

Abstract
Although the molecular pathways governing the development of the anterior pole of the heart have been the subject of intense investigation, little is understood about the molecular mechanisms underlying the morphogenesis of the posterior pole of the heart which generates the atria, pulmonary veins and portions of the atrio-ventricular canal. Here we show that Wnt2 is expressed specifically in the developing inflow tract mesoderm in a domain encompassing the dorsal mesocardium and dorsal mesenchymal protrusion which generates portions of the atria and atrio-ventricular cushions. Loss of Wnt2 results in defective development of the atrial myocardium, atrio-ventricular canal, and pulmonary veins resulting in a phenotype resembling the human congenital heart syndrome complete common atrio-ventricular canal. The dorsal mesocardium and dorsal mesenchymal protrusion overlaps spatially with posterior second heart field progenitors and we show that the number and proliferation of these progenitors is reduced in Wnt2-/- mutants. Remarkably, these defects can be rescued in vivo in a temporally restricted manner through pharmacological inhibition of Gsk-3β, indicating that Wnt2 regulates canonical Wnt signaling in the posterior cardiac mesoderm. Molecular and genetic analysis shows that Wnt2 works in a feed-forward transcriptional loop with Gata6 to regulate posterior cardiac development. These data reveal an important new molecular pathway regulating cardiac inflow tract development and demonstrates that such defects in the second heart field can be rescued pharmacologically in vivo.
Introduction
Cardiac morphogenesis occurs in a regionally distinctive manner with specific molecular programs directing the development of spatially distinct regions of the heart. The mammalian heart develops from a crescent shaped mesodermal population of cells that are readily apparent by E7.5 of mouse development. The heart displays distinctive anterior-posterior patterning which is most obvious in the divergent morphological origins and functions of the atria and ventricles. Gene expression analysis has demonstrated that atrial versus ventricular programs are active as early as the cardiac crescent stage of development (E7.5) suggesting that molecular pathways that define these different cardiac lineages are active before morphological distinction of these chambers. In addition to the anterior-posterior patterning events, the mesoderm that contributes to the heart can also be divided into two separate origins, the first heart field (FHF) and the second heart field (SHF). Important progress has been made in understanding the contribution of the SHF to development of the anterior portion of the heart including the outlflow tract and right ventricle (Cai et al., 2003).
In contrast to the advances made in understanding the mechanisms underlying development of the anterior pole and outflow tract of the heart, less is understood about the development of the posterior pole or inflow tract of the heart. Although there are important similarities in the derivation of anterior and posterior cardiac progenitors including Islet1 (Isl1) expression (Snarr et al., 2007a), several studies have described posterior cardiac progenitors differing from their more anterior neighbors at the morphological and molecular level (Anderson et al., 2006; Christoffels et al., 2006). Posterior cardiac progenitors are thought to generate portions of the inflow tract including the atria and atrio-ventricular (AV) canal (Anderson et al., 2006). In particular, the dorsal mesenchymal protrusion, a region that is encompassed within the posterior Isl1+ cardiac mesoderm population, generates portions of the atrial septum, AV canal, and inflow tract myocardium (Snarr et al., 2007b). The importance of the cardiac inflow tract and its derivatives is underscored by multiple congenital cardiovascular anomalies including complete common atrio-ventricular canal (CCAVC), atrial septal defects, and abnormal pulmonary venous return (reviewed in (Allwork, 1982; Strauss, 1998)). Thus, further investigation into both the morphological development as well as molecular pathways required for inflow tract development will have an important impact on our understanding of congenital disease associated with this region of the cardiovascular system.
Wnt signaling has been shown to regulate the development of the anterior portion of the heart including the outflow tract and right ventricle (Ai et al., 2007; Cohen et al., 2007; Kwon et al., 2007; Lin et al., 2007; Qyang et al., 2007). Loss of β-catenin leads to decreased numbers of anterior Isl1 positive (Isl1+) SHF progenitors whereas stabilization of β-catenin expands the number of these progenitors. Isl1+ SHF progenitors are also found in the posterior region of the developing cardiac mesoderm (Lin et al., 2007; Qyang et al., 2007). Although previous reports suggest that the posterior SHF progenitors contribute to the cardiac inflow tract mesoderm including the atria (Galli et al., 2008), the function of this subpopulation of SHF progenitors as well as the molecular pathways that regulate their expansion and differentiation remains unclear. Here we show that Wnt2, a ligand expressed specifically in the posterior pole of the developing heart, is essential for development and differentiation of posterior structures within the developing heart including the atria, pulmonary veins, and AV canal. The defects in Wnt2-/- mutants resemble the human congenital heart syndrome CCAVC and they can be rescued through pharmacological inhibition of Gsk-3β, indicating that Wnt2 signals through the β-catenin dependent Wnt pathway to regulate cardiac inflow tract development. Moreover, we show that Wnt2 signaling is necessary for Gata6 expression and acts epistatically with Gata6 to regulate cardiac inflow tract development. These data highlight a novel molecular pathway regulating development of the posterior pole of the heart including the cardiac inflow tract that can be pharmacologically rescued with Gsk-3β inhibitors.
Results
Expression of Wnt2 in the posterior pole and inflow tract of the heart
Previous reports have shown that Wnt2 is expressed in early cardiac mesoderm in the cardiac crescent at E7.5 (Monkley et al., 1996). To more fully address the pattern of Wnt2 expression during cardiovascular development, we performed in situ hybridization on whole embryos and tissue sections. From E8.5-E9.5, Wnt2 expression is observed in the posterior cardiac mesoderm surrounding the ventral aspect of the anterior foregut contiguous with the inflow tract of the developing heart (Fig. 1A-E). Expression of Wnt2 is also observed in the atria at E9.5 and E10.5 (Fig. 1E and F). Although expression of Wnt2 is observed in the developing inflow tract at these times, it is not observed in the ventricles or AV canal (Fig. 1A-C, G). Within the inflow tract mesoderm, high levels of Wnt2 are observed in a region overlapping with the dorsal mesocardium and dorsal mesenchymal protrusion (DMP) at E9.5 (Fig. 1D and Supplemental Figure 1). This tissue is contigious with the atrial myocardium including the primary atrial septum and the overlying mesenchymal cap, which in turn helps generate portions of the AV cushions (Fig. 1H and (Snarr et al., 2007a; Snarr et al., 2007b; Wessels et al., 2000). The dorsal mesocardium and DMP overlaps extensively with the posterior portion of the second heart field as shown by Isl1-cre fate mapping (Cai et al., 2003; Snarr et al., 2007a; Sun et al., 2007). Wnt2 expression is not observed in either epicardium or endocardium but is observed in septum transversum mesoderm in a pattern that overlaps with Tbx18 and Wilm's tumor-1 (WT1) at E9.5 and E10.5 (Fig. 1D and Supplemental Figure 2). These data are consistent with a previous report showing that Wnt2 is not expressed in the AV canal or ventricular myocardium at these times (Watanabe et al., 2006). By E10.5, expression of Wnt2 begins to decline and expression in the heart is very low to undetectable after E12.5 (data not shown).
Figure 1. Cardiac expression of Wnt2 and characterization of the CCAVC phenotype in Wnt2-/- mutants.
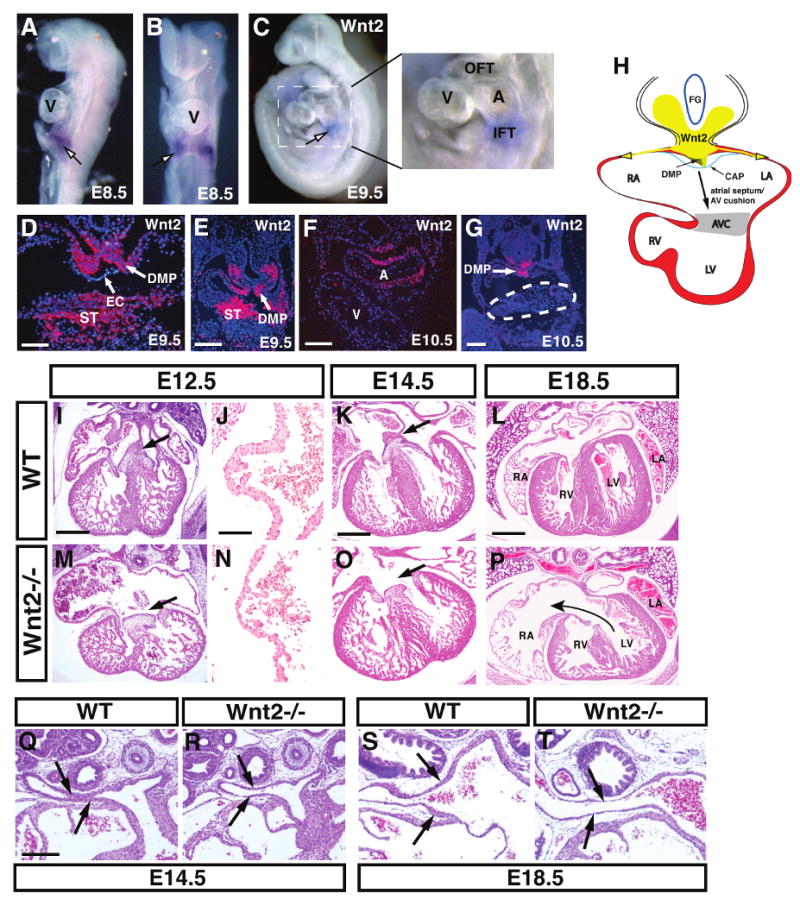
Wnt2 expression is observed in the posterior pole of the developing heart at E8.5 and E9.5 but not in the early ventricles as shown by whole mount in situ hybridization (A-C). Expression at E9.5 is found in the dorsal mesocardium, dorsal mesenchymal protrusion (DMP), and in the septum transversum (ST) but not in the overlying endocardium (EC) (D and E). At E10.5, expression of Wnt2 is also observed in the developing atria (F) but expression is absent in the AV canal (G, dotted line). Diagram showing relation of Wnt2 expression and the inflow tract region including the DMP and mesenhcymal cap (CAP) and the contribution of these tissues to the AV canal (H). Wild-type (I-L) and Wnt2-/- mutants (M-P) at the indicated ages showing reduced atrial and AV canal myocardial development (I and M, arrows) as well as loss of atrial septation (K and O, arrows). J and N are high magnification pictures of the atrial wall at E12.5. H+E staining of wild-type (Q and S) and Wnt2-/- mutant (R and T) sections at E14.5 and E18.5 show reduced wall thickness of pulmonary veins in Wnt2-/- mutants. Scale bars: D, J, N=50 μm, E and F=100 μm, G, I, M, K, O=200 μm, Q-T=250 μm, L and P=300 μm.
Wnt2 null mutants exhibit defects in cardiac inflow tract and AV canal morphogenesis
To determine the role of Wnt2 signaling in cardiac development, we generated and characterized Wnt2 null embryos. Approximately 85% of Wnt2 null mutants die at birth (Fig. 5A and (Goss et al., 2009)). Histological analysis shows that Wnt2-/- mutants have a thin atrial wall and defects in AV canal development including decreased development or complete loss of the primary atrial septum and a deficiency in development of the superior AV cushion and associated myocardium (Fig. 1I-P, Supplemental Figure 1). This defect is observed as early as E10.5 where there is an early loss of development of the primary atrial septum and the overlying mesenchymal cap which generates portions of the AV cushions (Supplemental Fig. 1). Overall myocardial differentiation in the atria and AV canal is deficient in Wnt2-/- mutants (Fig. 1I-P and Supplemental Figure 1). Together, these defects resemble the human congenital heart syndrome CCAVC. Pulmonary vein development was also significantly defective in Wnt2-/- mutants. At both E14.5 and E18.5, pulmonary veins in Wnt2-/- mutants had a thinner mural wall especially closer to the atria (Fig. 1Q-T). Ventricular and outflow tract development did not appear disrupted in Wnt2-/- mutants although we did observe some thinning of the ventricular wall later in development after E14.5 (Fig. 1P and Supplemental Figure 3). Signals from the epicardium have been shown to play a role in promoting myocardial proliferation and epicardial progenitors are derived from the septum transversum where Wnt2 is expressed (Fig. 1D and E and (Lavine et al., 2005). However, expression of the epicardial markers WT1 and Tbx18 are unchanged in Wnt2-/- mutants suggesting that epicardium is intact in these mutants (Fig. 3M and Supplemental Figure 2). Moreover, Wnt2 expression has been observed at E7.5 in the early cardiac mesoderm (Monkley et al., 1996). Thus, the ventricular thinning at later stages of Wnt2-/- mutants could be secondary to impending heart failure or to a role for Wnt2 in early cardiac mesoderm growth.
Figure 5. Pharmacological inhibition of Gsk-3β rescues cardiac defects in Wnt2-/- mutants.
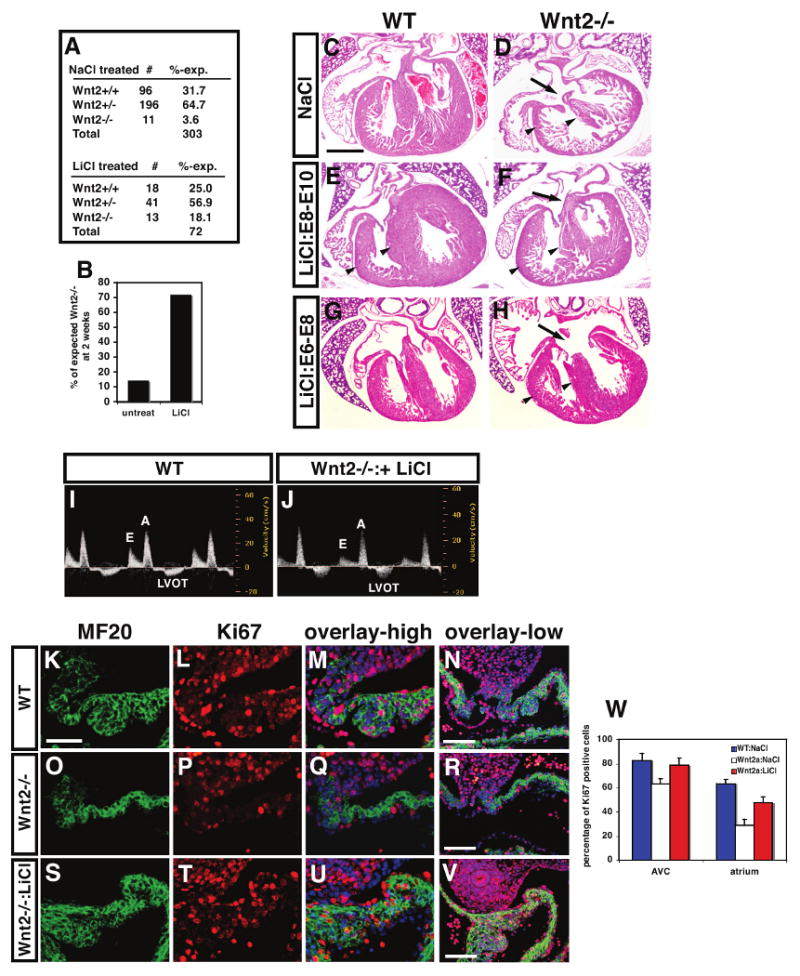
Embryos treated with LiCl from E8-E10 showed a significant increase in viability compared to untreated embryos (A and B). H+E stained sections show that E8-E10 LiCl treated Wnt2-/- mutants exhibited a reversal of the CCAVC phenotype at E17.5 including restoration of normal AV canal morphology (C-F, arrows). In contrast, treatment with LiCl between E6-E8 did not restore normal AV canal morphology but did increase ventricular myocardial mass (G and H). Fetal echocardiograms at E17.5 show normal blood flow across the AV canal in E8-E10 LiCl rescued Wnt2-/- mutants (I and J). MF20 and Ki67 immunostaining at E11.5 shows that the deficiency in inflow tract/atrial myocardial proliferation is rescued by E8-E10 LiCl treatment at high and low magnification (K-W). Scale bars: C-H=300 μm, K-S=25 μm, N, R, V=100 μm.
Figure 3. Loss of Wnt2 expression leads to a specific loss in atrial myocardial proliferation and differentiation.
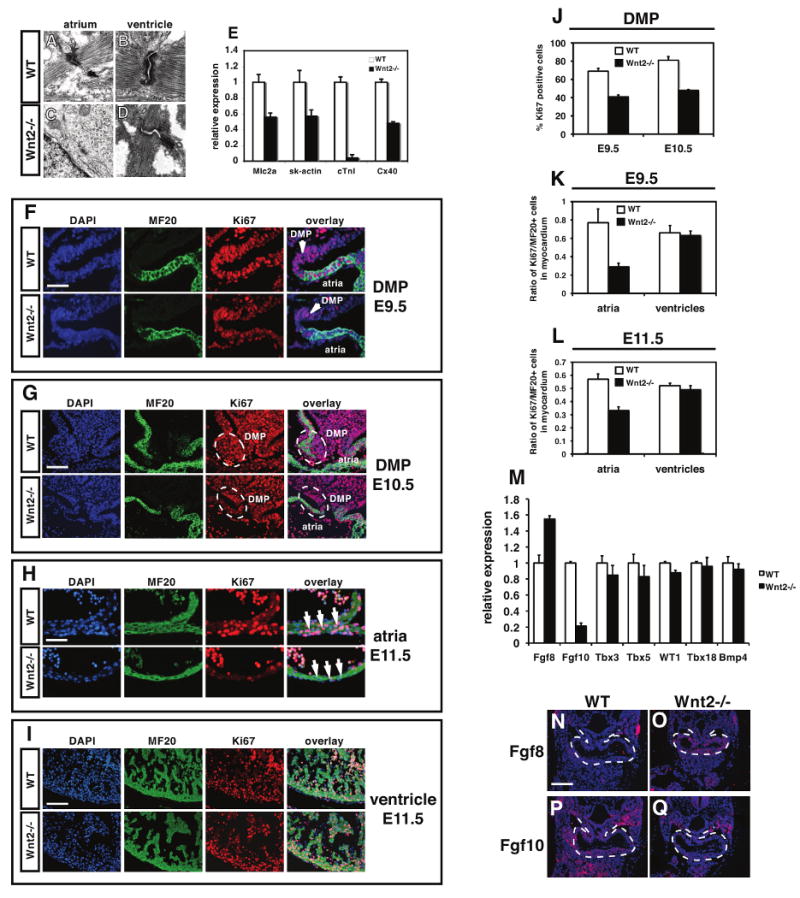
Transmission electron microscopy shows normal sarcomeric structure in the atria and ventricular myocardium of wild-type hearts at E11.5 (A and B). Although sarcomeric structure appears normal in the ventricles of Wnt2-/- mutants (D), there is little sarcomeric structure observed in atrial myocardium of Wnt2-/- mutants (C). Expression of several important myocardial genes is decreased in the atria of Wnt2-/- mutants (E). Proliferation is reduced in the dorsal mesocardium and DMP region at E9.5 (F) and E10.5 (G) as determined by Ki67 and MF20 immunostaining and quantitation (J). Myocardial proliferation is reduced in atrial (H) but not ventricular myocardium (I) of Wnt2-/- mutants as determined by Ki67 and MF20 immunostaining and quantitation (K and L). Expression of Fgf8, Fgf10, Tbx3, Tbx5, WT1, Tbx18, and Bmp4 in wild-type and Wnt2-/- mutants at E9.5 as determined by Q-PCR (M). In situ hybridization for Fgf8 and Fgf10 expression on E9.5 wild-type (N and P) and Wnt2-/- (O and Q) mutants. Scale bars: A-D=0.2 μm, F and H=25 μm, G and I=50 mm, N-Q=100 μm.
Endocardial cushions develop through a process of endothelial-mesenchymal transformation (EMT) where the endothelial cells respond to signals from the overlying myocardium to migrate into the extracellular matrix of the cardiac jelly (Nakajima et al., 2000). Wnt2 is expressed in the dorsal mesocardium and DMP and it may signal in a paracrine fashion to the overlying mesenchymal cap of the primary atrial septum and adjacent endocardium to promote proper development of the AV canal through regulation of EMT (Snarr et al., 2007a; Snarr et al., 2007b; Wessels et al., 2000). Thus, we wanted to determine whether EMT was perturbed in Wnt2-/- mutants. AV canal explants from E9.5 heterozygous and Wnt2-/- embryos were cultured in a collagen gel and the number of mesenchymal cells migrating out of the explant were quantitated. These results show that Wnt2-/- mutant explants had a significant decrease in EMT consistent with the observed defects in AV canal development (Fig. 2A-C).
Figure 2. Wnt2-/- mutants exhibit defects in AV canal development leading to abnormal AV blood flow.

EMT assays show that Wnt2-/- AV canal explants undergo EMT less efficiently than Wnt2+/- littermates (A-C). Echocardiograms of E16.5 wild-type (D) and Wnt2-/- mutants (E) reveal abnormal flow across the AV canal due to defective AV canal development. In wild-type embryo, E and A waves represent normal transvalvular Doppler flow from left atrium to left ventricle during early diastole (E) and left atrial contraction (A), respectively. LVOT wave represents normal flow from left ventricle to outflow tract. Wnt2-/- mutant embryo reveals abnormal flow across the AV canal (E). The waves below (red arrow) and above the baseline (green arrow) are the flows from left ventricle to right atria during systole and from right ventricle to left ventricle during diastole, respectively. Surviving adult Wnt2-/- mutants show regurgitant flow (3.8m/s) across the AV canal (G, arrow) with preserved cardiac systolic function whereas wild-type littermates show normal flow (F). Schematics represent placement of the cursor (black double line) for Doppler echocardiography measurements.
To assess whether cardiac function in Wnt2-/- mutants was compromised due to defective AV canal development, echocardiography was performed at E16.5 on wild-type and Wnt2-/- mutant littermates as well as adult mutants. These studies showed abnormal blood flow across the AV canal in embryonic Wnt2-/- mutants (Fig. 2D and E). Severe AV canal regurgitation was also observed in the small number of Wnt2-/- mutants that survive (Fig. 2F and G). Since we did not observe Wnt2 expression in AV canal cushions and valves, these data suggest that Wnt2 expression in the DMP in the posterior inflow tract region regulates AV canal morphogenesis.
Inflow tract and atrial myocardial defects in Wnt2 null mutants
To characterize the mechanism underlying defective inflow tract development in Wnt2-/- null mutants, we performed transmission electron microscopy (TEM) to assess sarcomeric structure in both atrial and ventricular myocardium. As expected, wild-type littermates exhibited normal sarcomeric structure in both atrial and ventricular myocardium at E12.5 (Fig. 3A and B). Wnt2-/- null ventricular myocytes also exhibited normal sarcomeric structure (Fig. 3D). However, sarcomeric structure was severely compromised observed in atrial myocytes of Wnt2-/- mutants as noted by the dramatic reduction of organized Z-bands (Fig. 3C). In agreement with these findings, expression of important cardiac contractile and cytoskeletal genes were reduced in the atria of Wnt2-/- mutants with cardiac troponin I (cTnI) expression reduced by more than 90% (Fig. 3E).
Given the poor myocardial development observed in the atria of Wnt2-/- mutants, we assessed proliferation in the dorsal mesocardium and DMP region as well as in the atria and ventricles at E9.5 and E11.5. Wnt2-/- mutants showed a considerable reduction in proliferation in the dorsal mesocardium, DMP, and atrial myocardium while ventricular proliferation was unaffected at E9.5 (Fig. 3F, G, J and Supplemental Figure 4). Wnt2-/- mutants continued to show reduced atrial but not ventricular myocardial proliferation at E11.5 (Fig. 3H, I, K and L).
We examined expression of several other key signaling and transcription factors known to be important for myocardial and AV canal development as well as markers of epicardium. Quantitative PCR (Q-PCR) shows that expression of Fgf10 is decreased in Wnt2-/- mutants while expression of Fgf8 is increased at E9.5 (Fig. 3M-Q). This result is consistent with previous studies showing that Fgf10 is a direct target of Wnt/β-catenin signaling (Cohen et al., 2007). Expression of Tbx3, Tbx5, Tbx18, WT1, Bmp2, and Bmp4 was not significantly altered in Wnt2-/- mutants at E9.5 (Fig. 3M and Supplemental Figure 2). Thus, loss of Wnt2 expression leads to disregulation of Fgf ligand expression but unaltered expression of other key factors in cardiac development.
Wnt2 regulates expansion of SHF progenitors through β-catenin dependent signaling
Wnt2 is expressed at significant levels in the posterior dorsal mesocardium and DMP region of the inflow tract which generates portions of the AV canal as well as inflow tract myocardium and overlaps extensively with posterior Isl1+ SHF progenitors (Snarr et al., 2007a; Snarr et al., 2007b; Wessels et al., 2000). To determine whether Wnt2 regulates the β-catenin dependant canonical Wnt pathway in the posterior inflow tract and AV canal, we generated Wnt2-/-:TOPGAL compound mutants. Previous reports have shown significant TOPGAL activity in the developing AV canal region in early cardiac development (Gitler et al., 2003). Wild-type TOPGAL animals revealed strong lacZ staining in the AV canal including both cushion mesenchyme and adjacent myocardium at E11.5 (Fig. 4A). However, Wnt2-/- mutants showed a dramatic down-regulation of lacZ staining in the AV canal region (Fig. 4B). To further assess Wnt signaling activity in Wnt2-/- mutants, we examined expression of the Wnt target genes axin2 and Lef1 at E9.5. Expression of axin2 and Lef1 were also significantly reduced in the inflow tract of Wnt2-/- mutants indicating a decrease in Wnt/β-catenin signaling (Fig. 4E and F). High levels of β-catenin protein expression as well as nuclear accumulation of β-catenin have been shown to reflect active Wnt staining in vivo (Zhang et al., 2008). High levels of β-catenin expression are observed both in the inflow tract mesoderm including both myocardium and endocardium as well as in the foregut endoderm of wild-type embryos while β-catenin expression was reduced in the inflow tract myocardium and endocardium of Wnt2-/- mutants at E9.5 (Fig. 4C and D). β-catenin expression in foregut endoderm of Wnt2-/- mutants was less affected (Fig. 4D). This is supported by decreased nuclear β-catenin expression in the dorsal mesocardium and DMP region of Wnt2-/- mutants at E9.5 in comparison to wild-type littermates (Fig. 4H-K).
Figure 4. Wnt/β-catenin signaling and Isl1+ SHF progenitor expansion is deficient in Wnt2-/- mutants.

TOPGAL (A) and Wnt2-/-:TOPGAL E11.5 compound mutants (B) were stained for β-galactosidase activity. β-catenin expression was assessed using immunohistochemistry on wild-type (C) and Wnt2-/- mutants (D) at E9.5. Q-PCR was used to determine expression of axin2 (E), Lef1 (F), and Isl1 (G) at E9.5 in wild-type and Wnt2-/- mutant inflow tract tissue. Nuclear β-catenin expression was assessed by fluorescent immunohistochemistry at E9.5 in the dorsal mesocardium and DMP region (region noted by a dotted square in C and D) of wild-type (H and J) and Wnt2-/- mutants (I and K). Cell nuclei are indicated with dotted outlines and arrowheads. Isl1 expression was assessed in wild-type and Wnt2-/- mutants using immunohistochemistry at E9.5 (L-N). Isl1 expression was assessed in wild-type (Nkx2.5cre/+) and Nkx2.5cre/+:Ctnnb1flox/flox mutants at E9.5 (O-Q). Arrowheads indicate sites of altered expression in each panel. Scale bars: A and B=150 μm, H-K=10 μm, L and M=50 μm, O and P=30 μm.
We and others have shown that Wnt/β-catenin signaling regulates expansion of SHF progenitors expressing the Isl1 transcription factor in the anterior portion of the developing heart (Ai et al., 2007; Cohen et al., 2007; Kwon et al., 2007; Lin et al., 2007; Qyang et al., 2007). Isl1 has also been shown to be a direct transcriptional target of Wnt/β-catenin signaling (Lin et al., 2007). Isl1 expression domain encompasses the majority of the mesoderm within the posterior pole of the heart including the DMP and Isl1-cre fate mapping has shown that most of the inflow tract of the heart is derived from Isl1+ progenitors (Cai et al., 2003; Galli et al., 2008; Snarr et al., 2007a; Sun et al., 2007). Moreover, the dorsal mesocardium and DMP region is the likely point of entry for Isl1+ SHF progenitors as they migrate into the posterior pole of the heart (Snarr et al., 2007a). However, the role for Wnt/β-catenin signaling in posterior SHF development is less clear. Isl1 gene expression was reduced in the posterior inflow tract region of Wnt2-/- mutants consistent with an effect on both Isl1 expression and number of Isl1+ SHF progenitors at E9.5 (Fig. 4G). Isl1 immunostaining confirmed that the number of Isl1+ SHF progenitors is significantly reduced in the dorsal mesocardium and DMP of Wnt2-/- mutants at E9.5 and E10.5 (Fig. 4L-N). Moreover, the posterior dorsal mesocardium of the heart including the DMP is hypoplastic suggesting that Isl1+ posterior progenitors in addition to the adjacent mesoderm requires Wnt2 for proper proliferation and development.
To demonstrate that reduced Isl1+ SHF progenitor number correlated, in part, with a loss of β-catenin dependent Wnt signaling we generated Ctnnb1flox/flox:Nkx2.5cre/+ mutants to delete β-catenin throughout early FHF and SHF progenitors (Moses et al., 2001). Loss of β-catenin expression in early cardiac progenitors resulted in a significant decrease in Isl1+ progenitors in the posterior inflow tract mesoderm (Fig. 4O-Q). These data indicate that Wnt2 regulates the expansion of Isl1+ SHF progenitors in the posterior pole of the heart in a region overlapping with the DMP, which likely accounts for the defects in myocardial and AV cushion development in the inflow tract region of these mutants and leads to the CCAVC phenotype.
Transient pharmacological activation of Wnt signaling leads to rescue of cardiac defects in Wnt2 mutants
Based on our expression data as well as previous studies, Wnt2 is expressed early in cardiac mesoderm at approximately E7.5-E8.0 while expression later between E8.5-E10.5 is confined to the cardiac inflow tract including the dorsal mesocardium, DMP, and developing atria (Fig. 1, Supplemental Figure 1 and (Monkley et al., 1996; Watanabe et al., 2006)). Therefore, we wanted to determine the temporal requirement of Wnt2 action in cardiac inflow tract development by activating Wnt/β-catenin signaling in a temporally restricted manner. Since previous reports using cre activated stabilization of β-catenin protein shows that persistent Wnt/β-catenin activation during cardiac development leads to severe cardiac defects and early embryonic demise (Cohen et al., 2007; Kwon et al., 2007), we chose a pharmacological approach using LiCl, an inhibitor of Gsk-3β and strong activator of Wnt/β-catenin signaling (Chu et al., 2004; Cohen et al., 2007; Klein and Melton, 1996; Nakamura et al., 2003). To determine when Wnt2 was required for cardiac development, we injected LiCl once a day in pregnant females from heterozygous intercrosses during three stages of development: E6-E8, E8-E10, and E11-E13. Since approximately 85% of Wnt2 mutants die at birth, we first asked whether LiCl treatment would increase survival of Wnt2-/- mutants. Wnt2-/- mutants treated with LiCl from E8-E10 had a significant increase in survival from 15% to 72% (Fig. 5A and B). All E8-E10 LiCl rescued Wnt2-/- mutants survived to at least 3 months of age and were normal as assessed by histology and echocardiography (data not shown). The lung hypoplasia previously reported in these mice was also rescued (data not shown and (Goss et al., 2009). Histological analysis of E18.5 hearts from NaCl (control) and LiCl E8-E10 treated Wnt2-/- mutants revealed a restoration of AV canal and inflow tract morphology reversing the CCAVC phenotype (Fig. 5C-F). Wnt2-/- mutants treated with LiCl from E6-E8 also exhibited an increase in survival up to 44% (Supplemental Figure 5). LiCl treatment between E6-E8 did not rescue the CCAVC phenotype but did rescue the thinned ventricular myocardium observed in many Wnt2-/- mutants (Fig. 5H). This correlates with previous studies showing low level expression of Wnt2 in the early cardiac crescent which suggests that Wnt2 acts at this earlier stage to promote general myocardial growth and loss of Wnt2 expression at this earlier stage may lead to the ventricular thinning we observe in some Wnt2-/- mutants in late development (Fig. 1P and (Monkley et al., 1996). LiCl also increased myocardial mass in wild-type embryos although this resolved itself after birth (Fig. 5E and data not shown). Echocardiograms at E16.5 and in adults showed normal AV canal function in E8-E10 LiCl treated Wnt2-/- mutants (Fig. 5I and J and data not shown). Wnt2-/- mutants treated with LiCl between E11-E13 did not show any significant increase in survival nor rescue of the CCAVC phenotype suggesting a lack of important Wnt2 activity after E11 (Supplemental Figure 5 and data not shown). Together, these data show that the E8-E10 LiCl rescued Wnt2-/- mutants are phenotypically indistinguishable from wild-type littermates and indicate that Wnt2 function is restricted to cardiac development before E11.
To determine whether the loss in inflow tract myocardial proliferation observed in Wnt2-/- mutants was rescued with LiCl activation of Wnt signaling, we performed MF20 and Ki67 immunostaining. These data show that DMP and atrial myocardial proliferation was largely rescued in E8-E10 LiCl treated Wnt2-/- mutants compared to controls at E11.5 (Fig. 5K-W). Proliferation in the AV canal, which is reduced in Wnt2-/- mutants, is also rescued by E8-E10 LiCl treatment (Fig. 5W). Together, these data reveal a significant reversal of the Wnt2-/- mutant CCAVC phenotype and perinatal lethality in a time dependent fashion by the Gsk-3β inhibitor LiCl. Moreover, these data indicate that Wnt2 acts primarily between E8-E10 of development to regulate cardiac inflow tract development.
Wnt2 regulates a transcriptional pathway important for cardiac progenitor development
To characterize the underlying molecular pathway that integrates Wnt2 signaling during cardiac inflow tract development, we performed microarray analysis on wild-type and Wnt2-/- mutant hearts at E12.5. These studies revealed a set of important transcription factors down-regulated in Wnt2-/- mutant hearts including Gata6, Sall1, and Sall3 (Fig. 6A). Decreased expression of Gata6, Sall1, and Sall3 was confirmed by Q-PCR (Fig. 6B). Moreover, expression of Gata6 in the posterior pole of the heart was decreased in Wnt2-/- mutants as assessed by in situ hybridization (Fig. 6C). Gata4/6 are important modulators of cardiac myocardial differentiation and proliferation (Rojas et al., 2008; Zeisberg et al., 2005). Sall genes are known to regulate important aspects of heart, kidney, and neural development as well as play important roles in stem/progenitor cells including embryonic stem cells (Harvey and Logan, 2006; Kohlhase et al., 2003; Koshiba-Takeuchi et al., 2006; Sweetman and Munsterberg, 2006; Warren et al., 2007).
Figure 6. Gata6 is a direct target of Wnt2 signaling in posterior cardiac mesoderm.
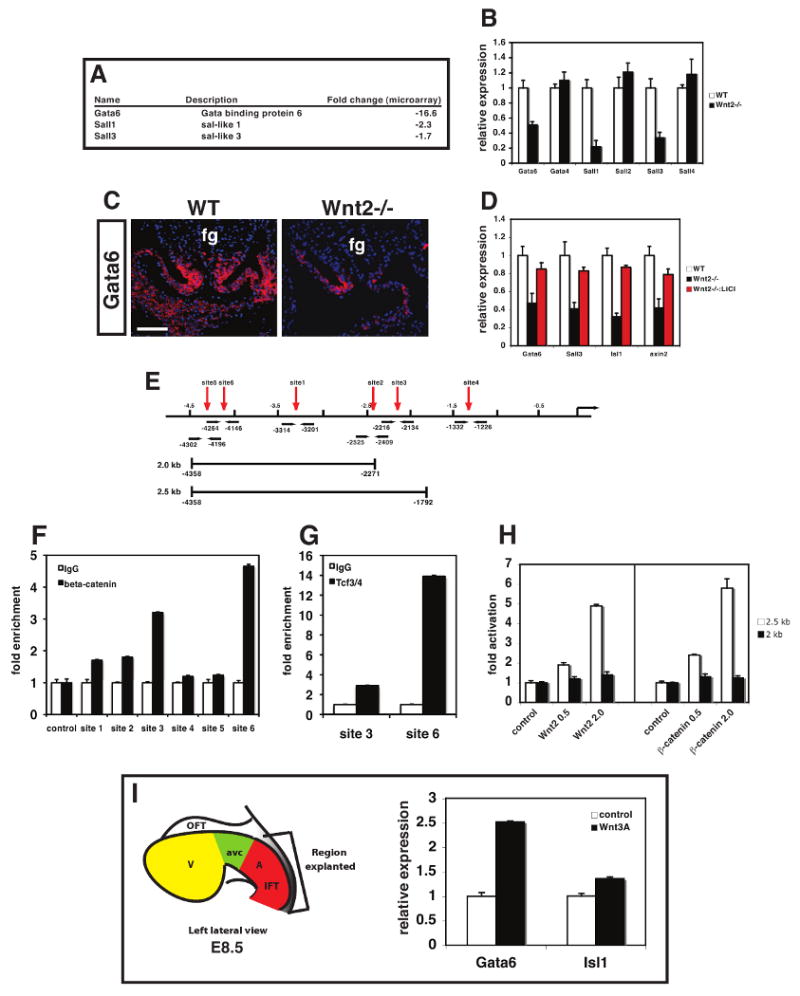
Microarray analysis revealed decreased cardiac expression of several transcriptional regulators including Gata6, Sall1, and Sall3 in Wnt2-/- mutants (A). Expression of these factors and related family members was assessed in wild-type and Wnt2-/- mutants at E10.5 by Q-PCR (B). Gata6 expression is decreased in the posterior pole and inflow tract of Wnt2-/- mutants by in situ hybridization (C). LiCl treatment between E8-E10 rescues expression of Gata6, Sall3, Isl1, and axin2 in Wnt2-/- mutants (D). Schematic of mouse Gata6 upstream regulatory region showing position of Lef/Tcf DNA binding sites (red arrows), primers used in ChIP experiments (black arrows), and regions used in luciferase reporter assays (E). ChIP assays using primers indicated in E and in Supplemental Table 1 showing enriched binding of β-catenin (F) and Tcf3/4 (G) to sites 3 and 6. Luciferase reporter assays using the 2 and 2.5 kb regions (E) and activated β-catenin or Wnt2 expression plasmids showing that the 2.5 kb region containing site 3 is trans-activated by active Wnt signaling (H). Diagram of explanted region of posterior pole of the heart at E8.5 that was cultured in the presence or absence of Wnt3a (200 ng/ml) showing that Wnt3a activates Gata6 and Isl1 expression (I). fg=foregut. Scale bars: C=50 μm.
We next tested whether LiCl treatment would rescue the decreased expression of these Wnt2 regulated factors. Q-PCR of E10.5 hearts from Wnt2+/- and Wnt2-/- mutants treated with LiCl from E8-E10 showed that LiCl rescued expression of Gata6, Sall3, and Isl1 in Wnt2-/- mutants (Fig. 6D). Given the critical role of Gata factors in cardiac development and progenitor specification (Zhao et al., 2008), we further characterized the regulation of Gata6 by Wnt2. Previous studies have identified an upstream regulatory region of the mouse Gata6 gene that can direct expression of lacZ to certain regions of the heart including the atria (Molkentin et al., 2000). We scanned the sequence of this enhancer as well as regions immediately flanking it and found six potential Lef/Tcf DNA binding sites (Fig. 6E). We performed chromatin immunoprecipitation (ChIP) analysis using both β-catenin and Tcf3/4 antibodies to assess association of these Wnt transcriptional effectors with the Lef/Tcf sites in vivo using chromatin from E9.5 hearts. These studies revealed that β-catenin and Tcf3/4 were associated with sites 3 and site 6 (Fig. 6F and G). Luciferase reporters containing different portions of this enhancer region revealed that of these two sites, site 3 was required for trans-activation by activated β-catenin and Wnt2 (Fig. 6H). To determine whether exogenous Wnt can activate Gata6 expression in the cardiac inflow tract, posterior pole cardiac explants were cultured in the presence or absence of Wnt3a. These data show that exogenous Wnt3a increases expression of both Gata6 and Isl1, a known target of Wnt signaling, in posterior cardiac explants (Fig. 6I). Together, these data indicate that Gata6 is a direct target of Wnt2 during cardiac development.
Wnt2 and Gata6 interact genetically to regulate inflow tract development in the heart
Although a role for Gata6 in cardiac neural crest dependent outflow tract development has been reported (Lepore et al., 2006), less is understood about its role in early cardiac development. Therefore, we inactivated Gata6 in early cardiac progenitors using the Nkx2.5cre/+ line (Moses et al., 2001). Approximately 50% of Nkx2.5cre/+:Gata6flox/flox mutants have distinct defects in AV canal development leading to a phenotype similar to that observed in Wnt2-/- mutants including CCAVC (Fig. 7A and B). The affected Nkx2.5cre/+:Gata6flox/flox mutants have more severe ventricular septal defects than Wnt2-/- mutants (Fig. 7B), demonstrating that Gata6 plays an important role in early myocardial development as has been reported (Zhao et al., 2008). To determine whether Wnt2 and Gata6 genetically interacted, we generated Wnt2/Gata6 compound mutants. The results from these studies revealed that Wnt2/Gata6 double heterozygous (Wnt2+/-:Gata6+/-) mutants are underrepresented at weaning (Fig. 7C). Histological analysis of Wnt2+/-:Gata6+/- animals shows defects similar to but less severe than Wnt2-/- null mutants including a thin atrial myocardial wall and decreased AV canal development including an atrial septal defect (Fig. 7D-F). To determine whether Wnt2 and Gata6 cooperated to regulate the same molecular pathway as Wnt2 alone, expression of genes found to be down-regulated by loss of Wnt2 were assessed in Wnt2+/-:Gata6+/- mutants. These data show that the transcriptional pathway downstream of Wnt2 is cooperatively decreased in Wnt2+/-:Gata6+/- mutants (Fig. 7G). The loss of atrial and AV canal myocardial development in Wnt2+/-:Gata6+/- mutants is illustrated by in situ hybridization with a Mlc2a probe (Fig. 7H and I). Moreover, atrial myocardial proliferation was decreased in Wnt2+/-:Gata6+/- mutants in comparison to Wnt2 and Gata6 single heterozygous mutants (Fig. 7J and K). Together with previous data showing that Wnt2 is a direct target of Gata6 (Alexandrovich et al., 2006), these data support a Wnt2-Gata6 feed-forward mechanism required for development of the cardiac inflow tract region including the DMP (Fig. 7L).
Figure 7. Wnt2 and Gata6 act in an epistatic fashion to regulate atrial and AV canal development.

Nkx2.5cre/+:Gata6flox/flox mutants exhibit defects in AV canal development and atrial septation at E17.5 (A and B, arrow). Wnt2+/-:Gata6+/- animals are underrepresented at weaning (C). Wnt2+/-:Gata6+/- mutants exhibit defects in AV canal and atrial myocardial development at E12.5 (D-F, arrows). Expression of important cardiac progenitor and myocardial genes is decreased in Wnt2+/-:Gata6+/- mutants by Q-PCR (G). Decreased atrial myocardial development in Wnt2+/-:Gata6+/- mutants as shown by Mlc2a in situ hybridization at E12.5 (H and I). Atrial myocardial proliferation is decreased in Wnt2+/-:Gata6+/- mutants as shown by MF20 and Ki67 immunostaining (J, arrows) and quantitation (K) at E12.5. Model of Wnt2/Gata6 feed-forward mechanism to promote posterior cardiac development through regulation of DMP and Isl1+ SHF progenitor proliferation and differentiation (L). Scale bars: A and B=300 μm, D-F, H and I=200 μm, J=25 μm.
Discussion
Development of the posterior pole of the heart including the cardiac inflow tract is poorly understood in comparison to the anterior or outflow region of the heart. In this report, we show that Wnt2, a ligand whose expression is restricted to the posterior pole of the developing heart including the cardiac inflow tract, is essential for expansion of cardiac mesoderm progenitors in this region and subsequent myocardial proliferation and differentiation. These defects in Wnt2-/- mutants results in a phenotype similar to that found in CCAVC, a congenital cardiac syndrome in humans. Importantly, these defects can be rescued in vivo through pharmacological activation of Wnt signaling. We also show that Wnt2 regulates cardiac inflow tract development, in part, through a feed-forward activation of Gata6. Together, these data identify a novel mechanism involving Wnt and Gata factors in regulating the development of posterior cardiac mesoderm and the cardiac inflow tract.
Little is understood about the development of cardiac mesoderm at the posterior pole of the developing vertebrate heart. Isl1-cre and cardiac explant studies using DiI labeling have shown that posterior Isl1+ mesoderm can contribute to the developing atria of the heart (Cai et al., 2003; Galli et al., 2008). This region encompasses the DMP which is thought to be an important contributor to the development of multiple structures within the cardiac inflow tract including atria myocardium, primary atrial septum, and AV canal cushions and myocardium (Snarr et al., 2007a; Snarr et al., 2007b). However, little is known about the molecular pathways that regulate the formation and development of the cardiac inflow tract. In contrast, multiple studies have defined specific spatially restricted roles for important transcription factors and signaling pathways in anterior pole development (Ai et al., 2006; Chen et al., 2007; Hamblet et al., 2002; High et al., 2007; Wang et al., 2004). Our data show that posterior cardiac mesoderm progenitors are regulated by Wnt2 and play an important role in development of the inflow tract including the atria, AV canal, and pulmonary veins. The clinical importance of the structures defined by the posterior cardiac mesoderm is well known and Wnt2-/- mutants phenotypically resemble humans with CCAVC, an important congenital heart anomaly. The finding that Wnt signaling plays a key role in expansion and differentiation of SHF progenitors both in the posterior pole (reported here) and in the anterior pole (Ai et al., 2007; Cohen et al., 2007; Kwon et al., 2007; Lin et al., 2007; Qyang et al., 2007) should spur interest in assessing whether congenital cardiac syndromes that share defects in both posterior and anterior related tissues are due to a common deficiency in this pathway.
We show that loss of Wnt2 can be rescued using LiCl, a known inhibitor of Gsk-3β (Klein and Melton, 1996). Although LiCl has been well studied as an inhibitor of Gsk-3β, it has also been shown to affect other signaling pathways including Akt and IP3 kinase signaling (Nemoto et al., 2008; Raffa and Martinez, 1992). The majority of known inhibitors of Wnt signaling impinge upon Gsk-3β activity and their specificity is largely unknown. Our data is the first to our knowledge of an example of LiCl being used to rescue a specific loss of Wnt signaling in mammalian development. These data strongly support the contention that, despite the ability of LiCl to affect multiple pathways, it can be used to inhibit Gsk-3β during development and activate Wnt signaling. Moreover, our data suggest that defects in tissue homoeostasis and development related to deficiencies in Wnt signaling could be positively impacted by treatment with small molecule modulators of Wnt.
The role for Wnt signaling in cardiac development has been studied primarily using loss and gain of function mutants in the critical Wnt pathway component β-catenin. However, β-catenin plays an important role in cell-cell adhesion as well as Wnt signaling thus it has been unclear whether the defects in cardiac loss of gain of function can be wholly attributed to Wnt signaling. Previous reports have described antagonistic affects of Wnt/β-catenin signaling in cardiogenesis. Most of these studies have utilized gain of function analysis in Xenopus and in pluripotent cell lines such as embryonic stem cells (Foley and Mercola, 2005; Liu et al., 2007; Marvin et al., 2001; Pandur et al., 2002; Schneider and Mercola, 2001; Terami et al., 2004; Ueno et al., 2007). Our data show that Wnt signaling plays a positive role in cooperation with Gata factors in promoting spatially and temporally specific cardiogenesis. The difference between our data and previous reports can most easily be attributed to a potent role for Wnt signaling in the SHF in a temporally restricted manner. Gain of function studies result in blocking cardiac mesoderm in a proliferative and undifferentiated state resulting in a loss of cardiac marker gene expression. A recent report suggested that Wnt signaling inhibited cardiogenesis by inhibiting Gata factor expression (Afouda et al., 2008). Our data show that Gata6 is a direct target of Wnt signaling, and along with previous data showing that Wnt2 is, in turn, a target of Gata6 in cardiac differentiation of P19 cells (Alexandrovich et al., 2006), suggests that Wnt and Gata factors act in a feed-forward mechanism to drive posterior cardiac progenitor expansion and differentiation and subsequent cardiac inflow tract development.
Although our data focus primarily on the SHF, Wnt2 is expressed at an earlier stage in the cardiac crescent mesoderm (Monkley et al., 1996). Expression of several Wnt ligands has been reported at this earlier stage including Wnt3a, Wnt8a, and Wnt11 (Eisenberg and Eisenberg, 1999; Jaspard et al., 2000; Nakamura et al., 2003). Conditional inactivation of β-catenin throughout the early cardiac mesoderm using the Mesp1-cre line results in loss of SHF markers Isl1 and Bmp4 (Klaus et al., 2007). In these studies, expression of the FHF markers Tbx5 and Hand1 were not affected by early loss of β-catenin in the precardiac mesoderm. These reports are consistent with our findings that the majority of defects observed in Wnt2 deficient embryos can be attributed to the affect of Wnt2 on SHF progenitors. Moreover, we also show that activation of Wnt signaling from E8-E10, when SHF progenitors are contributing to a large portion of the developing myocardium, is sufficient to rescue the majority of defects in these mutants and the lethal consequences.
We have shown that Wnt2 is required for proper development of the cardiac inflow tract mesoderm and in particular the myocardial component of the atria, AV canal and pulmonary veins resulting in a CCAVC phenotype. Moreover, we have shown that defects in Wnt signaling can be rescued using temporal specific application of pharmacological activators of this pathway in vivo during development. Given the emerging importance of Wnt signaling in cardiac progenitor development, our data indicate that this pathway may be a key integrator of multiple transcriptional and signaling networks required for proper development of the posterior pole of the heart including the cardiac inflow tract.
Materials and Methods
Animals
Generation and genotyping of the Wnt2, Gata6flox/flox, Ctnnb1flox/flox, Nkx2.5cre/+, and TOPGAL mouse lines has been reported (Brault et al., 2001; DasGupta and Fuchs, 1999; Goss et al., 2009; Lepore et al., 2006; Moses et al., 2001). Mice were kept on a mixed C57BL/6:129SVJ background. For the LiCl rescue studies, pregnant mice were injected interperitoneally with 200 mg/Kg once a day at the indicated times. All animal procedures were performed in accordance with the Institute for Animal Care and Use Committee at the University of Pennsylvania
Histology
Embryos and tissues were fixed in 4% paraformaldehyde for 24 hours, dehydrated in increasing concentrations of ethanol, and embedded in paraffin for tissue sectioning. In situ hybridization was performed as described (Morrisey et al., 1997a; Morrisey et al., 1997b). Immunohistochemistry was performed using the following antibodies: mouse anti-rat Isl-1 (University of Iowa Hybridoma Bank), rat anti-Ki67 (clone TEC-3, Dako), β-catenin (BD Biosciences, 1:50), and MF20 (University of Iowa Hybridoma Bank). For scanning electron microscopy, hearts from E12.5 embryos were fixed in 2% gluteraldehyde and processed as described (Shu et al., 2002). LacZ histochemical staining of embryos was performed as described (Shu et al., 2002).
EMT and posterior cardiac pole explant assays
AV canal regions of E9.5 hearts were dissected from the indicated genotypes and cultured for 48 hours on collagen/fibronectin-coated tissue culture plate as described (Wang et al., 2005). Photographs were taken using phase contrast microscopy. The posterior cardiac pole was dissected at E8.5 and cultured on collagen coated tissue culture plates for 48 hours as previously described (Cohen et al., 2007). Wnt3a (R&D Systems) was added at a concentration of 200 ng/ml.
Gene expression analysis
Total RNA was isolated from hearts at the indcated ages using Trizol reagent, reverse transcribed using SuperScript First Strand Synthesis System (Invitrogen), and used in quantitative real time PCR analysis using the oligonucleotides listed in Supplemental Table 1. Microarray analysis was performed with a Mouse Genome 430 2.0 array (Affymetrix, Santa Clara, CA). Total RNA isolated from E12.5 hearts from wild-type and Wnt2-/- null mutant littermates using three samples of each genotype. Data was analyzed using Microarray Suite 5.0 (MASS, Affymetrix) and Significance Analysis of Microarray (SAM). Genes with 1.5-fold and greater changes than the experimental mean and carrying out ANOVA with a p value less than 0.01 were considered significant.
Cell transfection and chromatin immunoprecipitation (ChIP) assays
The upstream enhancer region of mouse Gata6 has been described preciously (Molkentin et al., 2000). The region between -4kb and -1.5 kb was subcloned into the pGL3 promoter luciferase reporter vector to generate pGL3Gata6.luc vector. This plasmid was transfected with an active form of β-catenin (β-catenin41A/45A) into HEK293 cells using Fugene 6 and luciferase activities were determined using a commercially available kit (Promega). For ChIP assays, chromatin was extracted from 25 wild-type embryonic hearts at E9.5 using a ChIP assay kit (Upstate Biotechnology). Chromatin was immunoprecipated with either the β-catenin mAb (BD Biosciences), a Tcf3/4 antibody (Abcam ab12065), or a nonimmune control antibody mouse IgG (Sigma). Purified chromatin was subjected to PCR using the oligonucleotides listed in Supplemental Table 1.
Echocardiography
Pregnant mice were lightly anesthetized with 1-2% isoflurane and embryonic echocardiography was performed using a high-resolution Vevo 770 micro-ultrasound system (VisualSonics Inc.) equipped with a RMV-704 transducer with a center frequency of 40 MHz. For adult mouse echocardiography, a RMV-707 transducer with a center frequency of 30 MHz was used. Cardiac four-chamber view of the embryo was acquired by rotating the transducer about 45 degree counterclockwise from the longitudinal axis of the embryo body. End-diastolic and end-systolic left ventricular internal diameters (LVIDd, LVIDs) were measured from the left ventricular short axis view with 2D orientated M-mode imaging. Left ventricular systolic function was estimated by fractional shortening (FS, %), which was derived using the formula: (LVIDd-LVIDs) / LVIDd × 100, and ejection fraction (EF, %), which was calculated using the end-systolic and end-diastolic volumes as described (Stypmann et al., 2009). Doppler echocardiography was performed to detect flow velocities.
Supplementary Material
Supplemental Figure 1. Expression of Wnt2 at E10.5 and defects in atrial septal development and overall inflow tract myocardialization in Wnt2-/-mutants. Expression of Wnt2 is observed in the region where the primary atrial septum will form at E10.5 (A, boxed region). Arrow denotes lack of Wnt2 expression in the overlying endocardium (EC). H+E stained histology sections through the same region of wild-type (B and Wnt2-/- mutants (C) at E10.5 showing the lack of proper primary atrial septum development (arrows). H+E stained histology sections at E12.5 show the reduced development of the mesenchymal cap which overlies the poorly developed primary atrial septum of Wnt2-/- mutants (D and E). In situ hybridization using a probe to myosin light chain 2a (Mlc2a) shows overall reduced myocardial development in both the atria and AV canal region of Wnt2-/- mutants (F and G, arrows). Scale bars: A-C=50 μm, D and E=100 μm, F and G=150 μm.
Supplemental Figure 2. Expression of markers of epicardium and myocardium in Wnt2-/-mutants. Wnt2 expression is not observed in epicardium of E9.5 or E12.5 hearts (A and B). WT1 expression is observed in the epicardium of both wild-type and Wnt2-/- mutants at E10.5 (C-F, arrows). Tbx18 expression is observed in the septum transversum and epicardium of wild-type and Wnt2-/- mutants at E9.5 (H-K). Bmp2 expression is observed at normal levels in the atrial (L and M) and AV canal myocardium of wild-type and Wnt2-/- mutants at E9.5 (N and O, arrows). Tbx5 expression is observed at normal levels in the myocardium of wild-type and Wnt2-/- mutants at E9.5 (P and Q). Scale bars: A-F=50 μm, H-Q=100 μm.
Supplemental Figure 3. No outflow tract defects and normal expression of Isl1 in the anterior region of Wnt2-/-mutants. Histological sections of wild-type (A-C) and Wnt2-/- (D-F) mutants were stained with H+E. Sections from the anterior to posterior regions of the heart show normal septation of the aorta (ao) and pulmonary artery (pa). Isl1 immunostaining shows normal levels of Isl1 expression in the anterior region including the outflow tract region of Wnt2-/- mutants (G and H). Scale bar: A-F=250 μm, G and H=150 μm.
Supplemental Figure 4. Normal ventricular myocardial proliferation at E10.5 in Wnt2-/- mutants. Double immunofluorescent staining using MF20 and Ki67 antibodies shows normal levels of myocardial proliferation at E10.5 in the ventricles of Wnt2-/- mutants compared to wild-type littermates. Scale bar=25 μm.
Supplemental Figure 5. Survival of LiCl treated Wnt2-/-mutants at E6-8 and E11-13. Survival numbers and percentages of Wnt2-/- mutants treated with LiCl in vivo.
Acknowledgments
The authors would like to thank members of the Histology Core in the Penn Cardiovascular Institute for their exemplary work in these studies. Research in the Morrisey laboratory is supported by the National Institutes for Health (R01-HL064632, R01-HL087825, and U01-HL100405), an American Heart Association Jon Holden DeHaan Foundation Myogenesis Center Grant, and the Penn Institute for Regenerative Medicine. Y.T. was supported by a Postdoctoral Fellowship Grant from the American Heart Association.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Afouda BA, Martin J, Liu F, Ciau-Uitz A, Patient R, Hoppler S. GATA transcription factors integrate Wnt signalling during heart development. Development. 2008;135:3185–3190. doi: 10.1242/dev.026443. [DOI] [PubMed] [Google Scholar]
- Ai D, Fu X, Wang J, Lu MF, Chen L, Baldini A, Klein WH, Martin JF. Canonical Wnt signaling functions in second heart field to promote right ventricular growth. Proc Natl Acad Sci U S A. 2007;104:9319–9324. doi: 10.1073/pnas.0701212104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ai D, Liu W, Ma L, Dong F, Lu MF, Wang D, Verzi MP, Cai C, Gage PJ, Evans S, et al. Pitx2 regulates cardiac left-right asymmetry by patterning second cardiac lineage-derived myocardium. Dev Biol. 2006;296:437–449. doi: 10.1016/j.ydbio.2006.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrovich A, Arno M, Patient RK, Shah AM, Pizzey JA, Brewer AC. Wnt2 is a direct downstream target of GATA6 during early cardiogenesis. Mech Dev. 2006;123:297–311. doi: 10.1016/j.mod.2006.02.002. [DOI] [PubMed] [Google Scholar]
- Allwork SP. Anatomical-embryological correlates in atrioventricular septal defect. Br Heart J. 1982;47:419–429. doi: 10.1136/hrt.47.5.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson RH, Brown NA, Moorman AF. Development and structures of the venous pole of the heart. Dev Dyn. 2006;235:2–9. doi: 10.1002/dvdy.20578. [DOI] [PubMed] [Google Scholar]
- Brault V, Moore R, Kutsch S, Ishibashi M, Rowitch DH, McMahon AP, Sommer L, Boussadia O, Kemler R. Inactivation of the beta-catenin gene by Wnt1-Cre-mediated deletion results in dramatic brain malformation and failure of craniofacial development. Development. 2001;128:1253–1264. doi: 10.1242/dev.128.8.1253. [DOI] [PubMed] [Google Scholar]
- Cai CL, Liang X, Shi Y, Chu PH, Pfaff SL, Chen J, Evans S. Isl1 identifies a cardiac progenitor population that proliferates prior to differentiation and contributes a majority of cells to the heart. Dev Cell. 2003;5:877–889. doi: 10.1016/s1534-5807(03)00363-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YH, Ishii M, Sun J, Sucov HM, Maxson RE., Jr Msx1 and Msx2 regulate survival of secondary heart field precursors and post-migratory proliferation of cardiac neural crest in the outflow tract. Dev Biol. 2007;308:421–437. doi: 10.1016/j.ydbio.2007.05.037. [DOI] [PubMed] [Google Scholar]
- Christoffels VM, Mommersteeg MT, Trowe MO, Prall OW, de Gier-de Vries C, Soufan AT, Bussen M, Schuster-Gossler K, Harvey RP, Moorman AF, et al. Formation of the venous pole of the heart from an Nkx2-5-negative precursor population requires Tbx18. Circ Res. 2006;98:1555–1563. doi: 10.1161/01.RES.0000227571.84189.65. [DOI] [PubMed] [Google Scholar]
- Chu EY, Hens J, Andl T, Kairo A, Yamaguchi TP, Brisken C, Glick A, Wysolmerski JJ, Millar SE. Canonical WNT signaling promotes mammary placode development and is essential for initiation of mammary gland morphogenesis. Development. 2004;131:4819–4829. doi: 10.1242/dev.01347. [DOI] [PubMed] [Google Scholar]
- Cohen ED, Wang Z, Lepore JJ, Lu MM, Taketo MM, Epstein DJ, Morrisey EE. Wnt/beta-catenin signaling promotes expansion of Isl-1-positive cardiac progenitor cells through regulation of FGF signaling. J Clin Invest. 2007;117:1794–1804. doi: 10.1172/JCI31731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DasGupta R, Fuchs E. Multiple roles for activated LEF/TCF transcription complexes during hair follicle development and differentiation. Development. 1999;126:4557–4568. doi: 10.1242/dev.126.20.4557. [DOI] [PubMed] [Google Scholar]
- Eisenberg CA, Eisenberg LM. WNT11 promotes cardiac tissue formation of early mesoderm. Dev Dyn. 1999;216:45–58. doi: 10.1002/(SICI)1097-0177(199909)216:1<45::AID-DVDY7>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Foley AC, Mercola M. Heart induction by Wnt antagonists depends on the homeodomain transcription factor Hex. Genes Dev. 2005;19:387–396. doi: 10.1101/gad.1279405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galli D, Dominguez JN, Zaffran S, Munk A, Brown NA, Buckingham ME. Atrial myocardium derives from the posterior region of the second heart field, which acquires left-right identity as Pitx2c is expressed. Development. 2008;135:1157–1167. doi: 10.1242/dev.014563. [DOI] [PubMed] [Google Scholar]
- Gitler AD, Lu MM, Jiang YQ, Epstein JA, Gruber PJ. Molecular markers of cardiac endocardial cushion development. Dev Dyn. 2003;228:643–650. doi: 10.1002/dvdy.10418. [DOI] [PubMed] [Google Scholar]
- Goss AM, Tian Y, Tsukiyama T, Cohen ED, Zhou D, Lu MM, Yamaguchi TP, Morrisey EE. Wnt2/2b and β-catenin signaling are necessary and sufficient to specify lung progenitors in the foregut. Developmental Cell. 2009 doi: 10.1016/j.devcel.2009.06.005. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamblet NS, Lijam N, Ruiz-Lozano P, Wang J, Yang Y, Luo Z, Mei L, Chien KR, Sussman DJ, Wynshaw-Boris A. Dishevelled 2 is essential for cardiac outflow tract development, somite segmentation and neural tube closure. Development. 2002;129:5827–5838. doi: 10.1242/dev.00164. [DOI] [PubMed] [Google Scholar]
- Harvey SA, Logan MP. sall4 acts downstream of tbx5 and is required for pectoral fin outgrowth. Development. 2006;133:1165–1173. doi: 10.1242/dev.02259. [DOI] [PubMed] [Google Scholar]
- High FA, Zhang M, Proweller A, Tu L, Parmacek MS, Pear WS, Epstein JA. An essential role for Notch in neural crest during cardiovascular development and smooth muscle differentiation. J Clin Invest. 2007;117:353–363. doi: 10.1172/JCI30070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaspard B, Couffinhal T, Dufourcq P, Moreau C, Duplaa C. Expression pattern of mouse sFRP-1 and mWnt-8 gene during heart morphogenesis. Mech Dev. 2000;90:263–267. doi: 10.1016/s0925-4773(99)00236-1. [DOI] [PubMed] [Google Scholar]
- Klaus A, Saga Y, Taketo MM, Tzahor E, Birchmeier W. Distinct roles of Wnt/beta-catenin and Bmp signaling during early cardiogenesis. Proc Natl Acad Sci U S A. 2007;104:18531–18536. doi: 10.1073/pnas.0703113104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein PS, Melton DA. A molecular mechanism for the effect of lithium on development. Proc Natl Acad Sci U S A. 1996;93:8455–8459. doi: 10.1073/pnas.93.16.8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlhase J, Schubert L, Liebers M, Rauch A, Becker K, Mohammed SN, Newbury-Ecob R, Reardon W. Mutations at the SALL4 locus on chromosome 20 result in a range of clinically overlapping phenotypes, including Okihiro syndrome, Holt-Oram syndrome, acro-renal-ocular syndrome, and patients previously reported to represent thalidomide embryopathy. J Med Genet. 2003;40:473–478. doi: 10.1136/jmg.40.7.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshiba-Takeuchi K, Takeuchi JK, Arruda EP, Kathiriya IS, Mo R, Hui CC, Srivastava D, Bruneau BG. Cooperative and antagonistic interactions between Sall4 and Tbx5 pattern the mouse limb and heart. Nat Genet. 2006;38:175–183. doi: 10.1038/ng1707. [DOI] [PubMed] [Google Scholar]
- Kwon C, Arnold J, Hsiao EC, Taketo MM, Conklin BR, Srivastava D. Canonical Wnt signaling is a positive regulator of mammalian cardiac progenitors. Proc Natl Acad Sci U S A. 2007;104:10894–10899. doi: 10.1073/pnas.0704044104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavine KJ, Yu K, White AC, Zhang X, Smith C, Partanen J, Ornitz DM. Endocardial and epicardial derived FGF signals regulate myocardial proliferation and differentiation in vivo. Dev Cell. 2005;8:85–95. doi: 10.1016/j.devcel.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Lepore JJ, Mericko PA, Cheng L, Lu MM, Morrisey EE, Parmacek MS. GATA-6 regulates semaphorin 3C and is required in cardiac neural crest for cardiovascular morphogenesis. J Clin Invest. 2006;116:929–939. doi: 10.1172/JCI27363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L, Cui L, Zhou W, Dufort D, Zhang X, Cai CL, Bu L, Yang L, Martin J, Kemler R, et al. Beta-catenin directly regulates Islet1 expression in cardiovascular progenitors and is required for multiple aspects of cardiogenesis. Proc Natl Acad Sci U S A. 2007;104:9313–9318. doi: 10.1073/pnas.0700923104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Asakura M, Inoue H, Nakamura T, Sano M, Niu Z, Chen M, Schwartz RJ, Schneider MD. Sox17 is essential for the specification of cardiac mesoderm in embryonic stem cells. Proc Natl Acad Sci U S A. 2007;104:3859–3864. doi: 10.1073/pnas.0609100104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marvin MJ, Di Rocco G, Gardiner A, Bush SM, Lassar AB. Inhibition of Wnt activity induces heart formation from posterior mesoderm. Genes Dev. 2001;15:316–327. doi: 10.1101/gad.855501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molkentin JD, Antos C, Mercer B, Taigen T, Miano JM, Olson EN. Direct activation of a GATA6 cardiac enhancer by Nkx2.5: evidence for a reinforcing regulatory network of Nkx2.5 and GATA transcription factors in the developing heart. Dev Biol. 2000;217:301–309. doi: 10.1006/dbio.1999.9544. [DOI] [PubMed] [Google Scholar]
- Monkley SJ, Delaney SJ, Pennisi DJ, Christiansen JH, Wainwright BJ. Targeted disruption of the Wnt2 gene results in placentation defects. Development. 1996;122:3343–3353. doi: 10.1242/dev.122.11.3343. [DOI] [PubMed] [Google Scholar]
- Morrisey EE, Ip HS, Tang Z, Lu MM, Parmacek MS. GATA-5: a transcriptional activator expressed in a novel temporally and spatially-restricted pattern during embryonic development. Dev Biol. 1997a;183:21–36. doi: 10.1006/dbio.1996.8485. [DOI] [PubMed] [Google Scholar]
- Morrisey EE, Ip HS, Tang Z, Parmacek MS. GATA-4 activates transcription via two novel domains that are conserved within the GATA-4/5/6 subfamily. J Biol Chem. 1997b;272:8515–8524. doi: 10.1074/jbc.272.13.8515. [DOI] [PubMed] [Google Scholar]
- Moses KA, DeMayo F, Braun RM, Reecy JL, Schwartz RJ. Embryonic expression of an Nkx2-5/Cre gene using ROSA26 reporter mice. Genesis. 2001;31:176–180. doi: 10.1002/gene.10022. [DOI] [PubMed] [Google Scholar]
- Nakajima Y, Yamagishi T, Hokari S, Nakamura H. Mechanisms involved in valvuloseptal endocardial cushion formation in early cardiogenesis: roles of transforming growth factor (TGF)-beta and bone morphogenetic protein (BMP) Anat Rec. 2000;258:119–127. doi: 10.1002/(SICI)1097-0185(20000201)258:2<119::AID-AR1>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Sano M, Songyang Z, Schneider MD. A Wnt- and beta -catenin-dependent pathway for mammalian cardiac myogenesis. Proc Natl Acad Sci U S A. 2003;100:5834–5839. doi: 10.1073/pnas.0935626100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemoto T, Kanai T, Yanagita T, Satoh S, Maruta T, Yoshikawa N, Kobayashi H, Wada A. Regulation of Akt mRNA and protein levels by glycogen synthase kinase-3beta in adrenal chromaffin cells: effects of LiCl and SB216763. Eur J Pharmacol. 2008;586:82–89. doi: 10.1016/j.ejphar.2008.02.075. [DOI] [PubMed] [Google Scholar]
- Pandur P, Lasche M, Eisenberg LM, Kuhl M. Wnt-11 activation of a non-canonical Wnt signalling pathway is required for cardiogenesis. Nature. 2002;418:636–641. doi: 10.1038/nature00921. [DOI] [PubMed] [Google Scholar]
- Qyang Y, Martin-Puig S, Chiravuri M, Chen S, Xu H, Bu L, Jiang X, Laugwitz KL, Moon RT, Gruber P, et al. The Renewal and Differentiation of Isl1+ Cardiovascular Progenitors Are Controlled by a Wnt/beta-catenin Pathway. Cell Stem Cell. 2007;1:165–179. doi: 10.1016/j.stem.2007.05.018. [DOI] [PubMed] [Google Scholar]
- Raffa RB, Martinez RP. Morphine antinociception is mediated through a LiCl-sensitive, IP3-restorable pathway. Eur J Pharmacol. 1992;215:357–358. doi: 10.1016/0014-2999(92)90060-h. [DOI] [PubMed] [Google Scholar]
- Rojas A, Kong SW, Agarwal P, Gilliss B, Pu WT, Black BL. GATA4 is a direct transcriptional activator of cyclin D2 and Cdk4 and is required for cardiomyocyte proliferation in anterior heart field-derived myocardium. Mol Cell Biol. 2008;28:5420–5431. doi: 10.1128/MCB.00717-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider VA, Mercola M. Wnt antagonism initiates cardiogenesis in Xenopus laevis. Genes Dev. 2001;15:304–315. doi: 10.1101/gad.855601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu W, Jiang YQ, Lu MM, Morrisey EE. Wnt7b regulates mesenchymal proliferation and vascular development in the lung. Development. 2002;129:4831–4842. doi: 10.1242/dev.129.20.4831. [DOI] [PubMed] [Google Scholar]
- Snarr BS, O'Neal JL, Chintalapudi MR, Wirrig EE, Phelps AL, Kubalak SW, Wessels A. Isl1 expression at the venous pole identifies a novel role for the second heart field in cardiac development. Circ Res. 2007a;101:971–974. doi: 10.1161/CIRCRESAHA.107.162206. [DOI] [PubMed] [Google Scholar]
- Snarr BS, Wirrig EE, Phelps AL, Trusk TC, Wessels A. A spatiotemporal evaluation of the contribution of the dorsal mesenchymal protrusion to cardiac development. Dev Dyn. 2007b;236:1287–1294. doi: 10.1002/dvdy.21074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss AW. The molecular basis of congenital cardiac disease. Semin Thorac Cardiovasc Surg Pediatr Card Surg Annu. 1998;1:179–188. doi: 10.1016/s1092-9126(98)70021-3. [DOI] [PubMed] [Google Scholar]
- Stypmann J, Engelen MA, Troatz C, Rothenburger M, Eckardt L, Tiemann K. Echocardiographic assessment of global left ventricular function in mice. Lab Anim. 2009;43:127–137. doi: 10.1258/la.2007.06001e. [DOI] [PubMed] [Google Scholar]
- Sun Y, Liang X, Najafi N, Cass M, Lin L, Cai CL, Chen J, Evans SM. Islet 1 is expressed in distinct cardiovascular lineages, including pacemaker and coronary vascular cells. Dev Biol. 2007;304:286–296. doi: 10.1016/j.ydbio.2006.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweetman D, Munsterberg A. The vertebrate spalt genes in development and disease. Dev Biol. 2006;293:285–293. doi: 10.1016/j.ydbio.2006.02.009. [DOI] [PubMed] [Google Scholar]
- Terami H, Hidaka K, Katsumata T, Iio A, Morisaki T. Wnt11 facilitates embryonic stem cell differentiation to Nkx2.5-positive cardiomyocytes. Biochem Biophys Res Commun. 2004;325:968–975. doi: 10.1016/j.bbrc.2004.10.103. [DOI] [PubMed] [Google Scholar]
- Ueno S, Weidinger G, Osugi T, Kohn AD, Golob JL, Pabon L, Reinecke H, Moon RT, Murry CE. Biphasic role for Wnt/beta-catenin signaling in cardiac specification in zebrafish and embryonic stem cells. Proc Natl Acad Sci U S A. 2007;104:9685–9690. doi: 10.1073/pnas.0702859104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Weidenfeld J, Lu MM, Maika S, Kuziel WA, Morrisey EE, Tucker PW. Foxp1 regulates cardiac outflow tract, endocardial cushion morphogenesis and myocyte proliferation and maturation. Development. 2004;131:4477–4487. doi: 10.1242/dev.01287. [DOI] [PubMed] [Google Scholar]
- Wang J, Sridurongrit S, Dudas M, Thomas P, Nagy A, Schneider MD, Epstein JA, Kaartinen V. Atrioventricular cushion transformation is mediated by ALK2 in the developing mouse heart. Dev Biol. 2005;286:299–310. doi: 10.1016/j.ydbio.2005.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren M, Wang W, Spiden S, Chen-Murchie D, Tannahill D, Steel KP, Bradley A. A Sall4 mutant mouse model useful for studying the role of Sall4 in early embryonic development and organogenesis. Genesis. 2007;45:51–58. doi: 10.1002/dvg.20264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y, Kokubo H, Miyagawa-Tomita S, Endo M, Igarashi K, Aisaki K, Kanno J, Saga Y. Activation of Notch1 signaling in cardiogenic mesoderm induces abnormal heart morphogenesis in mouse. Development. 2006;133:1625–1634. doi: 10.1242/dev.02344. [DOI] [PubMed] [Google Scholar]
- Wessels A, Anderson RH, Markwald RR, Webb S, Brown NA, Viragh S, Moorman AF, Lamers WH. Atrial development in the human heart: an immunohistochemical study with emphasis on the role of mesenchymal tissues. Anat Rec. 2000;259:288–300. doi: 10.1002/1097-0185(20000701)259:3<288::AID-AR60>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Zeisberg EM, Ma Q, Juraszek AL, Moses K, Schwartz RJ, Izumo S, Pu WT. Morphogenesis of the right ventricle requires myocardial expression of Gata4. J Clin Invest. 2005;115:1522–1531. doi: 10.1172/JCI23769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Goss AM, Cohen ED, Kadzik R, Lepore JJ, Muthukumaraswamy K, Yang J, DeMayo FJ, Whitsett JA, Parmacek MS, et al. A Gata6-Wnt pathway required for epithelial stem cell development and airway regeneration. Nat Genet. 2008;40:862–870. doi: 10.1038/ng.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao R, Watt AJ, Battle MA, Li J, Bondow BJ, Duncan SA. Loss of both GATA4 and GATA6 blocks cardiac myocyte differentiation and results in acardia in mice. Dev Biol. 2008;317:614–619. doi: 10.1016/j.ydbio.2008.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Expression of Wnt2 at E10.5 and defects in atrial septal development and overall inflow tract myocardialization in Wnt2-/-mutants. Expression of Wnt2 is observed in the region where the primary atrial septum will form at E10.5 (A, boxed region). Arrow denotes lack of Wnt2 expression in the overlying endocardium (EC). H+E stained histology sections through the same region of wild-type (B and Wnt2-/- mutants (C) at E10.5 showing the lack of proper primary atrial septum development (arrows). H+E stained histology sections at E12.5 show the reduced development of the mesenchymal cap which overlies the poorly developed primary atrial septum of Wnt2-/- mutants (D and E). In situ hybridization using a probe to myosin light chain 2a (Mlc2a) shows overall reduced myocardial development in both the atria and AV canal region of Wnt2-/- mutants (F and G, arrows). Scale bars: A-C=50 μm, D and E=100 μm, F and G=150 μm.
Supplemental Figure 2. Expression of markers of epicardium and myocardium in Wnt2-/-mutants. Wnt2 expression is not observed in epicardium of E9.5 or E12.5 hearts (A and B). WT1 expression is observed in the epicardium of both wild-type and Wnt2-/- mutants at E10.5 (C-F, arrows). Tbx18 expression is observed in the septum transversum and epicardium of wild-type and Wnt2-/- mutants at E9.5 (H-K). Bmp2 expression is observed at normal levels in the atrial (L and M) and AV canal myocardium of wild-type and Wnt2-/- mutants at E9.5 (N and O, arrows). Tbx5 expression is observed at normal levels in the myocardium of wild-type and Wnt2-/- mutants at E9.5 (P and Q). Scale bars: A-F=50 μm, H-Q=100 μm.
Supplemental Figure 3. No outflow tract defects and normal expression of Isl1 in the anterior region of Wnt2-/-mutants. Histological sections of wild-type (A-C) and Wnt2-/- (D-F) mutants were stained with H+E. Sections from the anterior to posterior regions of the heart show normal septation of the aorta (ao) and pulmonary artery (pa). Isl1 immunostaining shows normal levels of Isl1 expression in the anterior region including the outflow tract region of Wnt2-/- mutants (G and H). Scale bar: A-F=250 μm, G and H=150 μm.
Supplemental Figure 4. Normal ventricular myocardial proliferation at E10.5 in Wnt2-/- mutants. Double immunofluorescent staining using MF20 and Ki67 antibodies shows normal levels of myocardial proliferation at E10.5 in the ventricles of Wnt2-/- mutants compared to wild-type littermates. Scale bar=25 μm.
Supplemental Figure 5. Survival of LiCl treated Wnt2-/-mutants at E6-8 and E11-13. Survival numbers and percentages of Wnt2-/- mutants treated with LiCl in vivo.