification of the nascent peptide (mcbBCD), MccB17 export (mcbEF), and host immunity (mcbG). The nascent ribosomal product McbA (preproMccB17) is a 69-residue peptide that contains a 26-residue N-terminal leader sequence, which is cleaved by a signal peptidase. The 43 amino acids that form the mature MccB17 peptide backbone include six glycine, four cysteine, and four serine residues that are converted to eight heterocycles, including two bis-heterocyclic oxazole-thiazole moieties, by McbBCD during maturation (Figure 8). As observed in lantibiotic biosynthesis, the N-terminal leader sequence of preproMccB17 must be intact for heterocycle production by McbBCD.[52,53] The 26-residue signal peptide is subsequently cleaved, presumably during export by the dedicated export pump McbEF, which is encoded by the MccB17 biosynthetic gene cluster.
Figure 8.

Post-translational processing of McbA. The leader peptide (residues 1–26) of McbA is underlined. Residues 27–38 are listed in italicized font. The cysteine and serine residues that undergo heterocyclization are labeled in bold font.
Biochemical characterization of the microcin synthetase McbBCD provided a detailed mechanistic description of heterocycle formation (Figure 9).[54] McbB is a zinc-containing cyclodehydratase that catalyzes the remarkable attack by the side chain SH of Cys residues and the OH of Ser residues on the immediately upstream amide carbonyl.[55] A tetrahedral adduct is formed, and subsequent expulsion of water creates the thiazoline or oxazoline ring as the cyclodehydration product. In principle, this dehydration reaction could be reversible. McbC is a flavoprotein dehydrogenase. It converts the thiazoline and oxazoline moieties to the aromatic thiazole and oxazole rings. McbD is essential for ATP-dependent cyclodehydration, but O-phosphoryl imtermediates (analogous to the lantibiotic maturation chemistry that is described in Section 2.2) have not been detected, so the exact role of McbD in MccB17 tailoring is not yet clarified.[56] Kinetic investigations employing purified McbBCD revealed that the rate of thiazole formation is approximately 100-times faster than oxazole formation,[57] which is consistent with the greater nucleophilicity and kinetic accessibility of a Cys thiolate anion compared to a Ser alcoholate anion. Mass spectrometry revealed that the eight heterocycles are introduced directionally and in a distributive manner.[58]
Figure 9.

Proposed mechanistic description of heterocycle formation in MccB17. A) Cyclodehydration of a Gly-Ser-Cys moiety provides an oxazoline-thiazoline. Dehydrogenation and aromatization affords the oxazole-thiazole pair. B) Mechanism for the McbB-catalyzed formation of thiazoline from a Gly-Cys dipeptide. C) Mechanism for McbC-catalyzed reduction of the thiazoline to thiazole.
The enzymatic cyclization of X-Cys and X-Ser to thiazolines and oxazolines and subsequent thiazoline-to-thiazole and oxazoline-to-oxazole conversions accomplish several things. First, heterocyclization dramatically alters the conformation of the peptide backbone, causing stabilization and rigidification, and provides resistance to hydrolysis and proteolysis. Second, the oxidation of thiazoline and oxazoline to thiazole and oxazole moieties, respectively, influences the equilibrium of the McbB-catalyzed cyclodehydration reactions and thereby ensures accumulation of the product with all eight heterocycles. One particularly remarkable transformation is the conversion of the Gly-Ser-Cys sequence to the tandem 4,2-connected oxazole-thiazole (and vice versa) (Figure 9). The bis-heterocycle has the dimensionality of a DNA base pair and intercalator. The target for MccB17 is DNA gyrase.[59-61] Blockade of its topoisomerase activity prevents unwinding of super-coilded DNA and halts DNA replication. DNA gyrase is a validated antibiotic target; the fluoroquinolones, which were the best-selling prescription antibiotics in 2006, inhibit DNA gyrase. It is not yet known what the smallest sequence of a microcin substrate can be or the most favorable identities and distribution(s) of embedded heterocycles. Further investigation in this area might yield a Microcin B17 analogue that is optimized to eradicate Gram-negative bacterial pathogens.
2.3.2 Patellamides
The patellamides constitute a family of ~60 heterocycle-containing cyclic octapeptide cytotoxins that were originally isolated from didemnid extracts.[62-68] Patellamides A and C (Figures 1 and 7) each contain two thiazole and two oxazoline rings. For many years, the biosynthetic origin of the patellamides was unclear.[62,69] The samples that provided the extracts were composed of two organisms, the ascidian Lissoclinum patella and its bacterial symbiont, a Prochloron species. Although initial isolation suggested patellamide production by Prochloron, the compounds were subsequently located in the didemnid tunic.[69] In the absence of genomic data, the octapeptides were predicted to be products of NRPS assembly lines (see Section 3). Schmidt and colleagues, however, discovered the patellamide biosynthetic gene cluster (pat) during the sequencing of the Prochloron didemni genome, which proved their ribosomal origin.[14,70] This work indicated that the linear octapeptide precursors to patellamides are embedded within a ribosomally synthesized 71-residue precursor peptide. Post-translational proteolytic cleavage, macrocyclization, epimerization, heterocyclization, and dehydrogenation yield the active cytotoxins (Figure 10). The pat gene cluster contains seven genes (patA–G). Several of these genes encode for the 71-residue precursor protein (patE), a protease (patA), and tailoring enzymes (patDG). PatDG are homologues of MccB17 maturation enzymes and are responsible for formation of the thiazole and oxazoline rings from X-Cys, X-Ser, and X-Thr dipeptides. Transfer of the pat gene cluster to E. coli confirmed that the genes were necessary and sufficient to make patellamide. Although the enzymology remains to be carried out, there is no doubt that the same logic of cyclodehydration and selective oxidation accounts for heterocyclization of the patellamide backbone. In addition to rigidification of the nascent peptide by heterocyclization, macrocyclization of the octapeptide affords a further conformational constraint. This double-barreled cyclization strategy is a dramatically efficient way in which Nature morphs a genetically encoded linear peptide into a compact biologically active small-molecule scaffold.
Figure 10.

Proposed post-translational processing steps for patellamide A, some of which parallel those in MccB17 maturation.
Schmidt and co-workers subsequently demonstrated that Prochloron spp. uses patE variants within a conserved pat gene cluster to produce a diversity of related metabolites, which include patellamide B, ulithiacyclamide, and the lissoclinamides (Figure 11).[71] Furthermore, mutagenesis of the octapeptide sequence within patE can yield novel variants. Eptidemnamide, which contains amino acids (Trp, Gly, Glu) not incorporated in known PatE products and is an analogue of the anticoagulant eptifibatide, was produced in E. coli that had been transformed with patABD(Edm)FG. (patEdm contains the octapeptide sequence for eptidemnamide). This work presages combinatorial biosynthesis to explore structure–activity relationships. The cloning of other cyanobacterial gene clusters (tru, lyn, trl, etc.) that encode heterocyclic peptides, including trichamide and members of the patellin family, indicated similar organization to pat; this establishes that such biosynthetic logic and machinery is a major route to “cyanobactin” assembly in cyanobacteria.[48]
Figure 11.

Additional cyanobactins generated by patE variants.
2.3.3 Streptolysin S
SLS is a hemolytic toxin and virulence factor that is produced by Streptococcus pyogenes.[72] This pathogenic bacterium is responsible for a range of human infections that range from pharyngitis to life-threatening necrotizing fasciitis. Despite a long-standing interest in its mechanism of action, the structure of SLS has remained unknown for decades. As part of an effort toward its elucidation, a SLS-associated gene locus was identified and cloned recently.[15,73] It has an organization sagABCD and three of the protein products, SagBCD, share sequence homology with McbBCD from the MccB17 gene cluster (SagB with McbC; SagC with McbB; SagD with McbD). The sagA gene encodes a 53-residue protoxin. Because of difficulties that were encountered in detecting over-expressed SagA by mass spectrometry, preproMccB17 was employed as a substrate to investigate SagBCD activity in vitro. These studies revealed that SagBCD installed up to four heterocycles into the preproMccB17 peptide backbone. Analysis of reaction mixtures by MALDI indicated the formation of four new species that differed from the substrate by a loss of 20, 40, 60, or 80 Da; these were attributed to formation of up to four thiazole/oxazole moieties. SagBCD also converted a maltose-binding protein fusion of SagA into a cytolytic product. These studies indicate that SagBCD will convert SagA into a thiazole and/or oxazole-containing membrane-disrupting toxin and provide the first in vitro reconstitution of SLS activity. Bioinformatic analysis of other prokaryotic genomes indicates the presence of homologous operons in a variety of species that include Clostridium botulinum, Listeria monocytogenes, and Staphylococcus aureus RF122. This examination suggests that antibiotic heterocyclic peptides are more ubiquitous than previously appreciated. It is also likely that other naturally occurring peptides with thiazoline/thiazole and oxazoline/oxazole rings will arise by comparable post-translational enzyme machinery. A subset of thiopeptide antibiotics that contain thiazoles attached to a trisubstituted dehydropiperidine or pyridine moiety (for example, thiostrepton,[47] GE 2270[74], as well as the micrococcins, thiocillins, and siomycins[75,76]) are likely to represent a convergence of lantibiotic- and heterocycle-type post-translational maturations on ribosomal peptide backbones, although nonribosomal routes have been suggested.[75] Microbial genome scanning should reveal new members of this ribosomal and heterocyclic peptide class, and set the stage for further elucidation and optimization of such scaffold modifications.
2.4 Microcins Without Heterocyclic Scaffolds
Post-translational modifications to microcin peptides are varied and, as exemplified above for heterocycle-containing MccB17, often required for antibiotic function. In this section, we highlight three additional and distinct post-translational tailoring capacities through consideration of the maturation pathways and structures of microcin J25 (MccJ25), microcin C7 (MccC7), and microcin E492m (MccE492m; Figure 12).
Figure 12.

Microcins that lack heterocyclic scaffolds. A) A depiction of MccE492m. Only the eleven C-terminal residues of the 84 aa MccE492m ribosomal peptide are shown. B) MccC7. The C-terminal post-translational modifications are shown in red. C) Sequence of MccJ25. The eight N-terminal residues of the lariat knot are shown in green and the 13-residue C-terminal tail in blue. The bond between Gly1 and Glu8 is depicted in pink. D) Three-dimensional structure of MccJ25 (PDB ID: 1Q71).
2.4.1 Microcin J25 (MccJ25)
MccJ25 is a hydrophobic 21-residue “lasso peptide” (GGAGHVPEYFVGIGTPISFYG) that is produced by E. coli strains that house the 60 kb pTUC100 plasmid (Figure 12).[77-80] It exhibits a connection between the Glu8 side-chain carboxylate and the amino group of Gly1, which forms a lariat ring. The 13 aa C-terminal tail is threaded into the ring and held into place by non-covalent interactions with Phe19 and Tyr20. This rigidified structure provides resistance to proteases and heat, allows for MccJ25 recognition by the outer membrane bacterial receptor FhuA, and is required for its antibiotic activity. MccJ25 targets the B’ subunit of RNA polymerase and blocks transcription.
The MccJ25 gene cluster contains four genes, mcjABCD, which encode for the ribosomal synthesis of the 58 aa precusor peptide McjA (mcjA), post-translational processing (mcjBC), MccJ25 export (mcjD), and immunity (mcjD).[81] Maturation of MccJ25 from McjA requires cleavage of the 37 aa N-terminal leader peptide and cyclization of the peptide backbone. In vitro reconstitution of MccJ25 from reactions containing McjA, McjB, and McjC was accomplished recently.[82] Both McjB and McjC are required to form the mature antibiotic and, as observed for tailoring of McbA, their action requires the 37 aa leader peptide. More detailed characterization of McjB and McjC was not reported, and such investigations will confirm their precise function(s) in MccJ25 maturation. Bioinformatic analysis indicates that McjC shares homology with class B asparagine synthetases and β-lactam synthetases, which suggests that it might be required for formation of the lactam bond between Gly1 and Glu8. McjB is homologous to several transglutaminases. Because many microbial transglutaminases are proteases, McjB may be the protease responsible for cleavage of the precursor peptide.
2.4.2 Microcin C7
MccC7 is a heptapeptide that is produced by strains of E. coli that carry the pMccC7 plasmid.[83-85] Post-translational modification of MccA, the MccC7 heptapeptide precursor, results in formation of a nonhydrolyzable N—P bond and installation of an adenosine monophosphate (AMP) moiety at its C terminus (Figure 12). Following entry into a susceptible bacterial cell through the transporter YejABEF,[86] E. coli peptidases, including PepA, PepB, and PepN,[87] degrade the heptapeptide and release a nonhydrolyzable aspartyl-AMP mimic (1, Figure 13), which is a potent inhibitor of the aspartyl-tRNA synthetase.[88] Blockade of protein synthesis and cell death result. MccC7 therefore employs a “Trojan horse” strategy to kill enterobacteria that lack immunity and modulate the microbial flora in the human gastrointestinal tract. A recent screening of E. coli isolated from human feces revealed that ~2 % of the strains produced MccC7.[89]
Figure 13.
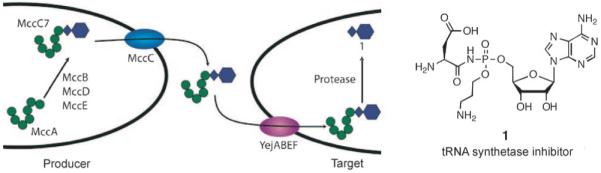
Left: Transport and antibiotic mechanism of action of MccC7. Right: Structure of the toxic aspartyl-AMP analogue. The N—P bond provides hydrolytic stability.
The MccC7 gene cluster contains six genes, mccABCDEF, five of which are necessary for microcin synthesis (mccA), post-translational modification (mccBDE), export (mccC), and immunity (mccF);[90] mccA is the smallest known protein-encoding gene with only 21 base pairs.[91] It encodes MccA, the MccC7 heptapeptide precursor (MRGTNAR). In contrast to other microcin precursors, MccA is not ribosomally translated with a N-terminal leader sequence that undergoes cleavage during maturation, and the N-terminal formyl group remains intact during export and entry into a neighboring cell. Post-translational tailoring of MccA results in several unique structural modifications: 1) conversion of the C-terminal Asn to isoAsn, 2) formation of a N—P bond, and 3) addition of propylamine to the AMP moiety. Recent in vitro characterization of MccB provides mechanistic insight into the former two modifications.
MccB is a ~40 kDa protein that shares homology with adenylating enzymes, including eukaryotic E1 ubiquitin ligases[92,93] and bacterial ThiF from thiamin pyrophosphate biosynthesis.[94] Overexpression, purification, and in vitro characterization indicated that MccB is necessary and sufficient for N—P bond formation in MccC7.[95] It activates and modifies the C-terminal Asn of MccA in a remarkable set of transformations that require two equivalents of ATP per peptide equivalent and involve formation and breakdown of a succinimide intermediate (Figure 14). The α-carboxylate of Asn7 attacks the first equivalent of ATP at Pα to generate a conventional peptidyl-CO–AMP intermediate (2). The β-carboxamido nitrogen atom of Asn7 intramolecularly captures the mixed acyl-AMP anhydride, which releases AMP and forms the heptapeptidyl succinimide (3). The succinimidyl nitrogen atom acts as a nucleophile and attacks Pα of a second ATP equivalent, yielding the hydrolytically stable N—P bond in 4. Addition of water to the carbonyl affords regioselective ring-opening of the succinimide and generation of the β-carboxylate moiety in 5 (isoAsn-type side chain). Maturation of MccC7 also requires addition of a propylamine arm to a phosphate oxygen moiety of AMP. From genetic studies that implicate MccDE in maturation,[90] we anticipate that one or both of these enzymes is required for installation of the propylamine moiety.
Figure 14.

Mechanism of MccB-catalyzed N—P bond formation and the Asn7 to isoAsn7 transformation in MccC7 maturation. The first six residues of MccA are removed for clarity. The first and second ATP equivalents are indicated in blue and red, respectively.
2.4.3 Microcin E492m
MccE492 is an 84-residue ribosomal protein produced by Klebsiella pneumoniae RYC492 that also undergoes post-translational modification at its C terminus (Figure 15).[96] This process results in attachment of an enterobactin derivative to the C-terminal serine residue of MccE492 to afford MccE492m (where “m” is for modified). Enterobactin is a nonribosomal peptide and iron chelator (siderophore) that is produced by Gram-negative bacteria during periods of nutrient deprivation.[97] The siderophore moiety allows for MccE492m recognition by siderophore permeases (FepA, Fiu, Cir) on the cell surface.[98,99] The ribosomal protein portion then inserts into the inner membrane, perhaps in complex with mannose permease proteins ManYZ,[100] and disrupts membrane integrity, forms pores, and induces bacterial cell death.[101] Like MccC7, MccE92m is also a “Trojan horse” toxin that uses a “smuggler strategy” because it masks itself as a beneficial iron chelator.
Figure 15.

Pathway for MccE492m maturation based on in vitro characterization of MceCDIJ by using a model system comprised of monoglycosylated enterobactin and the C-terminal decapeptide of MccE492. Only the last eleven residues of MccE492 are shown above. Abbreviations: Ent, enterobactin; MGE, monoglycosylated enterobactin; lin-MGE, linear monoglycosylated enterobactin; UDP-Glc, UDP-glucose.
The MccE492 gene cluster encodes ten genes, mceABCEDFGHIJ, that are involved in ribosomal synthesis (mceA), post-translational modification (mceCDIJ), host immunity (mceB), and export (mceGH).[102-104] The functions of mceE and mceF are undefined; the mceF gene product might be involved in export. MceA is a 99 or 103 aa precursor peptide that gets cleaved by a N-terminal signal peptidase at residue 15 or 19 to yield the 84-residue MccE492. Four proteins, MceCDIJ, carry out the modification steps, which result in attachment of the enterobactin derivative to the peptide C terminus through an ester linkage.
The structure of MccE492m isolated from producer strains reveals that the trilactone scaffold of enterobactin is linearized and that one of the three dihydroxybenzoate moieties is C-glycosylated at C5.[96] MccE492m analogues with the enterobactin dimer and monomer were also identified in bacterial cultures recently.[105] Linearized and glycosylated enterobactin derivatives are produced by virulent strains of enterobacteria that express homologues of MceC and MceD.[106] The glucose moiety of MccE492m functions as a bridge between the siderophore recognition element and the 84-residue peptide toxin. Overexpression of MceCDIJ in E. coli and in vitro characterization established that these four proteins are necessary and sufficient to produce MccE492m starting from enterobactin and the unmodified peptide.[107] MceC is a C-glycosyltransferase that carries out the unusual C-glycosylation by using UDP-glucose as electrophilic glucosyl donor. We presume that this reaction involves capture at the C1′ center by the C5 anion of the hexadienyl resonance contributor to the catecholate anion form of one dihydroxybenzolyl (DHB) moiety of enterobactin, although mechanistic analysis is required to verify this notion. This reaction yields a nonhydrolyzable C—C bond between glucose and DHB in the monoglycosylated product (MGE). MceD is a regio-selective hydrolase. It first cleaves one of the three trilactone ester linkages to give linear MGE (lin-MGE), and can also degrade lin-MGE further to yield the glycosylated DHB-serine dimer and monomer. MceIJ form a protein complex that exhibits ligase activity. It attaches MGE or lin-MGE to the MccE492 C terminus. In this transformation, the C-terminal serine carboxyl moiety is activated as the MccE492-CO-AMP through ATP cleavage at Pα. The peptidyl-CO–AMP is subsequently captured by the C4′-hydroxyl of the glucose moiety, and AMP is released (Figure 16).[108] This initial form of MccE492m, which has a C4′-glycosyl ester linkage, undergoes nonenzymatic and base-catalyzed rearrangement to yield MccE492m with a C6′-linkage. After assembly and export, the 84-residue ribosomal peptide can be smuggled into a neighboring cell and is taken up by enterobactin-selective outer-membrane permeases such as FepA.
Figure 16.

Mechanism of C-terminal activation and glycosyl ester formation by MceIJ. Only the C-terminal serine residue of MccE492 and a truncated depiction of MGE are shown for clarity.
The chemistry of MccE492 maturation, which proceeds through a peptidyl-CO-AMP intermediate, resembles that of E1 family members involved in ubiquitin conjugation,[92,93] ThiF from thiamin biosynthesis,[94] and also MoeB from molybdopterin assembly.[109] The glucose bridge in MccE492m is reminiscent of the C-glycosidic bridge in the antibiotic simocyclinone;[110] however, the use of glucose to afford a bifunctional antibiotic comprised of ribosomal and nonribosomal elements is novel for protein modification.
The examples of ribosomally derived antibiotic peptides described above provide an overview of the biosynthetic logic behind and structural features of this family. The fact that Nature produces such diverse structures from only 20 proteinogenic amino acid building blocks is remarkable. In the next section, we consider the nonribosomal route to natural product biosynthesis, which employs an extended set of building blocks and new tools for the production of antibacterial peptides (Figure 17).
Figure 17.
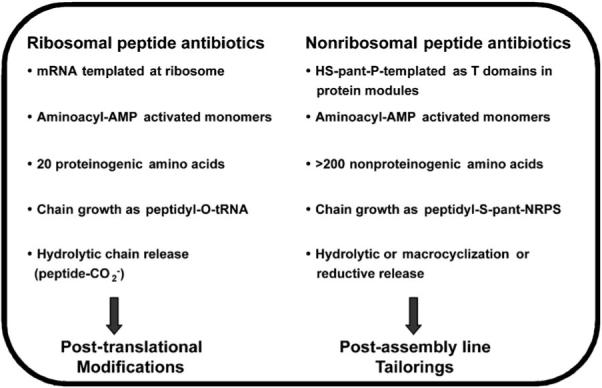
Comparison of ribosomal and nonribosomal peptide antibiotic synthesis. “Pant” stands for “phosphopantetheinyl.”
3. Nonribosomal Peptide Antibiotics
Prokaryotes and fungal eukaryotes can assemble amino acid building blocks into peptides that are 3–22 residues long in a sequence-specific manner without instructions from mRNA and without the intervention of the ribosome as a RNA-based condensation catalyst. In such instances, nonribosomal peptide synthesis (NRPS) machinery is employed (Figure 18).[16,111-113] This route provides microbes with an array of potent antibiotic and/or antifungal small molecules that can be employed to eradicate competing species in an ecological niche. In NRPS logic, a linear peptide oligomer is generated from a set of amino acid monomers, activated as aminoacyl-AMP mixed anhydrides, by iterative condensation reactions. The amino acid building blocks are activated as thioesters on phosphopantetheinyl arms that are prosthetic groups on ~10 kDa carrier protein domains. The carrier proteins are often called thiolation (T) domains and the phosphopantetheinyl arms, derived from coenzyme A,[114] are installed on specific Ser side chains, one in each carrier protein, by specific post-translational modifying enzymes (phosphopantetheinyl transferases) and each provides a terminal thiolate anion that captures the aminoacyl-AMP.[114] Carrier proteins in NRPS assembly lines are typically located in a protein module that contains two catalytic domains: 1) an adenylation (A) domain that selects a specific amino acid monomer to be activated by ATP to the aminoacyl-AMP and then transferred to the thiolate of the phosphopantetheinyl arm in the carrier protein, and 2) a condensation (C) domain that condenses the activated aminoacyl (peptidyl) thioesters, and makes amide bonds as they carry out chain elongation. Thus, each NRPS elongation module is comprised of a CAT trio (Figure 18). A NRPS assembly line contains one module for each amino acid that is selected and incorporated into the growing peptide chain. ACS synthetase in the β-lactam biosynthetic pathways has three modules to yield the l-δ-(α-amino-adipoyl)-l-cysteinyl-d-valine (ACV) tripeptide, and vancomycin synthetase has seven modules to construct the heptapeptidyl backbone of the antibiotic. The number of modules and the specificity of each A domain determines the length and sequence of the nonribosomal peptide produced. Tailoring enzymes (methyltransferases, halogenases, racemases, etc.) can modify the peptide backbone during chain elongation or after chain release from the assembly line.[17] The assembly line genes, genes for producing unusual amino acid building blocks, and genes for tailoring enzymes, are typically bundled together in NRP antibiotic biosynthetic gene clusters. This organization allows for coordinate regulation and probably reflects frequent horizontal gene transfer between microbes to introduce the full biosynthetic capacity in a single DNA transfer event.[115,116]
Figure 18.

Domains and modules in a prototypic NRPS assembly line. The initiation module (leftmost AT) in simple NRPS assembly lines lacks a C domain because there is no condensation with an upstream monomer unit. The TE domain provides hydrolytic release of the elongated peptide chain.
In this section, we consider four examples of NRPS assembly logic that are employed in the synthesis of 1) the simple ACV tripeptide backbone for penicillins and cephalosporins, 2) the glycopeptides vancomycin and teicoplanin,[19] 3) the lipopeptide daptomycin, and 4) the pseudopeptide antibiotic andrimid.[117-119] We note that other NRPS assembly lines exhibit additional variations;[120] however, these four examples provide an overview of many general assembly line features and also illustrate the versatility of this biosynthetic route to antibiotics.
3.1 ACV Synthetase: The gateway to penicillins and cephalosporins
The prototype for a simple NRPS is the enzyme ACV synthetase, which is found in prokaryotes and fungi that make penicillins and the ring-expanded cephalosporins.[18,121,122] ACV is the immediate precursor of penicillins. Ten domains are strung together in the >400 kDa ACV synthetase to convert aminoadipate, l-Cys, and l-Val into ACV. Three modules (one for each amino acid) are required, and the amino acids are selected and activated in an A1T1-CA2T2-CA3T3 framework (Figure 19). The initiation module in simple NRPS assembly lines lacks a C domain because there is no condensation with an upstream monomer unit. A1 is specific for making aminoadipoyl-O-AMP, A2 for Cys-O-AMP, and A3 for Val-O-AMP.
Figure 19.
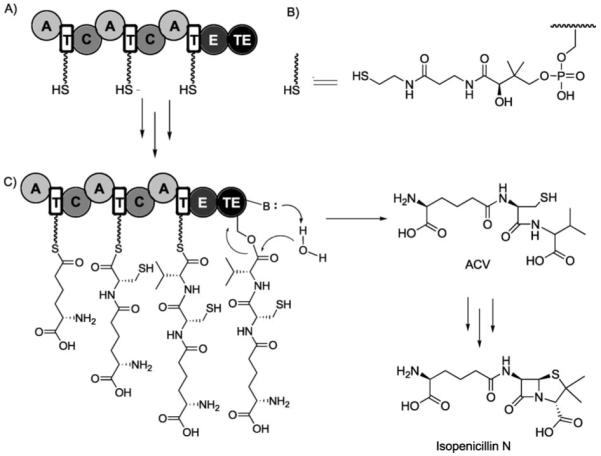
Chain growth and release of ACV on the ACV synthetase assembly line. A) Post-translationally primed ACV synthetase. B) Structure of the phosphopanthetheinyl arm. C) ACV Chain elongation and release. Further enzymatic modification of ACV affords isopenicillin N. “E” stands for epimerase domain.
ACV synthetase loads l-Val, which undergoes epimerization (to d-Val) on the assembly line. A dedicated epimerase (E) domain is embedded in module 3; this provides the organization AT1-CAT2-CAT3E. This nine-domain, three-module assembly line builds up a tripeptidyl chain that is covalently tethered by a thioester linkage to the phosphantetheinyl arm on T3 (aminoadipoyl-l-cysteinyl-d-valinyl-S-T3). NRPS assembly lines typically contain a C-terminal thioesterase (TE) domain, which provides hydrolytic release of the full-length peptide chain. Therefore, the ACS synthetase single-protein assembly line has the tendomain composition AT1-CAT2-CAT3E(TE) to produce and release ACV-CO2H in multiple catalytic turnovers.
The ACV tripeptide product has one nonproteinogenic residue (aminoadipate) and one d-isomer (d-Val), two hallmarks of NRP building block diversity. Of course, ACV is an acyclic tripeptide with no antibiotic activity. The tailoring enzyme isopenicillin N synthase (IPNS), a nonheme iron oxygenase, converts the acyclic ACV framework to the fused 4–5 bicyclic ring system of isopenicillin N in one catalytic cycle (Figure 20).[123] This metabolite harbors the β-lactam scaffold, which is the reactive chemical warhead of the penicillin/cephalosporin family. A second nonheme iron oxygenase, DAOC synthase, carries out ring-expansion and desaturation of the five-membered thiolane ring in penicillins to the six-membered olefinic system that provides cephalosporin antibiotics.[124] The oxygenative double tailoring morphs the linear ACV tripeptide framework into a dramatically altered and rigidified scaffold.
Figure 20.

Conversion of ACV to isopenicillin N and deacetoxycephalosporin C (DAOC) by the successive action of two nonheme iron oxygenases IPNS and DAOC synthase.
Thus, the penicillin/cephalosporin biosynthetic pathway uses a NRPS assembly line to first generate a NRP scaffold. Dedicated tailoring enzymes subsequently build in conformational constraints. The transformation of ACV to penicillin/cephalosporin affords the β-lactam suicide substrates for peptidoglycan crosslinking transpeptidases that inhibit bacterial cell-wall biosynthesis.
3.2 Vancomycin and teicoplanin glycopeptide antibiotics
The coupling of a NRP scaffold to tailoring enzyme maturations is also central to the biogenesis of vancomycin and teicoplanin, two glycopeptide antibiotics of last resort for life-threatening Gram-positive bacterial infections.[19] The core of these highly rigidified and crosslinked antibiotic scaffolds is a heptapeptide with either five (vancomycin) or seven (teicoplanin) nonproteinogenic amino acid building blocks. Vancomycin contains the nonproteinogenic amino acids β-OH-Tyr2, β-OH-Tyr6, 4-OH-PheGly4 (HPG), 4-OH-PheGly5, and 3,5-(OH)2-PheGly7 (DPG7).[19] Leu1 and Asn3 are the only proteinogenic amino acids and, in teicoplanin, they are replaced with 4-OH-PheGly1 and 3,5-(OH)2-PheGly3, respectively.
The NRPS assembly lines for vancomycin/teicoplanin contain seven modules (one for each amino acid that is activated and incorporated) that are spread over three distinct protein subunits. Four of the seven residues in vancomycin are d-stereoisomers (d-d-l-l-d-d-l-l) and three epimerase domains are found in the appropriate modules (Figure 21). After the peptide chain reaches the elongated and tethered heptapeptidyl-S-T7 stage, the electron-rich side chains of residues 2 and 4, 4 and 6, and 5 and 7 undergo regiospecific oxidative crosslinking.[125-127] Three cytochrome P450-type oxygenases are responsible for these transformations, which join β-OH-Tyr2/4-OH-PheGly4 and 4-OH-PheGly4/β-OH-Tyr6 by aryl–ether linkages and form a direct C—C bond between 4-OH-PheGly5 and DPG7.[128-132] This tailoring provides the dome-shaped architecture of the vancomycin aglycone, and creates a constrained structure and a high-affinity conformation for hydrogen-bonding interactions with the N-acyl-d-Ala-d-Ala termini of uncrosslinked peptidoglycan chains in bacterial cell wall assembly. Hydrolytic release of the heptapeptide scaffold by the NRPS TE domain follows.
Figure 21.

Chain growth and crosslinking on the vancomycin synthetase assembly line and post-assembly line glycosylation. The aryl ether and carbon–carbon bond crosslinks that are formed by the action of three P450 oxygenases are highlighted in grey.
N-methylation of Leu1 occurs and two successive glycosylations ensue that are catalyzed by tailoring enzymes that are encoded in the vancomycin biosynthetic gene cluster. The first glycosylation involves transfer of a glucosyl moiety from TDP-glucose to the phenolate of the 4-OH-PheGly4 residue. A dedicated vancosaminyl transferase subsequently transfers an l-vancosaminyl unit to the C2′-oxygen moiety of the newly introduced glucose unit. Thus, the vancomycin biosynthetic gene cluster encodes six tailoring enzyme genes along with the three NRPS subunits. The producing cell must also synthesize β-OH-Tyr and the nonproteinogenic building block HPG and DPGs for the NRPS assembly line to run; this requires nine additional genes that are also clustered with the NRPS genes. The actinomycete producer makes the necessary building blocks, including the unusual nucleotide deoxyamino sugar TDP-l-vancosamine, in a just-in-time fashion as it expresses the 24-domain seven-module NRPS assembly line and the seven tailoring enzymes in a coordinate manner.
Teicoplanin (Figure 22) production follows the same chemical blueprint and uses related enzymatic machinery.[133] None of the seven amino acid monomers are proteinogenic; they are made by diversion of primary metabolites (tyrosine, chorismate, and malonyl-CoA) when the microbe goes into production mode, expresses the seven-module teicoplanin NRPS assembly line, and converts all seven T domains from the apo to holo HS-pantetheinyl forms. In teicoplanin biosynthesis, all seven heptapeptide side chains are crosslinked (residues 1–3, 2–4, 4–6, 5–7) by four tailoring hemeprotein oxidases.[134,135] The conversion of the floppy acyclic and unconstrained heptapeptide into a fully rigidified template is an exquisite accomplishment and surely holds lessons for the purposeful semisynthetic morphing of peptide scaffolds. Teicoplanin is decorated with two N-acyl-d-glucosamines that are attached to residues 4 and 6 and a d-mannose moiety that is linked to residue 7; these glucosamines are installed by tailoring glycosyltransferases after chain release. A novel feature of teicoplanin maturation compared to that of vancomycin is the installation of a C10-acyl group, which is most likely derived from the β-oxidation of fatty acids, onto the glucosamine attached to residue 4.[135] Teicoplanin is therefore a lipoglycopeptide antibiotic with distinct physical and partitioning properties from vancomycin.
Figure 22.
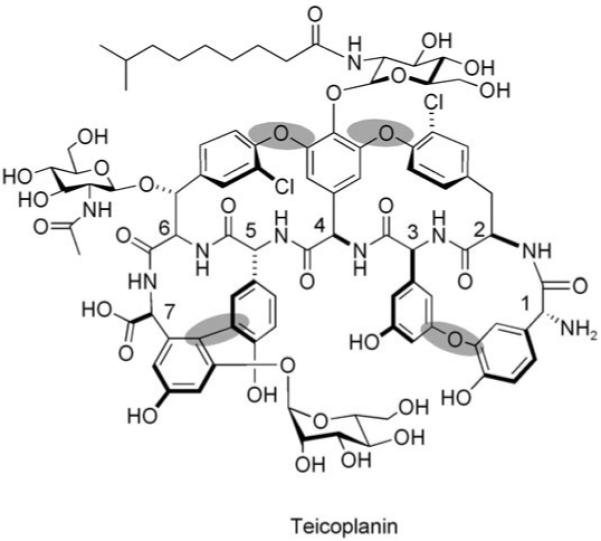
Structure of tecioplanin. The four crosslinks are highlighted by grey ovals.
3.3 Daptomycin, a lipopeptide macrolactone antibiotic
As vancomycin-resistant enterococci became important clinical pathogens, other antibiotics were developed to treat glycopeptide-resistant Gram-positive bacteria.[136,137] Among those recently approved is the NRP daptomycin, which has a 13-residue peptide scaffold and is macrocyclized between residues 4 and 13 (Figure 23).[20,138] It contains three d-amino acids (d-Asp2, d-Ala8, d-Ser11), three nonproteinogenic building blocks (ornithine6, (2S,3R)-3-methyl glutamic acid12, kynurenine13), and a fatty acid tail. The principles exemplified above for ACV and vancomycin biosynthesis also hold for the 13-module daptomycin NRPS assembly line, but it has two additional characteristics. First, module one is a C1A1T1 tridomain rather than an A1T1 didomain; this suggests that the first amino acid might be condensed by the action of C1 with an upstream acyl group. Indeed, daptomycin has a decanoyl (C10)-fatty acyl tail that is linked to its N terminus; this makes it a N-terminally acylated lipopeptide. Second, the last domain of the assembly line does not act as a thioesterase with hydrolytic release of the full-length and linear peptidyl chain. Rather, the TE domain is a macrocyclization catalyst. It specifically folds the full-length N-decanoyl-13-mer chain and catalyzes nucleophilic addition of the Thr4-OH side chain on the carbonyl of Kyn13 (Figure 23). The resulting macrolactone is conformationally constrained. This architecture, in addition to the N-terminal lipophilic group, is required for the antibiotic activity of daptomycin and its congeners, dozens of which have now been prepared by combinatorial reprogramming of modules within the NRPS assembly line.[136,137] Daptomycin itself interacts with CaII ions, which increases its amphipathicity, decreases its negative charge, allows for oligomerization, and facilitates its penetration of lipid membranes.[139,140] The cytoplasmic membrane targets of daptomycin are as yet determined. There are dozens, even hundreds, of known nonribosomal acylpeptidolactones that have been generated as antibiotics by microorganisms that have different physiological targets; this suggests that this assembly line and tailoring strategy creates a good balance of bioactive properties in a peptide framework.
Figure 23.

Truncated depiction of the daptomycin assembly line illustrating modules 1 and 13. Macrocyclization of the 13-mer peptide by the TE domain results in chain release and daptomycin formation.
3.4 Andrimid, a nonribosomal pseudopeptide–polyketide hybrid
The other major class of antibiotics in which architectural and functional group complexity is elaborated from simple monomer building blocks by thiotemplated, T domain-centered protein machinery are polyketides. The logic for polyketide synthase (PKS) assembly lines is highly cognate to that of NRPS albeit the elongation step is a decarboxylative thioclaisen C—C bond formation rather than construction of an amide bond.[16] We only note here the existence of many molecules, including bleomycin[141] and epothilone[142] that are hybrids of NRPS and PKS logic and assembly line machinery.[113,143] The NRPS and PKS modules might be interspersed within the hybrid assembly line. Nature’s ability to mix and match PKS and NRPS modules provides dramatic modulation of peptide backbone structures during assembly. The biogenesis of andrimid (Figure 24) provides one such example.[117-119]
Figure 24.
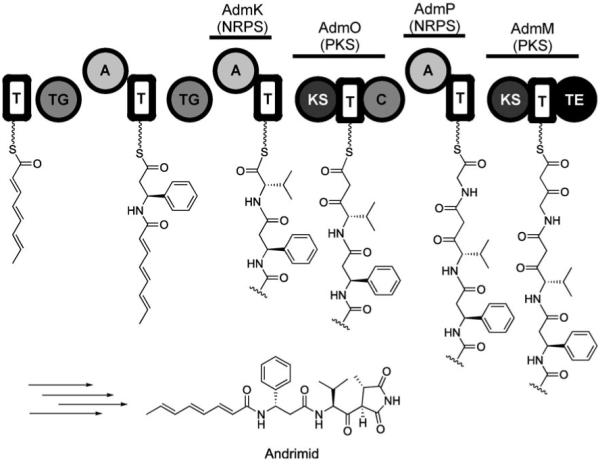
Depiction of the andrimid assembly line illustrating N-acylation of β-Phe and formation of the methylsuccinimide precusor chain. Abbreviations: TG=transglutaminase-like enzyme; KS=ketosynthase.
Andrimid is a NRPS–PKS hybrid antibiotic and nanomolar inhibitor of bacterial acetyl-CoA carboxylase, which catalyzes the first committed step in bacterial fatty acid biosynthesis. It consists of an elaborated β-phenylalanine-l-valine core. An octatrienyl moiety is linked to the amino group of β-phenylalanine by a transglutaminase-like enzyme (AdmF),[144] and the l-valine residue is C-capped with a methylsuccinimide moiety. Inspection of the andrimid gene cluster indicated that, after conversion of α-Phe to β-Phe by an aminomutase and assembly of the acylated β-Phe-l-Val core by NRPS logic, a PKS module (AdmO) uses malonyl-CoA to extend l-Val by a two-carbon unit.[119] The assembly line subsequently switches back to an NRPS module (AdmP) to install a glycine moiety. Further elongation occurs by addition of another two-carbon unit by the terminal PKS module (AdmM) that houses a chain-releasing TE domain (Figure 24). This densely hybrid NRPS–PKS organization therefore allows two of the three amino acid building blocks (l-Val, l-Gly) to undergo two-carbon Claisen extension chemistry, which generates the linear succinimide precursor.
Formation of the methylsuccinimide moiety, the details of which are not yet elucidated, occurs at some point during or after chain release. The C-terminal tetramic acid cap and the N-acyl cap protect both ends of the peptide scaffold. Mature andrimid is therefore a pseudopeptide. It is an elegant example of how assembly lines and tailoring enzymes can morph every constituent of a peptide backbone to create a hydrolytically stable framework with antibacterial action. The C-terminal methylsuccinimide moiety is the active pharmacophore for blockade of the carboxyltransferase subunit of bacterial acetyl-CoA carboxylase, presumably because it mimics the normal N-carboxybiotinyl substrate moiety.[145]
4. Comparison of Ribosomal and Nonribosomal Strategies for Nature’s Conversion of Peptide Scaffolds into Antibiotics
Both ribosomal and nonribosomal routes to peptide-based antibiotics have inherent advantages and limitations, some of which can be complementary. Whether or not an organism synthesizes NRP secondary metabolites can depend, at least to some degree, on its genome size. A recent correlation of 223 bacterial genomes indicated that microbes with larger genomes (>5 Mb) are more likely to produce many NRP (and polyketide) metabolites, whereas those with smaller genomes (< 3 Mb) produce no or very few NRPs.[146] Although further sequencing efforts are required to determine the strength of this correlation, the analysis suggests that Enterobacteriaceae require other, non-NRP strategies for self-protection because of their small genomes. The microcins exemplify one alternative approach.
Building block diversity and utilization are central to natural product biosynthesis. There are twenty common proteinogenic amino acids plus selenocysteine and, in a few methanogenic bacteria, pyrrolysine. This building block set for ribosomal machinery is limited when compared to the >200 nonproteinogenic amino acid monomers that are used by NRPS assembly lines. From the standpoint of synthesizing new antibiotics with unusual monomers, one must only evolve adenylation domains to select and activate the nonproteinogenic monomer(s) for NRPS-mediated incorporation into the growing peptide chain. Partner tRNAs, which would carry the monomers to the peptidyl transferase site of the ribosome, are not required.
On the other hand, the largest known nonribosomal peptide is 22 residues long whereas ribosomal proteins with 16 000 residues exist (for example, cyclosporine synthetase, a single protein NRPS assembly line). The energy expenditure that is required to build multimodular NRPS assembly lines can be prodigious. Consider cyclosporine, an eleven-residue cyclic peptide and potent immunosuppressant drug.[147] The cyclosporine synthetase is ~1.5 MDa and contains an eleven-module NRPS assembly line with 47 domains.[148] A huge investment of cellular energy (ATPs and GTPs) and a machine the size of the ribosome are required to make an eleven-residue peptide product. The 22-residue syringopeptins,[149] which are phytotoxins produced by strains of Pseudomonas syringae, require 3 MDa of protein machinery, which houses a 22-module assembly line. Such examples indicate that a crossover between the benefits of utilizing nonproteinogenic building blocks to diversify peptide scaffolds and the energetic benefits of RNA-directed peptide bond-forming machinery must have occurred.
Companion enzymes often morph nascent peptides. The resulting modifications are made to ribosomal peptides post-translationally. In NRPS pathways, the comparable tailoring enzymes might act at particular way stations during chain elongation or after release of the nascent peptide product. In both cases, the modifications can alter side chains, N and/or C termini, and backbone connectivity. Such alterations create constrained architectures, generate new functional groups, provide stability to proteolytic breakdown, and introduce the structural elements that are necessary for physiological function.
Post-translational modifications of ribosomal protein side chains by alkylation, phosphorylation, acylation, and glycosylation are ubiquitous, expand Nature’s inventory of genetically encoded protein structures, and are responsible for a myriad of functional consequences that range from signal transduction to histone code writing and rewriting.[150,151] N-terminal acylation (for example, myristoylation) and C-terminal amidation are relatively common ways to protect the ends of ribosomal peptide scaffolds from proteolysis. The lantibiotics provide a clear and well-established example of how stable crosslinks (thioethers rather than reducible disulfides) can be constructed post-translationally to generate constrained frameworks of great stability and high antibacterial potency. Little was known about how five-membered thiazoline/thiazole and oxazoline/oxazole moieties were introduced into ribosomal peptide backbones prior to deciphering the logic of MccB17 and, more recently, cyanobactin maturation. A convergence of lantibiotic and thiazole post-translational machinery might generate the rigid frameworks of the thiopeptide antibiotics.[75,76] Studies of other microcins, MccC7 and MccE492m in particular, provide rationale for modifications at the C terminus.
Nascent nonribosomal peptide scaffolds can undergo the same or related tailoring transformations. For instance, many siderophores contain thiazoline and oxazoline rings, which arise from tailoring enzyme-mediated cyclodehydrations.[152] The basic nitrogen atoms of the heterocycles are part of the ligand set for ferric ion chelation. The anticancer natural product bleomycin, a NRPS–PKS hybrid, exhibits a bithiazole moiety that allows for DNA intercalation. Modification of NRPs by dedicated acylation, alkylation, glycosylation, and oxidation enzymes also constitute typical maturation steps that are required for antibiotic activity. The oxygenative formation of the bicyclic 4–5 ring scaffold of penicillins and its subsequent expansion to the 4–6 ring system of cephalosporins, the oxygenative crosslinking of the vancomycin and teicoplanin heptapeptides and subsequent glycosylations, and the conversion of peptide C termini into tetramic acid rings with various oxidation states are particularly noteworthy examples. Continuing evaluation of the enzymatic tailoring steps to morph both ribosomal and nonribosomal peptide backbones and side chains should provide both insight and inspiration to medicinal chemistry efforts to make new antibiotic scaffolds.
5. Summary and Outlook
The emergence and selection of antibiotic-resistant pathogens in hospital wards across the globe necessitates the development of new antibiotic drugs to treat life-threatening infections. Deciphering the biosynthetic logic of naturally occurring antibiotics provides lessons for the design of new antibiotics with potential therapeutic applications. Hybrid NRPS–PKS assembly lines (for example, bleomycin, and andrimid) might be of special interest for re-engineering systems to produce different types of morphed peptides.[113,143] If NRP and PK modules can be readily mixed and matched by swapping constituent domains or by modifying domain specificity, a wide range of constrained and hydrolytically stable architectures should be accessible. The structural determination of MccE492m also points the way to engineering of convergent ribosomal–nonribosomal peptide hybrids. Lastly, the ability to evolve tRNA synthetases and cognate tRNA pairs to accept “unnatural” and nonproteinogenic amino acid monomers[153] further advances the strategies of purposeful modification of ribosomal peptide side chains and backbones to optimize a biological activity and design a scaffold for further maturation by tailoring enzymes.
6. Acknowledgements
Work in the authors’ laboratory was supported by NIH grants GM20011 and GM49338 (CTW), and a NIH post-doctoral fellowship (EMN). We thank Dr. Alex Koglin for preparing Figures 3 and 4, and Drs. Carl Balibar and Michael Acker for providing helpful comments on the manuscript.
Biography

Christopher T. Walsh is the Hamilton Kuhn professor of Biological Chemistry and Molecular Pharmacology (BCMP) at Harvard Medical School. He has served as the chair of the Department of Chemistry at MIT (1982–1987), the Department of BCMP at HMS (1987–1995), and the resident and CEO of the Dana Farber Cancer Institute (1992–1995). His research has focused on enzymes and enzyme inhibitors, with recent specialization on antibiotics.

Elizabeth M. Nolan received her Ph.D. in 2006 from MIT with professor Stephen J. Lippard. Her research there focused on small-molecule fluorescent sensors for detecting ZnII in biological samples and HgII in aqueous solution. She is currently a NIH Post-Doctoral Fellow in the laboratory of Christopher Walsh, where she is studying the post-translational tailoring of a microcin peptide.
References
- [1].Ganz T. Nat. Rev. Immunol. 2003;3:710–720. doi: 10.1038/nri1180. [DOI] [PubMed] [Google Scholar]
- [2].Selsted ME, Ouellette AJ. Nat. Immunol. 2005;6:551–557. doi: 10.1038/ni1206. [DOI] [PubMed] [Google Scholar]
- [3].Schneider JJ, Unholzer A, Schaller M, Schäfer-Korting M, Korting HC. J. Mol. Med. 2005;83:587–595. doi: 10.1007/s00109-005-0657-1. [DOI] [PubMed] [Google Scholar]
- [4].De Smet K, Contreras R. Biotechnol. Lett. 2005;27:1337–1347. doi: 10.1007/s10529-005-0936-5. [DOI] [PubMed] [Google Scholar]
- [5].Thomma BPHJ, Cammue BPA, Thevissen K. Suelo Planta. 2002;216:193–202. doi: 10.1007/s00425-002-0902-6. [DOI] [PubMed] [Google Scholar]
- [6].Mygind PH, Fischer RL, Schnorr KM, Hansen MT, Sönksen CP, Ludvigsen S, Raventós D, Buskov S, Christensen B, De Maria L, Taboureau O, Yaver D, Elvig-Jørgensen E, Sørensen MV, Christensen BE, Kjærulff S, Frimodt-Moller N, Lehrer RI, Zasloff M, Kristensen H-H. Nature. 2005;437:975–980. doi: 10.1038/nature04051. [DOI] [PubMed] [Google Scholar]
- [7].Destoumieux-Garzón D, Peduzzi J, Rebuffat S. Biochimie. 2002;84:511–519. doi: 10.1016/s0300-9084(02)01411-6. [DOI] [PubMed] [Google Scholar]
- [8].Duquesne S, Destoumieux-Garzón D, Peduzzi J, Rebuffat S. Nat. Prod. Rep. 2007;24:708–734. doi: 10.1039/b516237h. [DOI] [PubMed] [Google Scholar]
- [9].Duquesne S, Petit V, Peduzzi J, Rebuffat S. J. Mol. Microbiol. Biotechnol. 2007;13:200–209. doi: 10.1159/000104748. [DOI] [PubMed] [Google Scholar]
- [10].Severinov K, Semenova E, Kazakov A, Kazakov T, Gelfand MS. Mol. Microbiol. 2007;65:1380–1394. doi: 10.1111/j.1365-2958.2007.05874.x. [DOI] [PubMed] [Google Scholar]
- [11].McAuliffe O, Ross RP, Hill C. FEMS Microbiol. Rev. 2001;25:285–308. doi: 10.1111/j.1574-6976.2001.tb00579.x. [DOI] [PubMed] [Google Scholar]
- [12].Chatterjee C, Paul M, Xie L, van der Donk WA. Chem. Rev. 2005;105:633–683. doi: 10.1021/cr030105v. [DOI] [PubMed] [Google Scholar]
- [13].Patton GC, van der Donk WA. Curr. Opin. Microbiol. 2005;8:543–551. doi: 10.1016/j.mib.2005.08.008. [DOI] [PubMed] [Google Scholar]
- [14].Schmidt EW, Nelson JT, Rasko DA, Sudek S, Eisen JA, Haygood MG, Ravel J. Proc. Natl. Acad. Sci. USA. 2005;102:7315–7320. doi: 10.1073/pnas.0501424102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Lee SW, Mitchell DA, Markley AL, Hensler ME, Gonzalez D, Wohlrab A, Dorrestein PC, Nizet V, Dixon JE. Proc. Natl. Acad. Sci. USA. 2008;105:5879–5884. doi: 10.1073/pnas.0801338105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Fischbach MA, Walsh CT. Chem. Rev. 2006;106:3468–3496. doi: 10.1021/cr0503097. [DOI] [PubMed] [Google Scholar]
- [17].Walsh CT, Chen H, Keating TA, Hubbard BK, Losey HC, Luo L, Marshall CG, Miller DA, Patel HM. Curr. Opin. Chem. Biol. 2001;5:525–534. doi: 10.1016/s1367-5931(00)00235-0. [DOI] [PubMed] [Google Scholar]
- [18].Schofield CJ, Baldwin JE, Byford MF, Clifton I, Hajdu J, Hensgens C, Roach P. Curr. Opin. Struct. Biol. 1997;7:857–864. doi: 10.1016/s0959-440x(97)80158-3. [DOI] [PubMed] [Google Scholar]
- [19].Hubbard BK, Walsh CT. Angew. Chem. 2003;115:752–789. doi: 10.1002/anie.200390202. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2003;42:730–765. doi: 10.1002/anie.200390202. [DOI] [PubMed] [Google Scholar]
- [20].Miao V, Coeffet-LeGal M-F, Brian P, Brost R, Penn J, Whiting A, Martin S, Ford R, Parr I, Bouchard M, Silva CJ, Wrigley SK, Baltz RH. Microbiology. 2005;151:1507–1523. doi: 10.1099/mic.0.27757-0. [DOI] [PubMed] [Google Scholar]
- [21].Walsh C. Antibiotics: Actions, Origins, Resistance. ASM Press; Washington, DC: 2003. [Google Scholar]
- [22].Zanetti M. Curr. Issues Mol. Biol. 2005;7:179–196. [PubMed] [Google Scholar]
- [23].Pütsep K, Normark S, Boman HG. FEBS Lett. 1999;451:249–252. doi: 10.1016/s0014-5793(99)00582-7. [DOI] [PubMed] [Google Scholar]
- [24].Asthana N, Yadav SP, Ghosh JK. J. Biol. Chem. 2004;279:55042–55050. doi: 10.1074/jbc.M408881200. [DOI] [PubMed] [Google Scholar]
- [25].Maemoto A, Qu X, Rosengren KJ, Tanabe H, Henschen-Edman A, Craik DJ, Ouellette AJ. J. Biol. Chem. 2004;279:44188–44196. doi: 10.1074/jbc.M406154200. [DOI] [PubMed] [Google Scholar]
- [26].Mitta G, Vandenbulcke F, Hubert F, Roch P. J. Cell Sci. 1999;112:4233–4242. doi: 10.1242/jcs.112.23.4233. [DOI] [PubMed] [Google Scholar]
- [27].Yang Y-S, Mitta G, Chavanieu A, Calas B, Sanchez JF, Roch P, Aumelas A. Biochemistry. 2000;39:14436–14447. doi: 10.1021/bi0011835. [DOI] [PubMed] [Google Scholar]
- [28].Roch P, Yang Y, Toubiana M, Aumelas A. Dev. Comp. Immunol. 2008;32:227–238. doi: 10.1016/j.dci.2007.05.006. [DOI] [PubMed] [Google Scholar]
- [29].Tanabe H, Qu X, Weeks CS, Cummings JE, Kolusheva S, Walsh KB, Jelinek R, Vanderlick TK, Selsted ME, Ouellette AJ. J. Biol. Chem. 2004;279:11976–11983. doi: 10.1074/jbc.M310251200. [DOI] [PubMed] [Google Scholar]
- [30].Ouellette AJ. Best Pract. Res. Clin. Gastroenterol. 2004;18:405–419. doi: 10.1016/j.bpg.2003.10.010. [DOI] [PubMed] [Google Scholar]
- [31].Menendez A, Finlay BB. Curr. Opin. Immunol. 2007;19:385–391. doi: 10.1016/j.coi.2007.06.008. [DOI] [PubMed] [Google Scholar]
- [32].Zasloff M. Nature. 2002;415:389–395. doi: 10.1038/415389a. [DOI] [PubMed] [Google Scholar]
- [33].Gordon YJ, Romanowski EG, McDermott AW. Curr. Eye Res. 2005;30:505–515. doi: 10.1080/02713680590968637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Clarke DJ, Campopiano DJ. Biochem. Soc. Trans. 2006;34:251–256. doi: 10.1042/BST20060251. [DOI] [PubMed] [Google Scholar]
- [35].Verma C, Seebah S, Low SM, Zhou L, Liu SP, Li J, Beuerman RW. Biotechnol. J. 2007;2:1353–1359. doi: 10.1002/biot.200700148. [DOI] [PubMed] [Google Scholar]
- [36].Thevissen K, Kristensen H-H, Thomma BPHJ, Cammue BPA, François IEJA. Drug. Discov. Today. 2007;12:966–971. doi: 10.1016/j.drudis.2007.07.016. [DOI] [PubMed] [Google Scholar]
- [37].Cheigh C-I, Pyun Y-R. Biotechnol. Lett. 2005;27:1641–1648. doi: 10.1007/s10529-005-2721-x. [DOI] [PubMed] [Google Scholar]
- [38].Rodríquez JM, Dodd HM. Microbiologia. 1996;12:61–74. [PubMed] [Google Scholar]
- [39].Sen AK, Narbad A, Horn N, Dodd HM, Parr AJ, Colquhoun I, Gasson MJ. Eur. J. Biochem. 1999;261:524–532. doi: 10.1046/j.1432-1327.1999.00303.x. [DOI] [PubMed] [Google Scholar]
- [40].Koponen O, Tolonen M, Qiao M, Wahlström G, Helin J, Saris PEJ. Microbiology. 2002;148:3561–3568. doi: 10.1099/00221287-148-11-3561. [DOI] [PubMed] [Google Scholar]
- [41].Li B, Yu JP, Brunzelle JS, Moll GN, van der Donk WA, Nair SK. Science. 2006;311:1464–1467. doi: 10.1126/science.1121422. [DOI] [PubMed] [Google Scholar]
- [42].Hsu S-T, Breukink E, de Kruijff B, Kaptein R, Bonvin AMJJ, van Nuland NAJ. Biochemistry. 2002;41:7670–7676. doi: 10.1021/bi025679t. [DOI] [PubMed] [Google Scholar]
- [43].van Heusden HE, de Kruijff B, Breukink E. Biochemistry. 2002;41:12 171–12 178. doi: 10.1021/bi026090x. [DOI] [PubMed] [Google Scholar]
- [44].Hsu S-TD, Breukink E, Tischenko E, Lutters MA, de Kruijff B, Kaptein R, Bonvin AMJJ, van Nuland NAJ. Nat. Struct. Mol. Biol. 2004;11:963–967. doi: 10.1038/nsmb830. [DOI] [PubMed] [Google Scholar]
- [45].Hasper HE, de Kruijff B, Breukink E. Biochemistry. 2004;43:11567–11575. doi: 10.1021/bi049476b. [DOI] [PubMed] [Google Scholar]
- [46].Roy RS, Gehring AM, Milne JC, Belshaw PJ, Walsh CT. Nat. Prod. Rep. 1999;16:249–263. doi: 10.1039/a806930a. [DOI] [PubMed] [Google Scholar]
- [47].Anderson B, Hodgkin DC, Viswamitra MA. Nature. 1970;225:233–235. doi: 10.1038/225233a0. [DOI] [PubMed] [Google Scholar]
- [48].Donia MS, Ravel J, Schmidt EW. Nat. Chem. Biol. 2008;4:341–343. doi: 10.1038/nchembio.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Li Y-M, Milne JC, Madison LL, Kolter R, Walsh CT. Science. 1996;274:1188–1193. doi: 10.1126/science.274.5290.1188. [DOI] [PubMed] [Google Scholar]
- [50].Bayer A, Freund S, Nicholson G, Jung G. Angew. Chem. 1993;105:1410–1413. [Google Scholar]; Angew. Chem. Int. Ed. Engl. 1993;32:1336–1339. [Google Scholar]
- [51].Bayer A, Freund S, Jung G. Eur. J. Biochem. 1995;234:414–426. doi: 10.1111/j.1432-1033.1995.414_b.x. [DOI] [PubMed] [Google Scholar]
- [52].Madison LL, Vivas EI, Li YM, Walsh CT, Kolter R. Mol. Microbiol. 1997;23:161–168. doi: 10.1046/j.1365-2958.1997.2041565.x. [DOI] [PubMed] [Google Scholar]
- [53].Roy RS, Kim S, Baleja JD, Walsh CT. Chem. Biol. 1998;5:217–228. doi: 10.1016/s1074-5521(98)90635-4. [DOI] [PubMed] [Google Scholar]
- [54].Milne JC, Roy RS, Eliot AC, Kelleher NL, Wokhlu A, Nickels B, Walsh CT. Biochemistry. 1999;38:4768–4781. doi: 10.1021/bi982975q. [DOI] [PubMed] [Google Scholar]
- [55].Zamble DB, McClure CP, Penner-Hahn JE, Walsh CT. Biochemistry. 2000;39:16190–16199. doi: 10.1021/bi001398e. [DOI] [PubMed] [Google Scholar]
- [56].Milne JC, Eliot AC, Kelleher NL, Walsh CT. Biochemistry. 1998;37:13250–13261. doi: 10.1021/bi980996e. [DOI] [PubMed] [Google Scholar]
- [57].Belshaw PJ, Roy RS, Kelleher NL, Walsh CT. Chem. Biol. 1998;5:373–384. doi: 10.1016/s1074-5521(98)90071-0. [DOI] [PubMed] [Google Scholar]
- [58].Kelleher NL, Hendrickson CL, Walsh CT. Biochemistry. 1999;38:15623–15630. doi: 10.1021/bi9913698. [DOI] [PubMed] [Google Scholar]
- [59].Heddle JG, Blance SJ, Zamble DB, Hollfelder F, Miller DA, Wentzell LM, Walsh CT, Maxwell A. J. Mol. Biol. 2001;307:1223–1234. doi: 10.1006/jmbi.2001.4562. [DOI] [PubMed] [Google Scholar]
- [60].Zamble DB, Miller DA, Heddle JG, Maxwell A, Walsh CT, Hollfelder F. Proc. Natl. Acad. Sci. USA. 2001;98:7712–7717. doi: 10.1073/pnas.141225698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Pierrat OA, Maxwell A. Biochemistry. 2005;44:4204–4215. doi: 10.1021/bi0478751. [DOI] [PubMed] [Google Scholar]
- [62].Degnan BM, Hawkins CJ, Lavin MF, McCaffrey EJ, Parry DL, van den Brenk AL, Watters DJ. J. Med. Chem. 1989;32:1349–1354. doi: 10.1021/jm00126a034. [DOI] [PubMed] [Google Scholar]
- [63].McDonald LA, Ireland CM. J. Nat. Prod. 1992;55:376–379. doi: 10.1021/np50081a016. [DOI] [PubMed] [Google Scholar]
- [64].In Y, Doi M, Inoue M, Ishida T, Hamada Y, Shioiri T. Chem. Pharm. Bull. 1993;41:1686–1690. doi: 10.1248/cpb.41.1686. [DOI] [PubMed] [Google Scholar]
- [65].In Y, Doi M, Inoue M, Ishida T. Acta Crystallogr. 1994;C50:432–434. doi: 10.1107/s010827019300811x. [DOI] [PubMed] [Google Scholar]
- [66].Rashid MA, Gustafson KR, Cardellina JH, II, Boyd MR. J. Nat. Prod. 1995;58:594–597. doi: 10.1021/np50118a020. [DOI] [PubMed] [Google Scholar]
- [67].Fu X, Do T, Schmitz FJ, Andrusevich V, Engel MH. J. Nat. Prod. 1998;61:1547–1551. doi: 10.1021/np9802872. [DOI] [PubMed] [Google Scholar]
- [68].Schmidt EW, Sudek S, Haygood MG. J. Nat. Prod. 2004;67:1341–1345. doi: 10.1021/np049948n. [DOI] [PubMed] [Google Scholar]
- [69].Salomon CE, Faulkner DJ. J. Nat. Prod. 2002;65:689–692. doi: 10.1021/np010556f. [DOI] [PubMed] [Google Scholar]
- [70].Long PF, Dunlap WC, Battershill CN, Jaspars M. ChemBioChem. 2005;6:1760–1765. doi: 10.1002/cbic.200500210. [DOI] [PubMed] [Google Scholar]
- [71].Donia MS, Hathaway BJ, Sudek S, Haygood MG, Rosovitz MJ, Ravel J, Schmidt EW. Nat. Chem. Biol. 2006;2:729–735. doi: 10.1038/nchembio829. [DOI] [PubMed] [Google Scholar]
- [72].Datta V, Myskowski SM, Kwinn LA, Chiem DN, Varki N, Kansal RG, Kotb M, Nizet V. Mol. Microbiol. 2005;56:681–695. doi: 10.1111/j.1365-2958.2005.04583.x. [DOI] [PubMed] [Google Scholar]
- [73].Nizet V, Beall B, Bast DJ, Datta V, Kilburn L, Low DE, de Azavedo JCS. Infect. Immun. 2000;68:4245–4254. doi: 10.1128/iai.68.7.4245-4254.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Kurz M, Sottani C, Bonfichi R, Lociuro S, Selva E. J. Antibiot. 1994;47:1564–1567. doi: 10.7164/antibiotics.47.1564. [DOI] [PubMed] [Google Scholar]
- [75].Bagley MC, Dale JW, Merritt EA, Xiong X. Chem. Rev. 2005;105:685–714. doi: 10.1021/cr0300441. and references therein. [DOI] [PubMed] [Google Scholar]
- [76].Hughes RA, Moody CJ. Angew. Chem. 2007;119:8076–8101. [Google Scholar]; Angew. Chem. Int. Ed. 2007;46:7930–7954. doi: 10.1002/anie.200700728. [DOI] [PubMed] [Google Scholar]
- [77].Salomón RA, Farías RN. J. Bacteriol. 1992;174:7428–7435. doi: 10.1128/jb.174.22.7428-7435.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Rosengren KJ, Clark RJ, Daly NL, Göransson U, Jones A, Craik DJ. J. Am. Chem. Soc. 2003;125:12464–12474. doi: 10.1021/ja0367703. [DOI] [PubMed] [Google Scholar]
- [79].Bayro MJ, Mukhopadhyay J, Swapna GVT, Huang JY, Ma L-C, Sineva E, Dawson PE, Montelione GT, Ebright RH. J. Am. Chem. Soc. 2003;125:12382–12383. doi: 10.1021/ja036677e. [DOI] [PubMed] [Google Scholar]
- [80].Wilson K-A, Kalkum M, Ottesen J, Yuzenkova J, Chait BT, Landick R, Muir T, Severinov K, Darst SA. J. Am. Chem. Soc. 2003;125:12475–12483. doi: 10.1021/ja036756q. [DOI] [PubMed] [Google Scholar]
- [81].Solbiati JO, Ciaccio M, Farías RN, González-Pastor JE, Moreno F, Salomón RA. J. Bacteriol. 1999;181:2659–2662. doi: 10.1128/jb.181.8.2659-2662.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Duquesne S, Destoumieux-Garzón D, Zirah S, Goulard C, Peduzzi J, Rebuffat S. Chem. Biol. 2007;14:793–803. doi: 10.1016/j.chembiol.2007.06.004. [DOI] [PubMed] [Google Scholar]
- [83].Garcia-Bustos JF, Pezzi N, Asensio C. Biochem. Biophys. Res. Commun. 1984;119:779–785. doi: 10.1016/s0006-291x(84)80318-6. [DOI] [PubMed] [Google Scholar]
- [84].Novoa MA, Díaz-Guerra L, San Millán JL, Moreno F. J. Bacteriol. 1986;168:1384–1391. doi: 10.1128/jb.168.3.1384-1391.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Guijarro JI, González-Pastor JE, Baleux F, San Millán JL, Castilla MA, Rico M, Moreno F, Delepierre M. J. Biol. Chem. 1995;270:23 520–23 532. doi: 10.1074/jbc.270.40.23520. [DOI] [PubMed] [Google Scholar]
- [86].Novikova M, Metlitskaya A, Datsenko K, Kazakov T, Kazakov A, Wanner B, Severinov K. J. Bacteriol. 2007;189:8361–8365. doi: 10.1128/JB.01028-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Kazakov T, Vondenhoff GH, Datsenko KA, Novikova M, Metlitskaya A, Wanner BL, Severinov K. J. Bacteriol. 2008;190:2607–2610. doi: 10.1128/JB.01956-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Metlitskaya A, Kazakov T, Kommer A, Pavlova O, Praetorius-Ibba M, Ibba M, Krasheninnikov I, Kolb V, Khmel I, Severinov K. J. Biol. Chem. 2006;281:18033–18042. doi: 10.1074/jbc.M513174200. [DOI] [PubMed] [Google Scholar]
- [89].Šmajs D, Strouhal M, Matějková P, Čejková D, Cursino L, Chartone-Souza E, Šmarda J, Nascimento AMA. Plasmid. 2008;59:1–10. doi: 10.1016/j.plasmid.2007.08.002. [DOI] [PubMed] [Google Scholar]
- [90].González-Pastor JE, San Millán JL, Castilla MÁ, Moreno F. J. Bacteriol. 1995;177:7131–7140. doi: 10.1128/jb.177.24.7131-7140.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].González-Pastor JE, San Millán JL, Moreno F. Nature. 1994;369:281. doi: 10.1038/369281a0. [DOI] [PubMed] [Google Scholar]
- [92].Haas AL, Warms JVB, Hershko A, Rose IA. J. Biol. Chem. 1982;257:2543–2548. [PubMed] [Google Scholar]
- [93].Haas AL, Warms JVB, Rose IA. Biochemistry. 1983;22:4388–4394. doi: 10.1021/bi00288a007. [DOI] [PubMed] [Google Scholar]
- [94].Taylor SV, Kelleher NL, Kinsland C, Chiu H-J, Costello CA, Backstrom AD, McLafferty FW, Begley TP. J. Biol. Chem. 1998;273:16555–16560. doi: 10.1074/jbc.273.26.16555. [DOI] [PubMed] [Google Scholar]
- [95].Roush RF, Nolan EM, Löhr F, Walsh CT. J. Am. Chem. Soc. 2008;130:3603–3609. doi: 10.1021/ja7101949. [DOI] [PubMed] [Google Scholar]
- [96].Thomas X, Destoumieux-Garzón D, Peduzzi J, Afonso C, Blond A, Birlirakis N, Goulard C, Dubost L, Thai R, Tabet J-C, Rebuffat S. J. Biol. Chem. 2004;279:28233–28242. doi: 10.1074/jbc.M400228200. [DOI] [PubMed] [Google Scholar]
- [97].Raymond KN, Dertz EA, Kim SS. Proc. Natl. Acad. Sci. USA. 2003;100:3584–3588. doi: 10.1073/pnas.0630018100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Strahsburger E, Baeza M, Monasterio O, Lagos R. Antimicrob. Agents Chemother. 2005;49:3083–3086. doi: 10.1128/AAC.49.7.3083-3086.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Destoumieux-Garzón D, Peduzzi J, Thomas X, Djediat C, Rebuffat S. BioMetals. 2006;19:181–191. doi: 10.1007/s10534-005-4452-9. [DOI] [PubMed] [Google Scholar]
- [100].Bieler S, Silva F, Soto C, Belin D. J. Bacteriol. 2006;188:7049–7061. doi: 10.1128/JB.00688-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Destoumieux-Garzón D, Thomas X, Santamaria M, Goulard C, Barthélémy M, Boscher B, Bessin Y, Molle G, Pons A-M, Leteiller L, Peduzzi J, Rebuffat S. Mol. Microbiol. 2003;49:1031–1041. doi: 10.1046/j.1365-2958.2003.03610.x. [DOI] [PubMed] [Google Scholar]
- [102].Wilkens M, Villanueva JE, Cofré J, Chnaiderman J, Lagos R. J. Bacteriol. 1997;179:4789–4794. doi: 10.1128/jb.179.15.4789-4794.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Lagos R, Villanueva JE, Monasterio O. J. Bacteriol. 1999;181:212–217. doi: 10.1128/jb.181.1.212-217.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Lagos R, Baeza M, Corsini G, Hetz C, Strahsburger E, Castillo JA, Vergara C, Monasterio O. Mol. Microbiol. 2001;42:229–243. doi: 10.1046/j.1365-2958.2001.02630.x. [DOI] [PubMed] [Google Scholar]
- [105].Vassiliadis G, Peduzzi J, Zirah S, Thomas X, Rebuffat S, Destoumieux-Garzón D. Antimicrob. Agents Chemother. 2007;51:3546–3553. doi: 10.1128/AAC.00261-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Bister B, Bischoff D, Nicholson GJ, Valdebenito M, Schneider K, Winkelmann G, Hantke K, Süssmuth RD. BioMetals. 2004;17:471–481. doi: 10.1023/b:biom.0000029432.69418.6a. [DOI] [PubMed] [Google Scholar]
- [107].Nolan EM, Fischbach MA, Koglin A, Walsh CT. J. Am. Chem. Soc. 2007;129:14336–14347. doi: 10.1021/ja074650f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Nolan EM, Walsh CT. Biochemistry. 2008;47:9289–9299. doi: 10.1021/bi800826j. [DOI] [PubMed] [Google Scholar]
- [109].Lake MW, Wuebbens MM, Rajagopalan KV, Schindelin H. Nature. 2001;414:325–329. doi: 10.1038/35104586. [DOI] [PubMed] [Google Scholar]
- [110].Holzenkämpfer M, Walker M, Zeeck A, Schimana J, Fiedler HP. J. Antibiot. 2002;55:301–307. doi: 10.7164/antibiotics.55.301. [DOI] [PubMed] [Google Scholar]
- [111].Mootz HD, Schwarzer D, Marahiel MA. ChemBioChem. 2002;3:490–504. doi: 10.1002/1439-7633(20020603)3:6<490::AID-CBIC490>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- [112].Walsh CT. Acc. Chem. Res. 2008;41:4–10. doi: 10.1021/ar7000414. [DOI] [PubMed] [Google Scholar]
- [113].Walsh CT. Science. 2004;303:1805–1810. doi: 10.1126/science.1094318. [DOI] [PubMed] [Google Scholar]
- [114].Walsh CT, Gehring AM, Weinreb PH, Quadri LEN, Flugel RS. Curr. Opin. Chem. Biol. 1997;1:309–315. doi: 10.1016/s1367-5931(97)80067-1. [DOI] [PubMed] [Google Scholar]
- [115].Woese CR. Microbiol. Rev. 1987;51:221–271. doi: 10.1128/mr.51.2.221-271.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Riesenfeld CS, Schloss PD, Handelsman J. Annu. Rev. Genet. 2004;38:525–552. doi: 10.1146/annurev.genet.38.072902.091216. [DOI] [PubMed] [Google Scholar]
- [117].Fredenhagen A, Tamura SY, Kenny PTM, Komura H, Naya Y, Nakanishi K, Nishiyama K, Sugiura M, Kita H. J. Am. Chem. Soc. 1987;109:4409–4411. [Google Scholar]
- [118].Needham J, Kelly MT, Ishige M, Andersen RJ. J. Org. Chem. 1994;59:2058–2063. [Google Scholar]
- [119].Jin M, Fischbach MA, Clardy J. J. Am. Chem. Soc. 2006;128:10660–10661. doi: 10.1021/ja063194c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Sattely ES, Fischbach MA, Walsh CT. Nat. Prod. Rep. 2008;25:757–793. doi: 10.1039/b801747f. [DOI] [PubMed] [Google Scholar]
- [121].Banko G, Wolfe S, Demain AL. J. Am. Chem. Soc. 1987;109:2858–2860. [Google Scholar]
- [122].van Liempt H, Von Döhren H, Kleinkauf H. J. Biol. Chem. 1989;264:3680–3684. [PubMed] [Google Scholar]
- [123].Roach PL, Clifton IJ, Hensgens CMH, Shibata N, Schofield CJ, Hajdu J, Baldwin JE. Nature. 1997;387:827–830. doi: 10.1038/42990. [DOI] [PubMed] [Google Scholar]
- [124].Valegård K, van Scheltinga ACT, Lloyd MD, Hara T, Ramaswamy S, Perrakis A, Thompson A, Lee H-J, Baldwin JE, Schofield CJ, Hajdu J, Andersson I. Nature. 1998;394:805–809. doi: 10.1038/29575. [DOI] [PubMed] [Google Scholar]
- [125].Bischoff D, Pelzer S, Bister B, Nicholson GJ, Stockert S, Schirle M, Wohlleben W, Jung G, Süssmuth RD. Angew. Chem. 2001;113:4824–4827. doi: 10.1002/1521-3773(20011217)40:24<4688::aid-anie4688>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2001;40:4688–4691. doi: 10.1002/1521-3773(20011217)40:24<4688::aid-anie4688>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- [126].Vitali F, Zerbe K, Robinson JA. Chem. Commun. (Cambridge) 2003:2718–2719. doi: 10.1039/b308247d. [DOI] [PubMed] [Google Scholar]
- [127].Bischoff D, Bister B, Bertazzo M, Pfeifer V, Stegmann E, Nicholson GJ, Keller S, Pelzer S, Wohlleben W, Süssmuth RD. ChemBioChem. 2005;6:267–272. doi: 10.1002/cbic.200400328. [DOI] [PubMed] [Google Scholar]
- [128].Pelzer S, Süssmuth R, Heckmann D, Recktenwald J, Huber P, Jung G, Wohlleben W. Antimicrob. Agents Chemother. 1999;43:1565–1573. doi: 10.1128/aac.43.7.1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Zerbe K, Pylypenko O, Vitali F, Zhang W, Rouset S, Heck M, Vrijbloed JW, Bischoff D, Bister B, Süssmuth RD, Pelzer S, Wohlleben W, Robinson JA, Schlichting I. J. Biol. Chem. 2002;277:47476–47485. doi: 10.1074/jbc.M206342200. [DOI] [PubMed] [Google Scholar]
- [130].Pylypenko O, Vitali F, Zerbe K, Robinson JA, Schlichting I. J. Biol. Chem. 2003;278:46727–46733. doi: 10.1074/jbc.M306486200. [DOI] [PubMed] [Google Scholar]
- [131].Zerbe K, Woithe K, Li DB, Vitali F, Bigler L, Robinson JA. Angew. Chem. Int. Ed. 2004;43:6709–6713. doi: 10.1002/anie.200461278. [DOI] [PubMed] [Google Scholar]
- [132].Woithe K, Geib N, Zerbe K, Li DB, Heck M, Fournier-Rousset S, Meyer O, Vitali F, Matoba N, Abou-Hadeed K, Robinson JA. J. Am. Chem. Soc. 2007;129:6887–6895. doi: 10.1021/ja071038f. [DOI] [PubMed] [Google Scholar]
- [133].Li T-L, Huang F, Haydock SF, Mironenko T, Leadlay PF, Spencer JB. Chem. Biol. 2004;11:107–119. doi: 10.1016/j.chembiol.2004.01.001. [DOI] [PubMed] [Google Scholar]
- [134].Hadatsch B, Butz D, Schmiederer T, Steudle J, Wohlleben W, Süssmuth R, Stegmann E. Chem. Biol. 2007;14:1078–1089. doi: 10.1016/j.chembiol.2007.08.014. [DOI] [PubMed] [Google Scholar]
- [135].Borghi A, Edwards D, Zerili LF, Lancini GC. J. Gen. Microbiol. 1991;137:587–592. doi: 10.1099/00221287-137-3-587. [DOI] [PubMed] [Google Scholar]
- [136].Nguyen KT, Ritz D, Gu J-Q, Alexaxnder D, Chu M, Miao V, Brian P, Baltz RH. Proc. Natl. Acad. Sci. USA. 2006;103:17462–17467. doi: 10.1073/pnas.0608589103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Baltz RH, Miao V, Wrigley SK. Nat. Prod. Rep. 2005;22:717–741. doi: 10.1039/b416648p. [DOI] [PubMed] [Google Scholar]
- [138].Enoch DA, Bygott JM, Daly M-L, Karas JA. J. Infection. 2007;55:205–213. doi: 10.1016/j.jinf.2007.05.180. [DOI] [PubMed] [Google Scholar]
- [139].Jung D, Rozek A, Okon M, Hancock REW. Chem. Biol. 2004;11:949–957. doi: 10.1016/j.chembiol.2004.04.020. [DOI] [PubMed] [Google Scholar]
- [140].Scott WRP, Baek S-B, Jung D, Hancock REW, Straus SK. Biochim. Biophys. Acta, Biomembr. 2007;1768:3116–3126. doi: 10.1016/j.bbamem.2007.08.034. [DOI] [PubMed] [Google Scholar]
- [141].Umezawa H, Maeda K, Takeuchi T, Okami Y. J. Antibiot. 1966;19:200–209. [PubMed] [Google Scholar]
- [142].Gerth K, Bedorf N, Höfle G, Irschik H, Reichenbach H. J. Antibiot. 1996;49:560–563. doi: 10.7164/antibiotics.49.560. [DOI] [PubMed] [Google Scholar]
- [143].Du L, Sánchez C, Shen B. Metab. Eng. 2001;3:78–95. doi: 10.1006/mben.2000.0171. [DOI] [PubMed] [Google Scholar]
- [144].Fortin PD, Walsh CT, Margarvey NA. Nature. 2007;448:824–828. doi: 10.1038/nature06068. [DOI] [PubMed] [Google Scholar]
- [145].Freiberg C, Brunner NA, Schiffer G, Lampe T, Pohlmann J, Brands M, Raabe M, Häbich D, Ziegelbauer K. J. Biol. Chem. 2004;279:26066–26073. doi: 10.1074/jbc.M402989200. [DOI] [PubMed] [Google Scholar]
- [146].Donadio S, Monciardini P, Sosio M. Nat. Prod. Rep. 2007;24:1073–1109. doi: 10.1039/b514050c. [DOI] [PubMed] [Google Scholar]
- [147].Kleinkauf H, Dittmann J, Lawen A. Biomed. Biochim. Acta. 1991;50:S219–S224. [PubMed] [Google Scholar]
- [148].Weber G, Schörgendorfer K, Schneider-Scherzer E, Leitner E. Curr. Genet. 1994;26:120–125. doi: 10.1007/BF00313798. [DOI] [PubMed] [Google Scholar]
- [149].Ballio A, Barra D, Bossa F, Collina A, Grgurina I, Marino G, Moneti G, Paci M, Pucci P, Sergre A, Simmaco M. FEBS Lett. 1991;291:109–112. doi: 10.1016/0014-5793(91)81115-o. [DOI] [PubMed] [Google Scholar]
- [150].Walsh CT, Garneau-Tsodikova S, Gatto GJ. Angew. Chem. 2005;117:7508–7539. doi: 10.1002/anie.200501023. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2005;44:7342–7372. doi: 10.1002/anie.200501023. [DOI] [PubMed] [Google Scholar]
- [151].Walsh CT. Posttranslational Modification of Proteins: Exploring Nature’s Inventory. Roberts and Company Publishers; Englewood Colorado: 2006. [Google Scholar]
- [152].Crosa JH, Walsh CT. Microbiol. Mol. Biol. Rev. 2002;66:223–249. doi: 10.1128/MMBR.66.2.223-249.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Santoro SW, Wang L, Herberich B, King DS, Schultz PG. Nat. Biotechnol. 2002;20:1044–1048. doi: 10.1038/nbt742. [DOI] [PubMed] [Google Scholar]