

Abstract
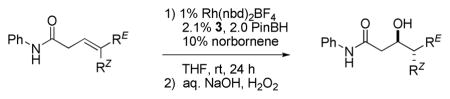
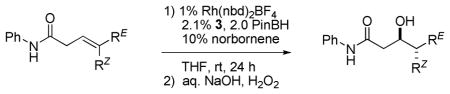
Rhodium-catalyzed hydroborations of trisubstituted alkenes are generally slow and often suffer from competing alkene isomerization. In contrast, the trisubstituted alkene moieties contained within the framework of a β,γ-unsaturated amide undergo facile reaction, perhaps facilitated by carbonyl directing effects and two-point binding of the substrate to the rhodium catalyst. Stereoisomeric substrates, for example, (E)- and (Z)-3, cleanly give rise to diastereomeric products and thus the rhodium-catalyzed reaction is stereospecific. In addition, simple TADDOL-derived phenyl monophosphite ligands in combination with Rh(nbd)2BF4 afford highly enantioselective catalysts (seven examples, 91–98% ee). These catalysts provide an alternative methodology to prepare Felkin or anti-Felkin acetate-aldol products and related derivatives that are obtainable from the intermediate chiral organoboranes.
Rhodium-catalyzed hydroboration1 often exhibits interesting chemo-, regio- and diastereoselectivity, at times nicely complementing that obtained via the non-catalyzed reaction. Chiral organoboranes are useful intermediates for a variety of subsequent transformations.2 Some efficient chiral catalysts have been developed for the catalytic asymmetric reaction, but they are largely limited to vinyl arene substrates.3,4 Furthermore, catalyzed hydroboration of trisubstituted alkenes is usually slow or suffers from competing rhodium-catalyzed alkene isomerization.5,6 Thus, the utility of catalytic asymmetric hydroboration is significantly compromised by the present lack of substrate scope.
Building on the Evans5c,d and Gevorgyan7 reports of carbonyl-directed hydroboration, we found that the amide-directed asymmetric hydroborations of (E)- and (Z)-disubstituted β,γ-unsaturated amides proceed with high regio- and enantioselectivity using (BINOL)PN(Me)Ph in conjunction with Rh(nbd)2BF4.8 However, this catalyst proves somewhat less applicable to similar trisubstituted alkene substrates, for example, (E)- and (Z)-1. Although the level of enantioselectivity obtained is good (89–90% ee), the reaction is slow and the yield rather modest (Table 1, entry 1). The results obtained using (TADDOL)POPh (3a) are more encouraging (entry 2).9
Table 1.
Catalyzed hydroborations of (E)- and (Z)-1 as a function of ligand 3a–d.a
| Entry | Ligand | (E)-1 ee | yield | (Z)-1 ee | yield |
|---|---|---|---|---|---|
| 1 | (BINOL)PN(Me)Ph | 90 | 55 | 89 | 50 |
| 2 | 3a | 89 | 65 | 90 | 81 |
| 3 | 3b | 91 | 72 | 87 | 76 |
| 4 | 3c | 98 | 79 | 96 | 80 |
| 5 | 3d | 91 | 76 | 95 | 80 |
| 6b | 3a | 87 | 66 | ||
| 7c | 3a | 5 | 35 |
Unless otherwise specified, the reaction was run as follows: 1.0% Rh(nbd)2BF4, 2.1% of the indicated ligand, 2.0 PinBH, 40 °C, 12–24 h.
Uses 1.0% Rh(cod)2BF4.
Uses 0.5% [Rh(cod)Cl]2.
Seebach showed that adding substituents to the four phenyl groups appended to the TADDOL core (e.g., structures 3a–d) subtly changes the topography defined by this versatile chiral scaffold.10 Screening a series of such ligands reveals that the tert-butyl-substituted derivative 3c affords both a good yield of product and high levels of enantioselectivity of the (E)- and (Z)-isomers of substrate 1 (Table 1, entry 4, 96–98% ee).11,12 Substituting [Rh(cod)Cl]2 as the source of the rhodium catalyst leads to markedly lower reactivity and poor asymmetric induction (entry 7). A trisubstituted alkene lacking the amide directing group, (E)-4, reacts only sluggishly under these conditions (<20% conversion) highlighting the role of the carbonyl directing group and apparent two-point binding of the unsaturated amide substrate to the catalyst.
Trisubstituted alkenes bearing non-identical alkenyl substituents generate two new stereocenters upon hydroboration, and therefore, syn/anti-diastereoselectivity is also a relevant concern. The rhodium-catalyzed asymmetric hydroboration of (E)-1 using ligand 3c affords the anti-diastereomer of 2 in good yield (79%) after oxidative workup (Figure 1). The level of diastereoselectivity is high; we see none of the corresponding syn-diastereomer which is easily recognized by 1H NMR analysis. The level of enantioselectivity is also excellent; anti-diastereomer (3R,4S)-2 is obtained in 98% ee as determined by chiral HPLC analysis. Using the same chiral ligand (i.e., 3c), (Z)-1 affords the syn-diastereomer (3R,4R)-2, again with high diastereo- and enantioselectivity (80% yield, 96% ee).13
Figure 1.

Selective formation of either Felkin or anti-Felkin acetate-aldol products via stereospecific rhodium-catalyzed asymmetric hydroboration.
While the reactions of (E)- and (Z)-1 are quite efficient, it was initially disappointing to find that other trisubstituted substrates gave slightly lower levels of enantioselectivity under similar conditions. Monitoring the course of the reaction of (E)-5 proved insightful. The blue data points in Figure 2 show the yield (squares) and enantiomeric excess (circles) of anti-6 as formed over time. In the initial stages of the reaction, the anti-6 produced is near racemic; only 10–15% ee in the first hour. However, the enantiomeric purity increases dramatically over time, and upon complete consumption of starting materials, anti-6 is obtained in good, but obviously not optimal, enantiopurity (80% yield, 92% ee). The improvement in enantioselectivity over time suggests that a transient rhodium complex is an active but poorly stereoselective catalyst at the early stages of the reaction. Once replaced by the more highly selective catalyst, the reaction proceeds with high levels of asymmetric induction.14
Figure 2.

Comparing the yield of anti-6 (indicated by squares) and its enantiomeric excess (indicated by circles) over time with (red data) and without (blue data) added norbornene.
Switching from Rh(nbd)2BF4 to Rh(cod)2BF4 or increasing the time for complexation with the chiral ligand (in this case, 3b) did not significantly improve the results. Several “sacrificial” alkene addends were screened based on the premise that a more reactive alkene might be preferentially consumed by the non-selective catalyst leaving the β,γ-unsaturated amide to react with the later formed selective catalyst. The red data points in Figure 2 show the yield for the formation of anti-6 (squares) and its enantiomeric excess (circles) for the reaction run in the presence of norbornene, a more reactive alkene under the conditions employed. In its presence (10 mole percent with respect to (E)-5), anti-6 is formed in similar yield, but somewhat higher enantiopurity (80%, 95% ee), than the reaction lacking norbornene.15
While more work is needed to understand the role of the addend, these modified reaction conditions prove useful for a number of substrates (Table 2). For example, both (E)-5 and its stereoisomer (Z)-5 (entries 1–2) undergo hydroboration/oxidation with a high degree of stereocontrol to afford anti-6 and syn-6 respectively, each in 95% ee. It is interesting to note that, while the end results are essentially identical, these stereoisomeric substrates each require a different ligand for optimal results.16 Additionally, (Z)-5 requires a higher catalyst load, 2% versus 1%, to effect complete conversion within 24 h. Other (E)-substrates also give the anti-product with good enantioselectivity (entries 3–4, 93 and 96% ee, respectively). Other (Z)-substrates, including ones bearing somewhat more sterically encumbering branched substituents, afford the syn-product in good yield and high enantioselectivity (entries 5–7, 80–82% yield, 91–95% ee).
Table 2.
Other trisubstituted alkene substrates undergo efficient catalytic asymmetric hydroboration.a
 | |||||
|---|---|---|---|---|---|
| Entry | Ligand | RE | RZ | Yield (%) | ee (%) |
| 1 | 3b | (CH2)3Ph | CH3 | 81 | 95 |
| 2b | 3d | CH3 | (CH2)3Ph | 83 | 95 |
| 3 | 3b | (CH2)4Ph | CH3 | 79 | 93 |
| 4c | 3c | (CH2)2CH3 | CH3 | 80 | 96 |
| 5b | 3b | CH3 | CH2CH(CH3)2 | 81 | 91 |
| 6 | 3c | CH3 | CH(CH3)2 | 80 | 95 |
| 7 | 3c | CH3 | c-C6H11 | 82 | 93 |
Unless otherwise specified the reaction conditions are as shown above the table.
Carried out using 2% Rh(nbd)2BF4, 4.1% ligand 3, and 10% norbornene.
Carried out in the absence of norbornene using 0.5% Rh(nbd)2BF4, 1.1% 3c, 40 °C.
In summary, the rhodium-catalyzed hydroborations of trisubstituted alkenes are generally slow or suffer competing isomerization. In contrast, the trisubstituted alkene moieties contained within the framework of a β,γ-unsaturated amide undergo facile reaction, perhaps facilitated by carbonyl directing effects and two-point binding of the substrate to the rhodium catalyst. The reactions of stereoisomer substrates, for example, (E)- and (Z)-3, cleanly give rise to diastereomeric anti- and syn-products, thus the rhodium-catalyzed reaction is stereospecific. In addition, simple TADDOL-derived phenyl monophosphite ligands in combination with Rh(nbd)2BF4 afford highly enantioselective catalysts. These catalysts provide an alternative methodology to prepare Felkin or anti-Felkin acetate-aldol products and related derivatives that are obtainable from the intermediate organoboranes. Further studies are in progress.
Supplementary Material
Acknowledgments
Financial support for this research from the NSF (CHE-0809637) and Nebraska Research Initiative is gratefully acknowledged. We thank T. A. George (UNL Chemistry) for the loan of equipment, N. C. Thacker (UNL Chemistry) for some preliminary experiments, and the NSF (CHE-0091975, MRI-0079750) and NIH (SIG-1-510-RR-06307) for the NMR spectrometers used in these studies carried out in facilities renovated under NIH RR016544.
Footnotes
Supporting Information Available: Experimental procedures and selected spectra. This information is available free of charge at http://pubs.acs.org.
References
- 1.Männig D, Nöth H. Angew Chem, Int Ed Engl. 1985;24:878–879. [Google Scholar]
- 2.(a) Ros A, Aggarwal VK. Angew Chem Int Ed. 2009;48:6289–6292. doi: 10.1002/anie.200901900. [DOI] [PubMed] [Google Scholar]; (b) Imao D, Glasspoole BW, Laberge VS, Crudden CM. J Am Chem Soc. 2009;131:5024–5025. doi: 10.1021/ja8094075. [DOI] [PubMed] [Google Scholar]; (c) Crudden CM, Glasspoole BW, Lata CJ. Chem Commun. 2009:6704–6716. doi: 10.1039/b911537d. [DOI] [PubMed] [Google Scholar]; (d) Crudden CM, Edwards D. Eur J Org Chem. 2003:4695–4712. [Google Scholar]; (e) Burks HE, Morken JP. Chem Commun. 2007:4717–4725. doi: 10.1039/b707779c. [DOI] [PubMed] [Google Scholar]
- 3.First reported by Hayashi T, Matsumoto Y, Ito Y. J Am Chem Soc. 1989;111:3426–3428.Reviews: Carroll AM, O’Sullivan TP, Guiry PJ. Adv Synth Catal. 2005;347:609–631.Vogels CM, Westcott SA. Curr Org Chem. 2005;9:687–699.
- 4.For recent progress toward alternative strategies to prepare chiral organoboranes, see: Lee Y, Hoveyda AH. J Am Chem Soc. 2009;131:3160–3161. doi: 10.1021/ja809382c.Sim HS, Feng X, Yun J. Chem Eur J. 2009;15:1939–1943. doi: 10.1002/chem.200802150.Fleming WJ, Müller-Bunz H, Lillo V, Fernández E, Guiry PJ. Org Biomol Chem. 2009;7:2520–2524. doi: 10.1039/b900741e.Lillo V, Prieto A, Bonet A, Díaz-Requejo MM, Ramírez J, Pérez PJ, Fernández E. Organometallics. 2009;28:659–662.Guiry PJ. ChemCatChem. 2009;1:233–236.
- 5.(a) Hadebe SW, Robinson RS. Tetrahedron Lett. 2006;47:1299–1302. [Google Scholar]; (b) Edwards DR, Crudden CM, Yam K. Adv Synth Catal. 2005;347:50–54. [Google Scholar]; (c) Evans DA, Fu GC, Hoveyda AH. J Am Chem Soc. 1992;114:6671–6679. [Google Scholar]; (d) Evans DA, Fu GC, Anderson BA. J Am Chem Soc. 1992;114:6679–6685. [Google Scholar]
- 6.For example, reaction in fluorous media; see: Juliette JJJ, Rutherford D, Horvth IT, Gladysz JA. J Am Chem Soc. 1999;121:2696–2704.
- 7.Rubina M, Rubin M, Gevorgyan V. J Am Chem Soc. 2003;125:7198–7199. doi: 10.1021/ja034210y. [DOI] [PubMed] [Google Scholar]
- 8.Smith SM, Thacker NC, Takacs JM. J Am Chem Soc. 2008;130:3734–3735. doi: 10.1021/ja710492q. [DOI] [PubMed] [Google Scholar]
- 9.Chiral monophosphites and monophosphoramidites are useful for the asymmetric hydroboration of styrenes: Moteki SA, Wu D, Chandra KL, Reddy DS, Takacs JM. Org Lett. 2006;8:3097–3100. doi: 10.1021/ol061117g.
- 10.Seebach D, Dahinden R, Marti RE, Beck AK, Plattner DA, Kuhnle FNM. J Org Chem. 1995;60:1788–1799. [Google Scholar]
- 11.A 2.1:1.0 P:Rh ratio is employed. At a 1:1 P:Rh ratio, the enantioselectivity is diminished while at a 4:1 P:Rh ratio, the yield suffers.
- 12.11B NMR experiments carried out on a related substrate indicate that rhodium-catalyzed decomposition of PinBH necessitates its use in excess, consistent with a report by Robinson; see: Hadebe SW, Robinson RS. Eur J Org Chem. 2006:4898–4904.
- 13.The enantiomeric syn- and anti-products are easily obtained using the enantiomeric TADDOL-derived ligand. For example, using (3aS,8aS)-3c (E)-1 gives the anti-diastereomer (3S,4R)-2 (80%, 98% ee) while (Z)-1 affords the syn-diastereomer (3S,4S)-2 (81%, 96% ee).
- 14.The extent to which the poorly-selective reaction competes is both a function of substrate and ligand. Substrate (E)-5 is particularly problematic.
- 15.Using 20 mol% norbornene does not further improve enantioselectivity, but 5 mol% gives slightly lower ee (93%). The reaction of norbornene itself (1.0% Rh(nbd)2BF4, 2.1% 3a, 2 equiv. PinBH) is complete within 1 h giving exo-norborneol quantitatively but only 20% ee.
- 16.For example, the reaction of (E)-5 in combination with 3d afforded anti-6 in only 84% ee; (Z)-5 in combination with 3b afforded syn-6 in only 91% ee.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.