

Abstract
Background/Aims
Stem cell factor (SCF) and stromal-derived factor-1 (SDF-1) regulate the regenerative response to liver injury, possibly through activation of liver progenitor ‘oval’ cells and recruitment of circulating, marrow-derived progenitors.
Methods
We performed a detailed analysis of SCF, SDF-1 and oval cell proliferation induced by tyrosinaemia, 3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC) or liver irradiation in mice by ELISA and immunofluorescence.
Results
Liver injury in the tyrosinaemia mouse is characterized by a dramatic decline in plasma SCF and absence of oval cell proliferation. In contrast, DDC induces bile duct (BD) and oval cell proliferation, and a modest decline in plasma SCF. Focal liver irradiation increases plasma SCF, but not oval cell density. In normal mouse liver, SCF is localized primarily to Kupffer cells, cholangiocytes and arterial smooth muscle, with little or no expression in hepatocytes. However, SCF appears in hepatocyte nuclei after injury, where its function is unknown. In all three models, SDF-1 is expressed exclusively in BD epithelium, indicating that tissue SDF-1 levels are proportional to the total mass of oval cells and cholangiocytes. However, increased plasma levels of SDF-1 in fumaryl acetoacetate hydroxylase-null mice were not accompanied by oval cell proliferation.
Conclusion
Changes in SCF and SDF-1 varied with the nature of liver injury and were not directly related to oval cell proliferation.
Keywords: liver injury, oval cells, stem cell factor, stromal-derived factor-1, tyrosinaemia
Murine models of hepatic injury have recently come to the forefront in hepatic progenitor cell research, including investigations of so-called ‘plasticity phenomena’, i.e. engraftment of circulating, sometimes marrow-derived cells as hepatocytes, cholangiocytes and oval cells. In this developing field, there have been many reports presenting seemingly contradictory data that have added to confusion. Some of these contradictions may involve differences in donor cells (whole bone marrow vs. haematopoietic stem cells vs. mesenchymal stem cells), strain, age and gender differences of animal subjects, detection technique and injury (1–19). Different models of injury have been used in many different studies and these different models may represent very different phenomena, that call forth different physiological responses to the injuries (20).
Commonly reported hepatic injuries have included damage related to marrow-lethal radiation in the preparation of marrow-transplanted animals (2), the fumaryl acetoacetate hydroxylase (FAH)-null mouse (11), a model of type I human tyrosinaemia and 3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC) biliary toxicity (4, 21). It is now known that engraftment phenomena may occur by at least two pathways: by fusion of circulating cells with pre-existing hepatobiliary cells and by direct differentiation of engrafting cells (6, 12, 13, 22, 23). The FAH-null mouse exemplifies the fusion pathway. Radiation-related injury may involve both processes (24). The DDC model of injury has been used to generate abundant oval cells, which appear to be of bone marrow origin in some reports but not in others (4, 22, 25).
When fusion is the underlying mechanism, it is probable that cells of the monocyte/macrophage lineage enter the liver and fuse directly with pre-existing hepatocytes (26). There is no apparent site specificity for this process and chemokine/cytokine influences on it have not yet been reported. On the other hand, engraftment by direct differentiation may occur by direct engraftment in the terminal branches of the biliary tree, the putative site of a hepatic progenitor cell niche, perhaps first differentiating into oval cells, before maturing into hepatocytes and/or cholangiocytes (5, 27).
Factors that may influence this process include stromal-derived factor-1 (SDF-1) (28) and the c-kit ligand, stem cell factor (SCF) (29–34). SDF-1 is upregulated in proliferating oval cells and recruits marrow-derived CD34+ cells to the site of injury (28). Exogenous delivery of SDF-1 by an adenoviral vector results in increased mobilization of haematopoietic stem and progenitor cells from the marrow compartment to blood in immunodeficient mice (35). SCF and SDF-1 act synergistically to increase the chemotaxis of CD34+ cells in vitro, suggesting that both play a role in the recruitment of stem cells to injured liver (28). In the 2-acetylaminofluorene (2-AAF)/partial hepatectomy model of oval cell induction, c-kit mRNA is upregulated in oval cells, while SCF transcripts are upregulated in both oval cells and stellate cells (31). SCF is also upregulated by hepatic stellate cells during myofibroblastic activation (32), perhaps contributing to fibrosis in chronic liver injury. Expression of c-kit, the SCF receptor, is markedly increased in paediatric human bile duct (BD) cells in the setting of fulminant hepatic failure, suggesting a role of SCF and c-kit in liver repair (34). Mice deficient in c-kit have impaired oval cell proliferation in response to BD ligation (30) and 2-AAF/partial hepatectomy (36). Exogenous administration of SCF coincident with acetaminophen dramatically reduced lethality and reduced histological liver injury in mice, while neutralizing antibodies against SCF increased both lethality and histological injury (33). It is not clear what role SCF plays, if any, in liver repair in injuries where oval cell proliferation is not a prominent feature.
Here we compare three types of liver injury to emphasize the importance of these differences in evaluating the role of stem and progenitor cells in tissue repair after liver injury. We use a well-established technique to quantify oval cell reactions (37), and evaluate SCF and SDF-1 in tissue (by immunofluorescence) and in serum (by ELISA). The observed differences may influence the comparison of data derived from experiments involving the different models. Understanding these differences may lead to greater clarity of interpretation of reported data and more precise experimental design for future experiments.
Materials and methods
Normal and FAH-null mice
Six- to 8-week-old C57Bl/6 mice were purchased from the Jackson Lab (Bar Harbor, ME, USA). FAH-null mice were kindly provided by Dr Marcus Grompe (Oregon Health & Science University, Portland, OR, USA) and were maintained as homozygotes. The FAH-null mouse was generated by targeted insertional mutagenesis, resulting in deletion of exon 5 as described in (38). In order to maintain survival and fertility, FAH-null mice were given drinking water supplemented with 10 mcg/ml of 2-(2-nitro-4-trifluoro-methylbenzoyl)-1,3-cyclohexanedione (NTBC, provided by Dr Ronald McClard, Portland, OR, USA), to prevent the accumulation of toxic metabolites of tyrosine (39). Upon removal of NTBC from drinking water, liver injury was rapidly induced and mice were sacrificed 3, 7, 10, 14 or 36 days after withdrawal of NTBC.
DDC diet
The compound DDC has been shown previously to induce severe ductular proliferation, reliably inducing oval cells in the liver (21). DDC was added at 0.1% w/w to a standard mouse diet (BioServ, Frenchtown, NJ, USA) and administered for 3, 7, 10 or 14 days. Additional animals were studied during the recovery period of 7 or 14 days on a standard diet after a 14-day course of DDC diet.
Irradiation
Mice were anaesthetized with intraperitoneal ketamine (100 mg/kg) and xylazine (10 mg/kg) and exposed to 1000 rads of X-ray irradiation (Siemens Stabilipan 250 kV; Siemens Medical Systems Inc., Avon, CT, USA) targeted to the liver over a 10.5-min period, calibrated previously by badge dosimetry using a fixed angle and distance from the radiation source. Radiation was restricted to the upper abdomen with lead shielding. Mice were sacrificed 3, 7, 14, 28 or 56 days after irradiation.
Animal handling and tissue processing
Experimental mice were treated with appropriate humane care under a protocol approved by the Institutional Care and Use Committee of the Yale University School of Medicine. Mice were sacrificed under ketamine (100 mg/kg) and xylazine (10 mg/kg) anaesthesia. The inferior vena cava was exposed by midline laparotomy, and blood was collected into a heparinized syringe using a 25-G needle. Plasma was separated by centrifugation and frozen for later measurement of SCF. The liver was excised, sectioned, fixed in 10% formalin for 4 h at room temperature, and then transferred into 70% ethanol. Sections were embedded in paraffin and then cut into 3-μm-thin sections and mounted on glass slides. Standard haematoxylin–eosin or diastase–PAS staining was performed.
Oval cell quantification
Immunoperoxidase staining to highlight oval cells was performed using rabbit anti-cow cytokeratin (CK) polyclonal antibodies (#Z0622; DakoCytomation, Carpinteria, CA, USA) on paraffin sections. This polyclonal antibody was raised against bovine epidermal keratin units of 58, 56 and 52 kDa, among others, and cross-reacts with a wide range of CKs, including the high-molecular-weight murine CKs present in cholangiocytes (DakoCytomation Data Sheet). We have validated the use of this approach previously by direct comparison with oval cell marker A6, showing high sensitivity and specificity of the anti-CK antibodies (37).
Liver sections were deparaffinized and rehydrated through graded alcohols to phosphate-buffered saline (PBS). Endogenous peroxidase was quenched with 3% H2O2 in PBS for 15 min. Slides were then treated with Proteinase K (Roche, Mannheim, Germany) 40 mcg/ml in PBS with 0.05% sodium dodecyl sulphate for 2 min at 42 °C, rinsed, blocked with serum-free protein (1% bovine albumin, Fraction V; Sigma, St Louis, MO, USA) in PBS for 20 min and incubated with rabbit anti-CK antibodies at 1:100 dilution as described above for 2 h at 37 °C. After washing with PBS, the secondary antibody (anti-rabbit–horseradish peroxidase complex, 1:100 dilution, DakoCytomation) was applied. Antigen was localized using a diaminobenzidine substrate (Animal Research Kit, DakoCytomation) according to the manufacturer's instructions, and then counterstained with Mayer's haematoxylin (Fisher, Fair Lawn, NJ, USA). In each fixed liver tissue sample, we examined up to 25 portal tracts (PT) of the appropriate size with visible BDs and portal vein (PV) and counted oval cells, defined as densely CK-positive cells with an oval/cuboidal morphology and a high nuclear to cytoplasmic ratio. Oval cells within a 50 μm radius of the PV were counted. Care was taken to identify those cells that did not border on lumina and were therefore not part of the pre-existing normal BD. Intermediate hepatocyte-like cells, i.e. cells with a hepatocyte-like morphology but CK-positive staining were rarely present in the regenerating mouse livers and were not included in quantifications. In normal mouse liver, the PV occupies nearly the entire portal tract area, with scant stroma. Thus, in order to evaluate the distribution of oval cells according to the PT size, we used the easily definable wall of the PV as a surrogate marker for PT size. Because the largest PT in any section (one or two) exhibited extensive branching, cross-sectional measurements could not be assessed; thus, these largest units were also excluded from analysis. The longest and shortest diameters of the sectioned PV were measured using an eyepiece with a measuring grid at the ×40 magnification. At this magnification, one scale unit of the measuring grid corresponds to 2.5 μm. The PV area was calculated according to the formula S (μm2) = 2.52πab/4, where a and b are the major and minor PV diameters. Thus, we calculated not only the number of oval cells per portal tract but also the density (D) of oval cells per portal tract using the formula D = n/ab, where n is the number of oval cells per portal tract, and a and b are the major and minor PV diameters.
Immunofluorescent detection of SCF, SDF-1 and CK
Three-micrometre paraffin sections were dewaxed and rehydrated as described above. Fixed antigens were unmasked using Retrieve-All-2, pH 10 (Signet Labs, Dedham, MA, USA), in a steam chamber for 30 min, cooled and washed in PBS. Nonspecific binding of anti-mouse secondary antibody to mouse tissues was blocked by incubation in a Mouse-on-mouse blocking reagent (Vector Labs, Burlingame, CA, USA). Slides were incubated with monoclonal mouse anti-human SDF-1 (Clone MAB350; R+D Systems, Minneapolis, MN, USA, final concentration of 10 mcg/ml) or monoclonal mouse anti-human SCF (#13126; Santa Cruz Biotechnology, Santa Cruz, CA, USA, final concentration 4 mcg/ml) in PBS supplemented with 1% w/v bovine serum albumin (Sigma). Following overnight incubation at 4 °C, the slides were washed in PBS and the primary antibody was detected using goat anti-mouse immunoglobulin G (IgG) conjugated to Alexafluor 488 (#A11001, 1:100 dilution; Invitrogen-Molecular Probes, Carlsbad, CA, USA). Isotype controls used an equal concentration of IgG1 (#X0931; DakoCytomation) or IgG2b (eBioscience #14-4732, San Diego, CA, USA) from nonimmunized mice in place of the primary antibody to SDF-1 or SCF respectively. CK was detected using rabbit anti-cow CKs (#Z0622; DAKOCytomation, 1:100 dilution) and donkey anti-rabbit Alexafluor 568 (#A31572, 1:100 dilution; Invitrogen-Molecular Probes).
Microscopy
Photomicrographs were taken using an Olympus BX51 fluorescence microscope using either brightfield illumination or appropriate fluorescence excitation and emission filters. Images were captured with a digital camera, using IPLab software (BD Biosciences, Rockville, MD, USA).
SCF and SDF-1 ELISA
Plasma SCF and SDF-1 were measured using ELISA kits (R+D Systems).
Results
Liver histology in the FAH-null, DDC-treated or liver-irradiated mouse
The FAH-knockout mouse model of hereditary tyrosinaemia provides compelling evidence that, under specific conditions of liver injury, macrophages derived from bone marrow stem cells can, on rare occasions, fuse with diseased hepatocytes to correct a metabolic defect functionally. Lethal hepatotoxicity due to accumulation of toxic metabolites of tyrosine is prevented by supplementing the drinking water with NTBC (39). Upon removal of NTBC from the drinking water, severe liver injury rapidly ensues. Even before the withdrawal of NTBC from FAH-null mice, there is irregularity of the hepatic cords, with focal enlargement of hepatocytes and their nuclei (Fig. 1B) compared with normal controls (Fig. 1A). Withdrawal of NTBC induced a spectrum of injury ranging from moderate hepatocyte injury to microvesicular steatosis, hepatic necrosis (Fig. 1C) with even greater nuclear pleomorphism and marked infiltration of macrophages adjacent to dying hepatocytes (Fig. 1D). DDC induced hepatocyte necrosis, cholestasis and proliferation of BDs within 3 days (Fig. 1E) and increased in severity by day 14 (Fig. 1F). In contrast with the FAH-null mouse and DDC injury, focal irradiation of the liver caused less dramatic histological changes, with no histological abnormality 3 days after exposure to 1000 rads (not shown). Focal hepatocyte necrosis was noted in some animals at day 7 (Fig. 1G), with appearance of mitotic hepatocytes (Fig. 1H, arrowheads) indicating recovery through hepatocyte replication rather than oval cell activation. These changes were reversible, with no abnormality at 28 or 56 days (not shown).
Fig. 1.
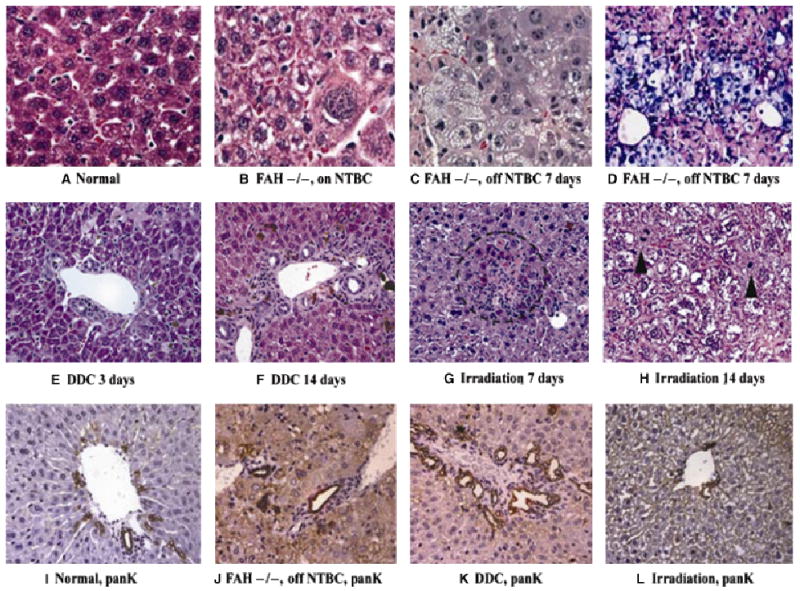
Histology of normal and injured liver. (A) Histology of normal mouse liver (haematoxylin and eosin, ×60 magnification). (B–D) Histology of FAH, nuclear pleomorphism and Kupffer cells in FAH-null liver. (B, C, haematoxylin and eosin; D, periodic acid-Schiff stain after diastase digestion) (B) age-matched control FAH before NTBC withdrawal (×60); demonstrates striking nuclear pleomorphism and probable aneuploidy. (C) FAH null 7 days after NTBC withdrawal (×60); marked vacuolization of hepatocyte cytoplasm, increased nuclear pleomorphism and infiltration of eosinophilic macrophages. (D) FAH null 7 days after NTBC withdrawal; dense infiltrates of macrophages (with PAS positive, diastase-resistant pink staining) expanding the sinusoids and coming in apparent direct contact with pre-existent diseased hepatocytes (×40). (E, F) Liver injury in mice treated with DDC (haematoxylin and eosin stain; ×40). (E) Hepatocyte necrosis, cholestasis and early ductal proliferation after 3 days of DDC diet. (F) Marked cholestasis, with ductal and oval cell proliferation after 14 days of DDC diet. Compare with normal liver histology in Fig. 1A. (G–H) Histology of liver injury after focal liver irradiation (haematoxylin and eosin stain; ×40 original magnification). Three days after irradiation, the liver appears normal (not shown). (G) 7 days after irradiation there are focal areas of hepatocyte necrosis, circled. (H) 14 days after irradiation there are numerous mitotic hepatocytes (arrowheads). (I–L) CK staining of bile ducts and oval cells (immunohistochemistry for biliary-type CKs; ×40). (I) Normal mouse bile duct with rare isolated (nonductal) CK-positive (brown) oval cells. (J) FAH null mouse 3 days after withdrawal of NTBC not only shows no oval cell proliferation, but appears to have diminished cholangiocytes at the limiting plate. Increased background stain is due to hepatocyte cholestasis and necrosis. (K) Marked ductal and oval cell proliferation and cholestasis after 7 days of DDC diet. (L) Normal-appearing bile ducts, ductules and canal of Hering profiles 14 days after focal liver irradiation. NTBC, 2-(2-nitro-4-trifluoro-methylbenzoyl)-1,3-cyclohexanedione; DDC, 3,5-diethoxycarbonyl-1,4-dihydrocollidine; FAH, fumaryl acetoacetate hydroxylase.
Oval cell appearance in the FAH-null, DDC-treated or liver-irradiated mouse
When hepatocytes are incapable of dividing rapidly enough to restore damaged liver, either owing to overwhelming injury or biochemical inhibition of mitosis, intrahepatic progenitors known as oval cells proliferate in response to hepatic injury and give rise to both hepatocytes and mature cholangiocytes. We studied the appearance of oval cells in each of these injury models by staining for biliary-type CKs, which stain all segments of the biliary tree (canals of Hering, ductules, BDs of all sizes) and reactive proliferations of oval cells (Fig. 1I–L). FAH-null mice, either before or after withdrawal of NTBC, not only had no increase in the number or density of oval cells in PT, but the most proximal branches of the biliary tree, the canals of Hering and ductules, also appeared diminished (Figs 1J and 2B). In contrast, DDC treatment induced marked upregulation of both BDs and oval cells (Fig. 1K). In DDC-treated mice, the density of oval cells (Fig. 2A) increased modestly by day 3 and more markedly after discontinuation of the DDC diet after 14 days. The peak of BD density appeared before the peak of oval cell density (data not shown), indicating that ductal proliferation preceded the appearance of oval cells. Radiation liver injury induced neither BD or oval cell proliferation nor loss (Figs 1L and 2B) compared with normal controls.
Fig. 2.
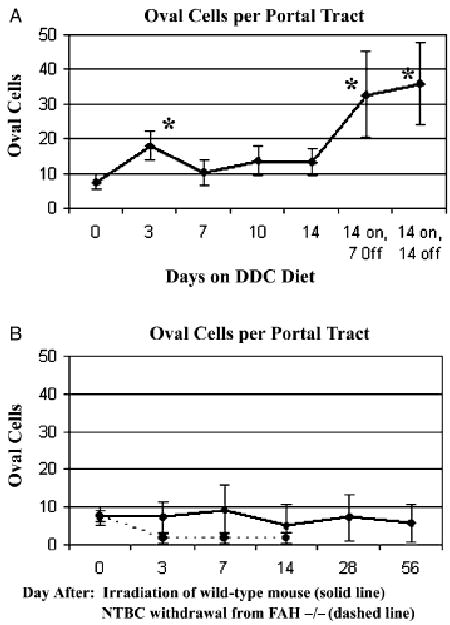
Time course of oval cell proliferation (mean ± SE, *P < 0.05 by student's t-test). Normal oval cell density is indicated as a dotted horizontal line. (A) DDC diet – there was a modest increase in oval cell density by day 3, becoming more marked during the recovery period 7–14 days after stopping DDC (stars). (B) Liver irradiation and FAH-null mouse after NTBC withdrawal. There were no significant deviations from normal in oval cell density after focal liver irradiation; FAH-null mouse shows diminished proximal biliary structures and no oval cell proliferation. DDC, 3,5-diethoxycarbonyl-1,4-dihydrocollidine; FAH, fumaryl acetoacetate hydroxylase.
Expression of SCF in normal and injured liver
Immunofluorescent localization of SCF in normal mice revealed that most SCF is expressed in Kupffer cells, peribiliary cells and arterial smooth muscle (Fig. 3A–D). SCF is weakly expressed in cholangiocytes (Fig. 3B) but absent from normal hepatocytes. Surprisingly, SCF is detected in hepatocyte nuclei of FAH-null mice after NTBC withdrawal (Fig. 3G) and in mice treated with DDC (Fig. 3I). The absence of green nuclear staining in the isotype controls indicates that this finding is not an artefact of autofluorescence induced by liver injury. FAH-null mice had normal levels of circulating SCF while being treated with NTBC (Fig. 4A). Upon withdrawal of NTBC, SCF levels declined significantly at day 3, then returned to normal at day 7 and increased to significantly elevated levels by day 14. In DDC-treated mice (Fig. 4B), there was a more modest decrease in plasma SCF by the 10th day of DDC diet, followed by a return to normal levels at day 14 through the recovery period off DDC from day 14 through 28. Despite the absence of overt histological injury in mice treated with focal liver irradiation, plasma SCF increased significantly at day 14, and then returned to normal by day 56 (Fig. 4C).
Fig. 3.
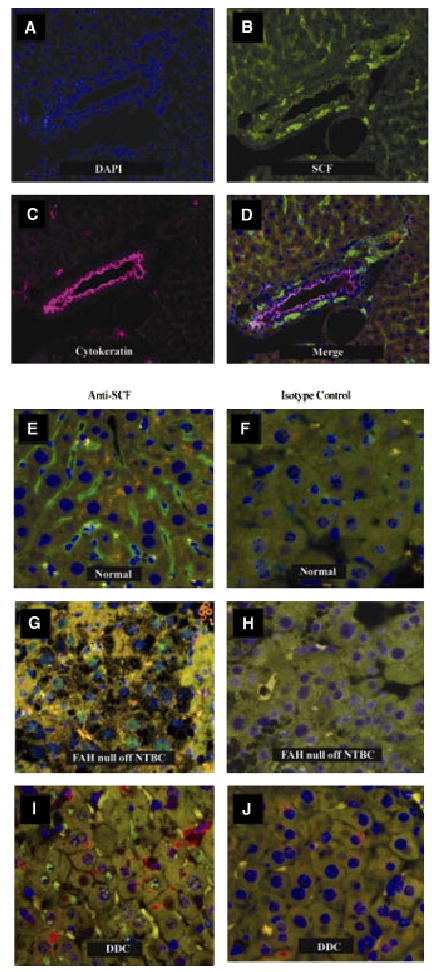
Immunofluorescent localization of stem cell factor (SCF) in normal and injured liver. (A–D) Colocalization of SCF and biliary CKs in normal liver. (A) Nuclear DNA visualized with DAPI. (B) SCF (green). (C) Biliary CKs (pink). (D) Merged image of A–C. SCF is highly expressed in Kupffer cells and in the cells surrounding bile ducts. SCF is seen in arterial smooth muscle (white arrowhead), but appears to be absent from both the arterial and the venous endothelium. SCF is expressed at low levels in cholangiocytes (best seen in 3B) and is absent from hepatocytes. (E–J) Appearance of SCF in hepatocyte nuclei after liver injury. (E) SCF is absent from hepatocyte nuclei in normal liver. (F) Isotype control, normal liver. (G) Following NTBC withdrawal from FAH null mice, SCF (green) appears in hepatocyte nuclei. (H) Isotype negative control, using a serial section from the same liver shown in 3G. (I) SCF also appears in hepatocyte nuclei in mice treated with DDC. (J) Isotype-negative control for Fig. 3I using a serial section from the same animal. DDC, 3,5-diethoxycarbonyl-1,4-dihydrocollidine; FAH, fumaryl acetoacetate hydroxylase; SCF, stem cell factor.
Fig. 4.
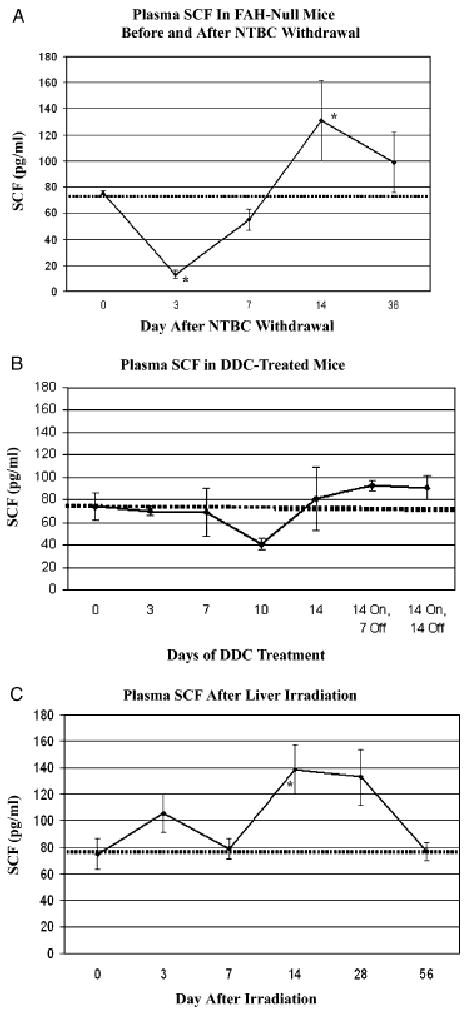
Time course of plasma stem cell factor (SCF) in response to liver injury (mean ± SE, *P < 0.05 by student's t-test). The SCF level in normal mice is indicated by a dashed horizontal line. (A) Plasma SCF in FAH null mice while on NTBC (day 0) was equal to normal controls. After induction of liver injury by withdrawal of NTBC, there was a significant decrease in SCF at 3 days, followed by normalization at 7 days and elevation at 14 days. (B) Plasma SCF in mice treated with DDC-diet for 14 days to induce ductal proliferation and oval cells. The modest decrease in SCF at 10 days was not significant. (C) Plasma SCF in controls (Day 0) or 3–56 days after focal liver irradiation. By day 14, there was a significant increase in SCF, followed by return to normal. CK cytokeratin; DDC, 3,5-diethoxycarbonyl-1,4-dihydrocollidine; FAH, fumaryl acetoacetate hydroxylase.
Expression of SDF-1 in normal and injured liver
Stromal-derived factor-1 was expressed only in BDs in normal mice (Fig. 5A). Colocalization of SDF-1 with biliary CKs 8, 18 and 19 in normal (Fig. 5A–C), FAH-null (5D–F), DDC-treated (5G–I), and liver-irradiated mice (5J–L) indicated that all biliary and oval cells express SDF-1, and SDF-1 is not expressed elsewhere. Because SDF-1 expression is proportional to the total number of oval cell and duct proliferations, the overall level of hepatic SDF-1 was increased by DDC, but not by irradiation or FAH deficiency. Circulating SDF-1 was significantly elevated in FAH-null mice before NTBC withdrawal (Fig. 6A). After NTBC withdrawal, SDF-1 levels increased dramatically and then returned towards normal by day 14 (Fig. 6A). SDF-1 levels increased above baseline within 3 days of initiating the DDC diet, and remained at or above normal even after the DDC diet was stopped for 14 days (Fig. 6B). Liver irradiation induced a significant increase in SDF-1 within 3 days (Fig. 6C). SDF-1 levels at later time points returned to normal.
Fig. 5.
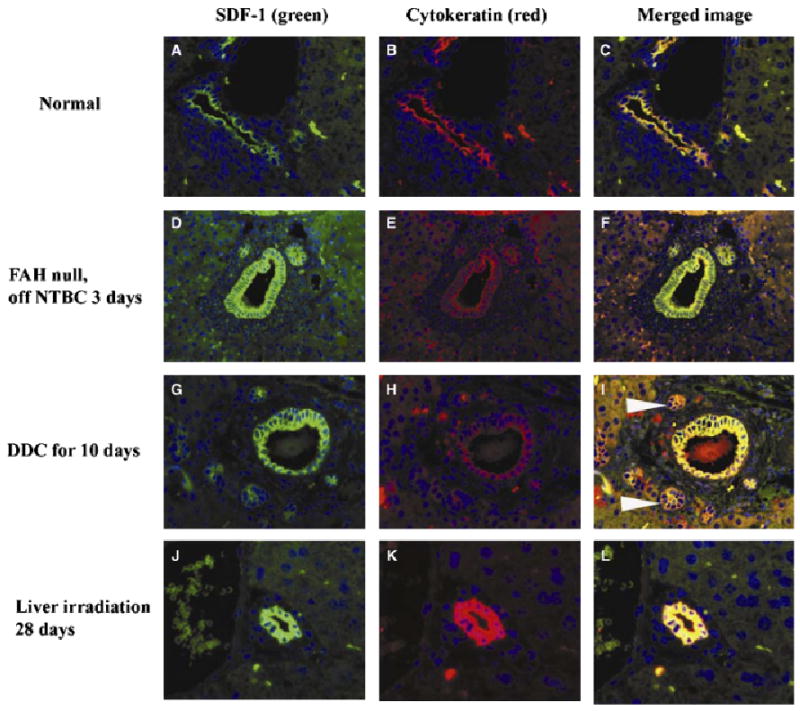
Immunofluorescent colocalization of stromal-derived factor-1 and pancytokeratin (biliary-type cytokeratins). Yellow colour confirms colocalization in the merged images. The extent of overall SDF-1 expression in the liver is therefore diminished by the paucity of bile ducts in the FAH-null mouse after NTBC withdrawal, increased by the ductal proliferation induced by DDC injury (white arrowheads) and normal in radiation injury, in parallel with the presence of cholangiocytes/oval cells. The DDC-injured liver has increased parenchymal autofluorescence in the FITC and rhodamine channels owing to cholestasis, partially obscuring the intensity of SDF-1 staining recorded in digital photographs. DDC, 3,5-diethoxycarbonyl-1,4-dihydrocollidine; FAH, fumaryl acetoacetate hydroxylase.
Fig. 6.

Time course of plasma stromal-derived factor (SDF)-1 levels in response to liver injury. Plasma SDF-1 levels were determined by ELISA for each liver injury model. In each case, normal controls (0.64 ± 0.13 ng/ml, n = 7) are indicated by the dashed line. *P < 0.05 vs. normal control, **P < 0.001 vs. normal control. (A) FAH null mice before (n = 3) and 3 days (n = 3), 7 days (n = 3) or 14 days (n = 2) after withdrawal of NTBC. (B) Normal mice treated with a DDC diet for up to 14 days (n = 3 per group). Additional mice (n = 3 per group) were allowed to recover for 7 or 14 days after a 14-day course of DDC diet. (C) Normal mice treated with focal liver irradiation (n = 4 per group). DDC, 3,5-diethoxycarbonyl-1,4-dihydrocollidine; FAH, fumaryl acetoacetate hydroxylase.
Discussion
The striking phenomenon of cellular proliferation and compensatory growth after liver injury suggests that a more detailed understanding of these processes might lead to novel treatments for liver disease. The cellular response is determined by the nature of the injury and its effect on hepatocytes, liver progenitor cells and, possibly, extrahepatic progenitor cells including those in the bone marrow. Our work illustrates the complex relationships among liver injury, oval cell proliferation, SCF and SDF-1.
Tyrosinaemia injury is not characterized by an impressive oval cell response. Rather, there is marked infiltration of macrophages in response to severe necrosis. It has been suggested (22, 23, 29) that macrophage infiltration, rather than circulating progenitor cells, leads to infrequent fusion events between macrophages and hepatocytes, in effect delivering the missing FAH gene and generating a chimeric cell that is at least tetraploid and perhaps higher order. The hepatocyte phenotype must predominate to induce expression of the FAH gene from the macrophage nucleus, where it was previously silent. Metabolic selection apparently allows such cells to then expand in vivo. Thus, the tyrosinaemia mouse liver injury is ameliorated through nonhepatic cells originating outside the liver. The dependence on dense macrophage infiltration for the rare fusion events in the FAH-null model is echoed by our own recent findings regarding recurrent acute injury from carbon tetrachloride injury (40). In that model as well, macrophages are present in dense clusters located close to residual hepatic parenchyma. In both the FAH-null mouse and carbon tetrachloride injury, endothelial damage is also present (39, 41). We have therefore suggested that the propensity for fusion in these models, over engraftment by direct differentiation, may relate to the marked density of macrophage infiltrate that increases direct membrane–membrane contact with hepatocytes, under pressure, when the sinusoidal endothelial barrier between them is abrogated.
We observed a transient decrease in SCF in the FAH mice immediately after withdrawal of NTBC, followed by rebound to supranormal levels by day 14. The abrupt decline may reflect an earlier, massive release of SCF from dying hepatocytes. In contrast with the early decrease in blood levels of SCF, followed by elevation that we observed in the FAH-null mouse, increased circulating levels of SCF were reported after acetaminophen injury, (33) accompanied by declining liver tissue levels of SCF. Seventy per cent hepatectomy was accompanied by a rapid increase in serum SCF, followed by a rapid return to normal levels (29). These differences in the SCF level may reflect differences in the onset, duration and recovery from acute liver injury, in relation to the timing of measurements. We found upregulation of SCF in the nuclei of damaged, but not normal, hepatoctyes. The source and function of nuclear SCF are not known.
The chemokine SDF-1 plays a key role in the homing of stem cells via its receptor, CXCR4. SDF-1 is known to be expressed in BD cells and may also be important in liver regeneration. We localized SDF-1 in normal and injured liver by immunofluorescence. Consistent with previous reports, we observed SDF-1 highly expressed in BD epithelium, as well as in oval cells (Fig. 5). Thus, the overall level of tissue SDF-1 appears to be proportional to the number of BDs and oval cells and, therefore, when these proliferations were more prominent, SDF-1 expression was considerably upregulated. On the other hand, in the FAH-null mouse, after withdrawal of NTBC, with the absence of oval cell proliferation and apparent reduction in canals of Hering and ductules, SDF-1 expression was quite limited. SDF-1 levels in FAH null before NTBC withdrawal were elevated relative to normal controls, consistent with ongoing subclinical liver injury in this model despite NTBC administration. In each liver injury model, there was a significant increase in circulating SDF-1 at early time points, followed by reduction towards normal levels. The magnitude of the increase in SDF-1 was proportional to the degree of acute liver injury. The subsequent reduction in plasma SDF-1 occurred despite ongoing liver injury, in the case of FAH nulls withdrawn from NTBC or normal mice treated with DDC. Ductal proliferation induced by DDC increased the tissue levels of SDF-1 in proportion to the mass of proliferating ducts, and this was matched by elevated plasma SDF-1. In contrast, liver injury in the FAH or liver irradiation model was not associated with ductal proliferation and only a transient increase in circulating SDF-1. Thus, circulating SDF-1 may be released acutely from injured liver. When the injury induces ductal proliferation, circulating SDF-1 levels remain high, while injuries that do not induce ductal proliferation (FAH null and liver irradiation) are not associated with persistently elevated circulating SDF-1. While SDF-1 and SCF may play a role in the recruitment of marrow-derived cells, neither fully accounts for the appearance of oval cells from any source in the models we describe.
Total body irradiation is used to ablate endogenous bone marrow in preparation for bone marrow transplantation in mice. In the process, the liver may be subtly injured. Further, focal irradiation of the liver in combination with acute ischaemic injury has been shown to increase the efficiency of engraftment of transplanted hepatocytes dramatically (24, 42, 43). It was suggested that focal radiation of the liver inhibits the ability of native hepatocytes to proliferate in response to injury, allowing greater engraftment of transplanted hepatocytes. In our experiments, there was only mild histological hepatocyte injury after 1000 rads to the liver. The presence of mitotic hepatocytes and the absence of oval cells after hepatic irradiation suggest that hepatocyte replication was not significantly inhibited at this dose of radiation. Repair appeared to occur primarily through replication of hepatocytes rather than proliferation of intrahepatic progenitors.
The results we describe underscore the critical importance of detailed characterization of liver injury in models that have been used to assess bone marrow stem cell plasticity in the liver. Thus, as we have suggested previously (1), the phenomenon of engraftment of circulating cells into the liver depends on the nature and severity of the injury. Conclusions about which parenchymal and nonparenchymal cells may derive from circulating progenitors and by which pathway (fusion vs. direct differentiation) they engraft are always model-dependent. Conclusions about plasticity events should not be generalized from single experiments. The mechanisms by which marrow-derived cells become integrated into the liver as epithelial cells may be as diverse as the injuries themselves.
Acknowledgments
Financial Support was provided by NIH DK073404, Yale Center of Excellence in Molecular Hematology; PI–Swenson NIH DK61846, HL073742 and PI – Krause NIH DK58559, PI-Theise.
Abbreviations
- DDC
3,5-diethoxycarbonyl-1,4-dihydrocollidine
- FAH
fumaryl acetoacetate hydrolase
- SCF
stem cell factor
- SDF-1
stromal-derived factor-1
References
- 1.Theise ND, Krause DS, Sharkis S. Comment on ‘Little evidence for developmental plasticity of adult hematopoietic stem cells’. Science. 2003;299:1317. doi: 10.1126/science.1078412. [DOI] [PubMed] [Google Scholar]
- 2.Theise ND, Badve S, Saxena R, et al. Derivation of hepatocytes from bone marrow cells in mice after radiation-induced myeloablation. Hepatology. 2000;31:235–40. doi: 10.1002/hep.510310135. [DOI] [PubMed] [Google Scholar]
- 3.Schwartz RE, Reyes M, Koodie L, et al. Multipotent adult progenitor cells from bone marrow differentiate into functional hepatocyte-like cells. J Clin Invest. 2002;109:1291–302. doi: 10.1172/JCI15182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Menthena A, Deb N, Oertel M, et al. Bone marrow progenitors are not the source of expanding oval cells in injured liver. Stem Cells. 2004;22:1049–61. doi: 10.1634/stemcells.22-6-1049. [DOI] [PubMed] [Google Scholar]
- 5.Petersen BE, Bowen WC, Patrene KD, et al. Bone marrow as a potential source of hepatic oval cells. Science. 1999;284:1168–70. doi: 10.1126/science.284.5417.1168. [DOI] [PubMed] [Google Scholar]
- 6.Newsome PN, Johannessen I, Boyle S, et al. Human cord blood-derived cells can differentiate into hepatocytes in the mouse liver with no evidence of cellular fusion. Gastroenterology. 2003;124:1891–900. doi: 10.1016/s0016-5085(03)00401-3. [DOI] [PubMed] [Google Scholar]
- 7.Krause DS, Theise ND, Collector MI, et al. Multi-organ, multi-lineage engraftment by a single bone marrow-derived stem cell. Cell. 2001;105:369–77. doi: 10.1016/s0092-8674(01)00328-2. [DOI] [PubMed] [Google Scholar]
- 8.Theise ND, Nimmakayalu M, Gardner R, et al. Liver from bone marrow in humans. Hepatology. 2000;32:11–6. doi: 10.1053/jhep.2000.9124. [DOI] [PubMed] [Google Scholar]
- 9.Wang X, Ge S, Mcnamara G, Hao QL, Crooks GM, Nolta JA. Albumin expressing hepatocyte-like cells develop in the livers of immune-deficient mice transmitted with highly purified human hematopoietic stem cells. Blood. 2003;30:30. doi: 10.1182/blood-2002-05-1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Korbling M, Katz RL, Khanna A, et al. Hepatocytes and epithelial cells of donor origin in recipients of peripheral-blood stem cells. N Engl J Med. 2002;346:738–46. doi: 10.1056/NEJMoa3461002. [DOI] [PubMed] [Google Scholar]
- 11.Lagasse E, Connors H, Al-Dhalimy M, et al. Purified hematopoietic stem cells can differentiate into hepatocytes in vivo. Nat Med. 2000;6:1229–34. doi: 10.1038/81326. [DOI] [PubMed] [Google Scholar]
- 12.Jang YY, Collector MI, Baylin SB, Diehl AM, Sharkis SJ. Hematopoietic stem cells convert into liver cells within days without fusion. Nat Cell Biol. 2004;6:532–9. doi: 10.1038/ncb1132. [DOI] [PubMed] [Google Scholar]
- 13.Harris RG, Herzog EL, Bruscia EM, Grove JE, Van Arnam JS, Krause DS. Lack of a fusion requirement for development of bone marrow-derived epithelia. Science. 2004;305:90–3. doi: 10.1126/science.1098925. [DOI] [PubMed] [Google Scholar]
- 14.Wang X, Foster M, Al-Dhalimy M, Lagasse E, Finegold M, Grompe M. The origin and liver repopulating capacity of murine oval cells. Proceedings of the National Academy of Sciences of the United States of America. 2003 doi: 10.1073/pnas.1734199100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jiang Y, Jahagirdar BN, Reinhardt RL, et al. Pluripotency of mesenchymal stem cells derived from adult marrow. Nature. 2002;20:20. doi: 10.1038/nature00870. [DOI] [PubMed] [Google Scholar]
- 16.Almeida-Porada G, Porada CD, Chamberlain J, Torabi A, Zanjani ED. Formation of human hepatocytes by human hematopoietic stem cells in sheep. Blood. 2004;104:2582–90. doi: 10.1182/blood-2004-01-0259. [DOI] [PubMed] [Google Scholar]
- 17.Wagers AJ, Sherwood RI, Christensen JL, Weissman IL. Little evidence for developmental plasticity of adult hematopoietic stem cells. Science. 2002;297:2256–9. doi: 10.1126/science.1074807. [DOI] [PubMed] [Google Scholar]
- 18.Grompe M. The role of bone marrow stem cells in liver regeneration. Semin Liver Dis. 2003;23:363–72. doi: 10.1055/s-2004-815560. [DOI] [PubMed] [Google Scholar]
- 19.Zeng L, Rahrmann E, Hu Q, et al. Multi-potent adult progenitor cells from swine bone marrow. Stem Cells. 2006;24:2355–66. doi: 10.1634/stemcells.2005-0551. [DOI] [PubMed] [Google Scholar]
- 20.Thorgeirsson SS, Grisham JW. Hematopoietic cells as hepatocyte stem cells: a critical review of the evidence. Hepatology. 2006;43:2–8. doi: 10.1002/hep.21015. [DOI] [PubMed] [Google Scholar]
- 21.Preisegger KH, Factor VM, Fuchsbichler A, Stumptner C, Denk H, Thorgeirsson SS. Atypical ductular proliferation and its inhibition by transforming growth factor beta1 in the 3,5-diethoxycarbonyl-1,4-dihydrocollidine mouse model for chronic alcoholic liver disease. Lab Invest; J Tech Methods Pathol. 1999;79:103–9. [PubMed] [Google Scholar]
- 22.Wang X, Willenbring H, Akkari Y, et al. Cell fusion is the principal source of bone-marrow-derived hepatocytes. Nature. 2003;422:897–901. doi: 10.1038/nature01531. [DOI] [PubMed] [Google Scholar]
- 23.Vassilopoulos G, Wang PR, Russell DW. Transplanted bone marrow regenerates liver by cell fusion. Nature. 2003;422:901–4. doi: 10.1038/nature01539. [DOI] [PubMed] [Google Scholar]
- 24.Guha C, Parashar B, Deb NJ, et al. Liver irradiation: a potential preparative regimen for hepatocyte transplantation. Int J Rad Oncol Biol Phys. 2001;49:451–7. doi: 10.1016/s0360-3016(00)01495-4. [DOI] [PubMed] [Google Scholar]
- 25.Wulf G, Luo K, Jackson K, Brenner M, Goodell M. Cells of the hepatic side population contribute to liver regeneration and can be replenished with bone marrow stem cells. Haematologica. 2003;88:368–78. [PubMed] [Google Scholar]
- 26.Camargo FD, Finegold M, Goodell MA. Hematopoietic myelomonocytic cells are the major source of hepatocyte fusion partners. J Clin Invest. 2004;113:1266–70. doi: 10.1172/JCI21301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Crosby HA, Kelly DA, Strain AJ. Human hepatic stem-like cells isolated using c-kit or CD34 can differentiate into biliary epithelium. Gastroenterology. 2001;120:534–44. doi: 10.1053/gast.2001.21175. [DOI] [PubMed] [Google Scholar]
- 28.Kollet O, Shivtiel S, Chen YQ, et al. HGF, SDF-1, and MMP-9 are involved in stress-induced human CD34+ stem cell recruitment to the liver. J Clin Invest. 2003;112:160–9. doi: 10.1172/JCI17902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ren X, Hogaboam C, Carpenter A, Colletti L. Stem cell factor restores hepatocyte proliferation in IL-6 knockout mice following 70% hepatectomy. J Clin Invest. 2003;112:1407–18. doi: 10.1172/JCI17391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Satake M, Shimano K, Yamamoto T, et al. Role of c-kit receptor tyrosine kinase-mediated signal transduction in proliferation of bile epithelial cells in young rats after ligation of bile duct: a study using Ws/Ws c-kit mutant rats. J Hepatol. 2003;39:86–92. doi: 10.1016/s0168-8278(03)00149-1. [DOI] [PubMed] [Google Scholar]
- 31.Fujio K, Evarts RP, Hu Z, Marsden ER, Thorgeirsson SS. Expression of stem cell factor and its receptor, c-kit, during liver regeneration from putative stem cells in adult rat. Lab Invest; J Tech Methods Pathol. 1994;70:511–6. [PubMed] [Google Scholar]
- 32.Gaca MD, Pickering JA, Arthur MJ, Benyon RC. Human and rat hepatic stellate cells produce stem cell factor: a possible mechanism for mast cell recruitment in liver fibrosis. J Hepatol. 1999;30:850–8. doi: 10.1016/s0168-8278(99)80139-1. [DOI] [PubMed] [Google Scholar]
- 33.Simpson K, Hogaboam CM, Kunkel SL, Harrison DJ, Bone-Larson C, Lukacs NW. Stem cell factor attenuates liver damage in a murine model of acetaminophen-induced hepatic injury. Lab Invest; J Tech Methods Pathol. 2003;83:199–206. doi: 10.1097/01.lab.0000057002.16935.84. [DOI] [PubMed] [Google Scholar]
- 34.Baumann U, Crosby HA, Ramani P, Kelly DA, Strain AJ. Expression of the stem cell factor receptor c-kit in normal and diseased pediatric liver: identification of a human hepatic progenitor cell? Hepatology. 1999;30:112–7. doi: 10.1002/hep.510300140. [DOI] [PubMed] [Google Scholar]
- 35.Hattori K, Heissig B, Tashiro K, et al. Plasma elevation of stromal cell-derived factor-1 induces mobilization of mature and immature hematopoietic progenitor and stem cells. Blood. 2001;97:3354–60. doi: 10.1182/blood.v97.11.3354. [DOI] [PubMed] [Google Scholar]
- 36.Matsusaka S, Tsujimura T, Toyosaka A, et al. Role of c-kit receptor tyrosine kinase in development of oval cells in the rat 2-acetylaminofluorene/partial hepatectomy model. Hepatology. 1999;29:670–6. doi: 10.1002/hep.510290304. [DOI] [PubMed] [Google Scholar]
- 37.Kofman AV, Morgan G, Kirschenbaum A, et al. Dose- and time-dependent oval cell reaction in acetaminophen-induced murine liver injury. Hepatology. 2005;41:1252–61. doi: 10.1002/hep.20696. [DOI] [PubMed] [Google Scholar]
- 38.Grompe M, Al-Dhalimy M, Finegold M, et al. Loss of fumarylacetoacetate hydrolase is responsible for the neonatal hepatic dysfunction phenotype of lethal albino mice. Genes Dev. 1993;7:2298–307. doi: 10.1101/gad.7.12a.2298. [DOI] [PubMed] [Google Scholar]
- 39.Grompe M, Lindstedt S, Al-Dhalimy M, et al. Pharmacological correction of neonatal lethal hepatic dysfunction in a murine model of hereditary tyrosinaemia type I. Nat Genet. 1995;10:453–60. doi: 10.1038/ng0895-453. [DOI] [PubMed] [Google Scholar]
- 40.Quintana-Bustamante O, Alvarez-Barrientos A, Kofman AV, et al. Hematopoietic mobilization in mice increases the presence of bone marrow-derived hepatocytes via in vivo cell fusion. Hepatology. 2006;43:108–16. doi: 10.1002/hep.21005. [DOI] [PubMed] [Google Scholar]
- 41.Laskin DL. Sinusoidal lining cells and hepatotoxicity. Toxicol Pathol. 1996;24:112–8. doi: 10.1177/019262339602400115. [DOI] [PubMed] [Google Scholar]
- 42.Malhi H, Gorla GR, Irani AN, Annamaneni P, Gupta S. Cell transplantation after oxidative hepatic preconditioning with radiation and ischemia-reperfusion leads to extensive liver repopulation. Proc Natl Acad Sci USA. 2002;99:13114–9. doi: 10.1073/pnas.192365499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guha C, Deb NJ, Sappal BS, Ghosh SS, Roy-Chowdhury N, Roy-Chowdhury J. Amplification of engrafted hepatocytes by preparative manipulation of the host liver. Artif Organs. 2001;25:522–8. doi: 10.1046/j.1525-1594.2001.025007522.x. [DOI] [PubMed] [Google Scholar]