

Abstract
Purpose
Liver transplant recepients (LTRs) have an increased risk of colorectal neoplasia. The mechanism responsible for this is unknown. JCV encodes for TAg and has been implicated in colorectal carcinogenesis. We hypothesized that the use of immunosuppression in LTRs facilitates activation of JCV and is responsible for the increased risk of neoplasia.
Experimental Design
JCV TAg DNA and protein expression were determined in normal colonic epithelium (n = 15) and adenomatous polyps (n = 26) from LTRs and compared with tissue samples from control patients (normal colon, n = 21; adenomas, n = 40). Apoptosis and proliferation were determined by M30 and Ki-67 immunoreactivity, respectively.
Results
JCV TAg DNA was found in 10 of 15 (67%) of normal colonic mucosa from LTRs compared with 5 of 21 (24%) of control normal mucosa (P = 0.025). JCV TAg DNA was detected in 16 of 26 (62%) of the adenomas from LTRs and in 20 of 40 (50%) of control adenomas. JCV TAg protein was expressed in 13 of 26 (50%) adenomas from LTRs versus 2 of 40 (5%) of adenomas from controls (P < 0.001). In adenomas from LTRs, the mean proliferative activity was higher compared with controls (60.3 ± 3.2% versus 42.7 ± 2.8%, P < 0.001), whereas mean apoptotic indices were lower in LTRs (0.29 ± 0.08% versus 0.39 ± 0.06%, P = 0.05).
Conclusions
The presence of JCV in the colorectal mucosa and adenomas from LTRs, in concert with the use of immunosuppressive agents, suggests that JCV may undergo reactivation, and the subsequent TAg protein expression might explain the increased risk of colorectal neoplasia in LTRs.
Liver transplant recipients (LTR) are at an increased risk of developing de novo malignancies after orthotopic liver transplantation (LT; ref. 1). De novo malignancies include skin cancers, solid tumors, and lymphoproliferative disorders (2). The overall incidence of colorectal neoplasia in this population is controversial (3); however, most studies have identified an increased risk of colorectal neoplasia in this setting (2, 4, 5). Other groups, such as patients with primary sclerosing cholangitis (PSC) and inflammatory bowel disease (IBD), are especially at increased risk for colorectal neoplasia, and screening colonoscopy is recommended before LT (3). Importantly, colorectal neoplasia in transplant recipients appears at an earlier age and is associated with a diminished 5-year survival, and the tumors have more aggressive clinical behaviors (6). In previous studies, asymptomatic LTRs were found to have a significant increase in risk of colorectal neoplasia compared with asymptomatic average risk cohorts (2, 4).
Risk factors for cancer in this cohort may include long-term exposure to alcohol, but the most important factor is the posttransplantation immunosuppressive treatment regimens. Immunosuppressive medications decrease immune surveillance and enable the reactivation of potentially oncogenic viruses, such as human papillomavirus, EBV, and human herpes virus-8 (1). An oncogenic virus might also explain the earlier onset and higher risk for advanced colorectal neoplasia in LTRs.
JC virus (JCV) is a polyomavirus that infects most humans worldwide. We have previously provided evidence for the presence of JCV in the normal gastrointestinal tract and its involvement in colorectal cancer in humans (7–9). About 90% of the adult population carries antibodies to the virus, and it seems that in most people, the virus remains latent. However, in immunocompromised patients, JCV may become reactivated and can cause the lethal demyelinating disease, progressive multifocal leukoencephalopathy (10).
Translational Relevance.
The present study shows that 50% of the adenomatous polyps developing after liver transplantation express JC virus (JCV) Tantigen (TAg) protein compared with 5% of the adenomas of the controls. JCV is a human polyomavirus recently linked to human cancers, particularly those affecting the colon and rectum. We also found that normal tissues from liver transplant recipients (LTR) harbor the viral DNA more frequently compared with controls. Furthermore, LTR adenomas showed increased proliferation activity and less apoptosis compared with adenomas from controls, which is consistent with the TAg activity found in vitro in previous studies. We hypothesize that the use of immunosuppression in LTRs facilitates activation of the oncogenic JCV and is responsible for the increased risk of neoplasia. We believe that these results open new perspectives in understanding the increased susceptibility to colon cancer in LTRs, leading to possible future vaccination and prevention of cancer in this specific population.
The suspicion that JCV might be oncogenic arose because studies showed that the virus causes aneuploid tumors after injection into the brain of rodents and primates (11) and the virus can be detected in high-grade human brain tumors (12). The oncogenic potential of the virus is a consequence of the potent transforming oncoprotein, T antigen (TAg). TAg is a multifunctional protein and is able to bind and inactivate the tumor suppressor proteins p53 and pRb, leading to dysregulation of the cell cycle, permitting replication of cells with damaged chromosomes, which is normally censored by the G1-S cell cycle checkpoint (13, 14). More importantly in the context of colorectal carcinogenesis, TAg interacts with β-catenin, which can dysregulate the WNT signaling pathway (15), and JCV can bind to p53 and β-catenin and induce chromosomal instability in the colon cancer cell line RKO (16). Finally, there is a strong association between JCV TAg expression and the methylator phenotype in colorectal cancer, suggesting that aberrant methylation in colorectal cancer may also be related to JCV infection (17). Because of the strong correlation between TAg expression and colorectal cancer, we proposed that the virus might be detectable more frequently in adenomatous polyps of patients who underwent LT and hypothesized that the use of immunosuppression leads to reactivation of the virus, resulting in accelerated adenoma development in LTRs. Our results support this conclusion.
Materials and Methods
Inclusion criteria and patients
Between March 1979 and October 2000, 381 adult patients had a LT in the Department of Gastroenterology and Hepatology of the University Medical Center of Groningen (Groningen, the Netherlands). Patients surviving at least 5 y following LT were eligible for this study. In 147 of these patients, one or more colonoscopies had been done following LT. Data were collected on the age, gender, diagnosis of liver disease, diagnosis of pretransplant IBD, and the use of immunosuppressive medications after LT. The endoscopic and pathologic findings in a subset of those patients have been previously published (4). For this study, we included all neoplastic lesions found at least 3 y after LT, whether or not the patients were symptomatic.
We obtained formalin-fixed, paraffin-embedded samples of 15 normal colonic epithelial tissues and 26 adenomatous polyps from LTRs (Table 1). Samples were collected during colonoscopy done after LT. Posttransplant colonoscopy had been done at a mean interval of 11.1 y (range, 3.6-23) after LT. Nonpathologic colonic mucosal biopsies from LTRs on long-term immunosuppressive therapy who had undergone screening colonoscopy were included (18). Following informed consent, mucosal biopsies were taken from the ascending and transverse colon between 2003 and 2005. Biopsies of normal mucosa from patients with a history of adenomas or carcinomas were excluded.
Table 1.
Clinical and histopathologic findings in the studied populations
| LT (n = 41) | Controls (n = 61) | P | |||
|---|---|---|---|---|---|
| Adenomas (n = 26) |
Normal colon (n = 15) |
Adenomas (n = 40) |
Normal colon (n = 21) |
||
| Male sex | 13 (50%) | 6 (40%) | 24 (58%) | 8 (38%) | n.s. |
| Age at colonoscopy, mean (range) | 58 (48-71) | 50.9 (31-66) | 64.7 (47-89) | 40.1 (18-65) | n.s. |
| Mean age at LT (range) | 47 (27-61) | 43.4 (21-63) | n.s. | ||
| Reasons for transplant | PSC 6 | HCV 2 | |||
| PBC 10 | PBC 1 | ||||
| AIC 3 | PSC 1 | ||||
| ALC 3 | NRH 1 | ||||
| HBC 1 | AIC 3 | ||||
| BCS 2 | ALC 4 | ||||
| HCC 1 | HBV 1 | ||||
| PCD 1 | |||||
| Wilson 1 | |||||
| Characteristics of adenomatous polyps | |||||
| Tubular growth | 80.8% | 72.5% | n.s. | ||
| Villous growth | 19.2% | 27.5% | n.s. | ||
| HGD | 19.2% | 25% | n.s. | ||
| Size (mm) | 6 (mean) | 9.8 (mean) | n.s. | ||
| Proliferative activity, % (mean ± SE) | 60.3 ± 3.2 | 42.7 ± 2.8 | <0.001 | ||
| Apoptotic index, % (mean ± SE) | 0.29 ± 0.08 | 0.39 ± 0.06 | <0.05 | ||
Abbreviations: PBC, primary biliary cirrhosis; AIC, autoimmune cirrhosis; ALC, alcoholic cirrhosis; HBC, hepatitis B cirrhosis; BCS, Budd-Chiari syndrome; HCC, hepatocellular carcinoma; HCV, hepatitis C cirrhosis; HGD, high-grade dysplasia; NRH, nodular regenerative hyperplasia; PCD, polycystic liver disease; n.s., not significant.
A series of normal colonic samples obtained from 21 subjects were used as controls. Samples were selected from patients who underwent colonoscopy in 2005 with normal endoscopic findings and from whom random biopsies had been taken, which were histologically normal. Forty adenomas, matched for size, degree of dysplasia, and growth pattern with LTR adenomas, were selected as controls from a large series of previously characterized adenomas (19). In both groups, patients with a previous history of adenomas or carcinomas, as well as patients with immunosuppressive conditions in the past, were excluded.
All pathologic specimens of colorectal adenomas were reviewed by one pathologist for confirmation of diagnosis and assessment of the degree of dysplasia and the presence of villous architecture according to criteria established by WHO (20). An advanced adenoma was defined as an adenoma at least 1 cm in size, and/or (tubular) villous architecture, and/or high-grade dysplasia. Institutional Review Board approval was granted for this study and informed consent was given by the patients included in the study.
DNA extraction and detection of JCV genomic sequences
DNA was obtained from normal colonic and adenomatous polyps samples microdissected from 10-μm tissue slices. DNA isolation was done using the QIAmp DNA extraction kit (Qiagen) according to the manufacturer's recommendations. The DNA extraction was done in a room completely isolated from any post-PCR samples to avoid contamination with PCR-amplified products. To avoid contamination with JCV genomes, we did not include any positive controls carrying JCV DNA sequences; rather, each positive sample was sequenced in both directions to verify the authenticity of the JCV TAg DNA.
For the detection of JCV TAg gene sequences, PCR was done using gene-specific primers for TAg, which amplified a 154-bp NH2-terminal region of JCV TAg, as previously described (21). Five microliters of the PCR products were analyzed on an ethidium bromide–stained 1% agarose gel. A positive PCR product was confirmed through automated sequencing using an ABI PRISM BigDye Terminator v1.1 Cycle Sequencing kit on an ABI PRISM 3100 Avant Genetic Analyzer (Applied Biosystems). The sequencing results were aligned with JCV sequences from GenBank (National Center for Biotechnology Information, Bethesda, MD).
Immunohistochemical staining
Paraffin-embedded tissue specimens were sectioned at a thickness of 5 μm and mounted onto positively charged slides. The sections were placed in an oven at 65°C to melt the paraffin, deparaffinized with xylene, and rehydrated through an alcohol gradient. Nonenzymatic antigen retrieval was done in 0.01 mol/L sodium citrate buffer (pH 6.0) at 102°C to 104°C for 15 min. After cooling for a period of 20 min and rinsing with PBS, endogenous enzyme blocking was done by incubating the slides in 5% normal goat serum for 1 h at room temperature. Primary antibodies against viral and cellular proteins were incubated with the hydrated tissue sections overnight at room temperature in a humidified chamber. The primary antibodies used to detect JCV proteins were a mouse monoclonal antibody against SV40 large TAg that cross-reacts with JCV TAg (clone PAb416, 1:100 dilution; Oncogene Research Products) and a mouse monoclonal antibody against JCV TAg (clone PAb2003, 1:100 dilution; kindly provided by Dr. Richard Frisque, Professor of Molecular Virology, Pennsylvania State University). Monoclonal antibodies against the caspase-cleaved portion of cytokeratin 18 (M30; Boehringer Mannheim GmbH) and Ki-67 (MIB-1; Immunotech) were used to determine apoptosis and proliferative activity at final concentrations of 1:50 and 1:400, respectively. After washing in PBS, incubation with Dako EnVision-labeled polymer (DakoCytomation, Inc.) was done for 1 h at room temperature. Staining was developed by a reaction with 3,3′-diaminobenzidine chromogen for 1 to 5 min and then counterstained for 45 s with hematoxylin, dehydrated, dehydrated in xylene, and mounted. Slides were reviewed by two pathologists who were blinded to the other results.
For M30 and MIB-1 staining, quantitative analyses were done on complete crypts from normal mucosa and adenomas. M30 positivity was identified as brown cytoplasmic staining. Morphologic characteristics, including the presence of apoptotic bodies, nuclear condensation, cytoplasmic shrinkage, and membrane blebbing, were assessed in H&E-stained tissue sections. In all cases, at least 1,000 epithelial cells were counted and morphologic apoptotic and M30-positive cells were expressed as a percentage of the total number of cells counted (apoptotic index). Apoptotic cells located in the lumen, assessed by either morphologic criteria or M30 positivity, were excluded. For evaluation of MIB-1 staining, complete crypts were counted.
Statistical analysis
Mean M30 and MIB-1 counts were compared between LTRs and controls using the Mann-Whitney test. The Student's t test, χ2 test, and Fisher's exact test were used to compare LTR adenomas with non-LTR adenomas. The confounding effect of age was further controlled using regression models that included a continuous term for age. Statistical analysis was done using Statistical Package for the Social Sciences software 11.0 (SPSS, Inc.). P values were considered significant when <0.05.
Results
Clinical characteristics
Clinical features of the patients investigated in this study are summarized in Table 1. The mean age at LT was 45.6 years (SD ± 9.8; range, 21-61). Colonoscopy in the LTRs with adenomatous polyps was done later (128.2 ± 74.9 months after LT; mean ± SD) compared with the LTRs with normal endoscopic findings (89.5 ± 36.5 months after LT; mean ± SD). Most LTR patients had received immunosuppression consisting of cyclosporin A, prednisolone, and azathioprine (n = 28), whereas eight patients received prednisolone and tacrolimus and five patients received prednisolone plus azathioprine and tacrolimus. The adenomatous polyps in the LTR group were similar to those obtained from the control group.
High proliferative activity and low apoptotic rates in adenomatous polyps from LTRs
The proliferative activity (mean ± SE) was higher in the adenomas obtained from LTRs compared with that obtained from controls (60.3 ± 3.2% versus 42.7 ± 2.8%; P < 0.001). Furthermore, the apoptotic indices were lower in LTRs (0.29 ± 0.08% versus 0.39 ± 0.06%; P = 0.05) than in controls. In normal colonic epithelium, no differences in apoptosis or proliferative activity were found between LTRs and controls (Table 1).
JCV TAg DNA sequences are frequently present in normal colonic mucosa and adenomatous polyps obtained from LTRs
We tested the hypothesis that JCV TAg DNA would be more prevalent in adenomatous polyps from LTRs compared with controls. First, we used JCV TAg-specific primers to detect viral DNA sequences (Fig. 1A). JCV TAg DNA sequences were found more frequently in the normal colonic mucosa obtained from LTRs than controls [10 of 15 (67%) versus 5 of 21 (24%); P = 0.025], whereas no significant differences were found between adenomatous polyps obtained from either group [16 of 26 (62%) versus 20 of 40 (50%); P = 0.74; Fig. 2]. Each of the PCR amplicons was confirmed to be JCV TAg DNA by sequencing.
Fig. 1.
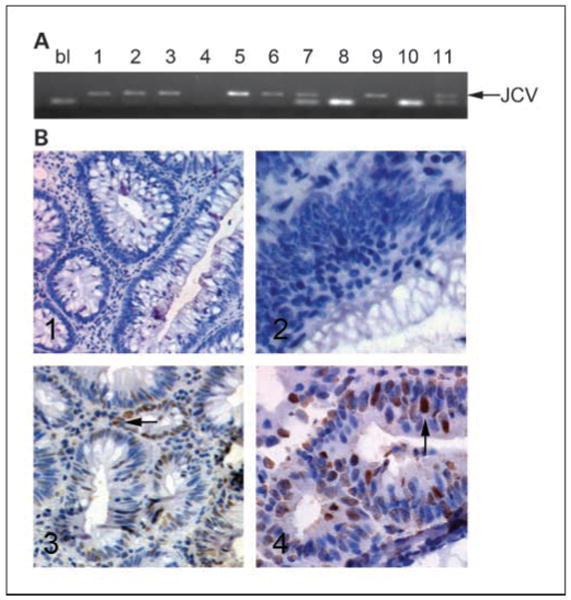
PCR amplification of JCV TAg DNA sequences (A) and expression of JCV TAg in adenomatous polyps. A, example of PCR amplification of JCV TAg DNA. Arrow, size of the JCV TAg DNA fragment. 1 to 4, control adenomas; 5 to 7, LTR adenomas; 8 and 9, control normal mucosa; 10 and 11, LTR normal mucosa; bl, blank (water control). B, immunohistochemistry showing samples positive and negative for JCV TAg protein. Samples 1 and 2 (corresponding to samples 1 and 2 in A) are adenomas obtained from controls, whereas samples 3 and 4 (corresponding to samples 5 and 6 in A) are adenomas obtained from LTRs. Arrow in sample 5, positive staining; arrow in sample 6, positive nuclear staining for JCV TAg.
Fig. 2.
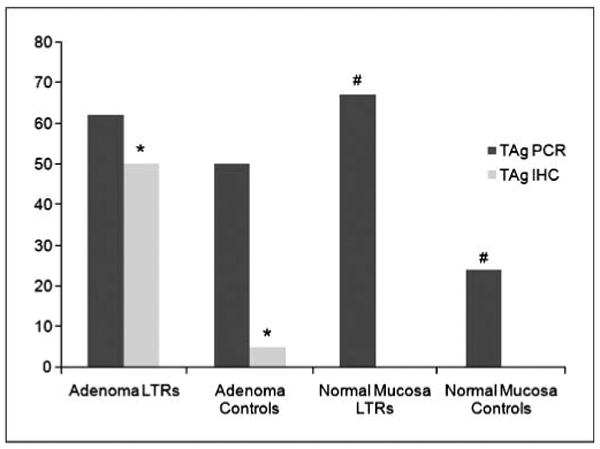
Prevalence of JCV TAg DNA sequences and protein expression among the studied groups. A significant higher expression of JCV TAg was found in adenomas from LTRs than controls. Interestingly, JCV TAg DNA was significantly more amplifiable in normal mucosa samples obtained from LTRs than controls. No differences in JCV TAg DNA amplification were found in adenomas among the two groups, whereas no JCV TAg expression was present in any of the normal mucosa samples. *, P = 0.0002; #, P = 0.025.
JCV TAg is primarily expressed in adenomatous polyps obtained from LTRs
We next examined JCV TAg protein expression in our samples. Although JCV DNA sequences are readily amplified from either neoplastic or nonneoplastic tissues, TAg expression would be a more convincing finding if one were to implicate the virus in carcinogenesis. Figure 1B shows positive and negative immunohistochemical staining for TAg in the dysplastic areas of the adenomatous polyps from LTRs and control patients. In total, 13 of 26 (50%) of the adenomatous polyps from LTRs showed positive nuclear staining for JCV TAg compared with 2 of 40 (5%) of the adenomas obtained from controls (P = 0.0002). Furthermore, all 15 TAg-positive samples harbored JCV DNA sequences. Positive staining for JCV TAg was restricted to the nuclei of the dysplastic domains in colonic samples but was never present in the cytoplasm or adjacent nonneoplastic cells. No TAg protein expression was present in any of the normal colonic mucosa samples, whether a LTR or not.
Discussion
In our study, we show that JCV TAg is frequently expressed in adenomatous polyps of patients who had undergone LT. Our results suggest that the expression of JCV TAg in premalignant lesions in concert with the use of immunosuppressive agents might be involved in the risk of colorectal neoplasia in LTRs.
The overall incidence of neoplasms in LTRs is reported to be higher compared with the general population (22). Moreover, several published studies describe an increased incidence of colorectal neoplasia in LTRs (3, 22). In certain high-risk patients, such as those with PSC and IBD, the incidence of colorectal cancer is substantially higher (23–26). However, our study cohort contained just two patients with PSC and ulcerative colitis. The increasing number of LTs both in the United States and Europe, and the significant improvement in survival after transplant, have created a steadily increasing population of living LTRs who may be at increased risk for malignancies.7
The question remains whether the increased prevalence of colorectal neoplasia in LTRs is because liver transplant candidates have an intrinsically increased predisposition for developing colorectal neoplasia or whether tumorigenesis in these patients is triggered by factors associated with transplantation, such as the use of immunosuppressive medications. Studies done on renal, lung, and cardiac transplant recipients are not conclusive on the role played by immunosuppression in increasing the risk of colon cancer (27 – 32). However, the frequent finding of JCV DNA in nonneoplastic colonic tissue from LTRs opens the speculation that the immunosuppression facilitates reactivation of oncogenic viruses, such as JCV, accelerating adenoma formation. The finding that JCV TAg is highly expressed in adenomatous polyps from LTRs adds fuel to this discussion.
In our study, apoptotic rates were significantly lower in adenomatous polyps from LTRs compared with control adenomas. This is consistent with a recent study, which suggested that JCV infection promotes antiapoptotic mechanisms in immunosuppressed patients (33). Additionally, we found significantly increased proliferative activity in adenomas from LTRs compared with sporadic adenomas. It has been shown that JCV TAg can form a complex with β-catenin, an integral component of the Wnt signaling pathway (15). JCV Tag could chaperone β-catenin to the nucleus and directly stimulate the proliferative program in the colonic epithelial cell (15, 16).
In sporadic adenomatous polyps, JCV TAg expression was found in 2 of 40 adenomas in this study and in 16% in a previous series using different samples studied by different examiners (21). We have previously reported that JCV TAg DNA was detected in a substantially high proportion of normal colonic epithelium. The detection of JCV in tissues is quite sensitive to the techniques used, and in our previous study, we used fresh-frozen tissues, two rounds of (nested) PCR, and degenerate primers to increase sensitivity (9). In this study, DNA was extracted from formalin-fixed, paraffin-embedded tissues microdissected from neoplastic domains on glass slides, which would be expected to limit detection of JCV. The JCV copy number is quite low in normal colonic tissues.
Interestingly, although there was a significant difference in the prevalence of JCV DNA in the normal mucosa of LTRs compared with controls, there was no significant difference in the prevalence of JCV DNA sequences in the adenomas from either group. We believe that the best explanation for these results might be that low-level JCV infection is nearly universal among adults, and proliferating adenomatous cells with latent viral infection would make the virus easier to detect by PCR. The higher rate of finding amplifiable JCV DNA from the normal tissues of LTRs suggests that these patients may be experiencing reactivation of the virus and increased copy numbers of viral DNA in normal tissues. Another explanation for the higher rate of JCV DNA from the normal tissues of LTRs might be a higher infection rate in these patients. But this seems to be unlikely because the virus infects humans in their adolescence, and >90% of the population carries antibodies to the virus by adult life (7). However, the important issue is not simply the presence of the virus in these tissues, which has been shown previously by our group and others, but the reactivation and subsequent expression of TAg that is associated with the adenomas.
The association between JCV infection and adenoma development in liver transplant patients leads to the hypothesis that this oncogenic virus may undergo activation in the gut as a consequence of the immunosuppression from a latent state to an active infection, which might trigger or accelerate the adenoma-to-carcinoma sequence in this group of patients. Future studies will be required to determine the exact molecular and virological mechanisms of viral activation and viral-induced carcinogenesis in LTRs.
Acknowledgments
Grant support: NIH/National Cancer Institute grant R01CA98572 (C.R. Boland).
Footnotes
Organ Procurement and Transplantation Network. U.S. transplantations 1998-2004. http://www.optn.org/latestData/rptData.asp.
Disclosure of Potential Conflicts of Interest: No potential conflicts of interest were disclosed.
References
- 1.Sanchez W, Talwalkar JA, Gores GJ. Will all liver transplantation patients eventually die from cancer? J Hepatol. 2006;44:13–8. doi: 10.1016/j.jhep.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 2.Haagsma EB, Hagens VE, Schaapveld M, et al. Increased cancer risk after liver transplantation: a population-based study. J Hepatol. 2001;34:84–91. doi: 10.1016/s0168-8278(00)00077-5. [DOI] [PubMed] [Google Scholar]
- 3.Delco F, Mullhaupt B. Should we screen for colorectal cancer in liver transplantation? J Hepatol. 2006;44:32–8. doi: 10.1016/j.jhep.2005.10.011. [DOI] [PubMed] [Google Scholar]
- 4.Koornstra JJ, Wesseling J, de Jong AE, Vasen HF, Kleibeuker JH, Haagsma EB. Increased risk of colorectal neoplasia in asymptomatic liver-transplant recipients. Gut. 2007;56:892–3. doi: 10.1136/gut.2007.120121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Buell JF, Papaconstantinou HT, Skalow B, Hanaway MJ, Alloway RR, Woodle ES. De novo colorectal cancer: five-year survival is markedly lower in transplant recipients compared with the general population. Transplant Proc. 2005;37:960–1. doi: 10.1016/j.transproceed.2004.12.122. [DOI] [PubMed] [Google Scholar]
- 6.Johnson EE, Leverson GE, Pirsch JD, Heise CP. A 30-year analysis of colorectal adenocarcinoma in transplant recipients and proposal for altered screening. J Gastrointest Surg. 2007;11:272–9. doi: 10.1007/s11605-007-0084-4. [DOI] [PubMed] [Google Scholar]
- 7.Knowles WA. Discovery and epidemiology of the human polyomaviruses BK virus (BKV) and JC virus (JCV) Adv Exp Med Biol. 2006;577:19–45. doi: 10.1007/0-387-32957-9_2. [DOI] [PubMed] [Google Scholar]
- 8.Laghi L, Randolph AE, Chauhan DP, et al. JC virus DNA is present in the mucosa of the human colon and in colorectal cancers. Proc Natl Acad Sci U S A. 1999;96:7484–9. doi: 10.1073/pnas.96.13.7484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ricciardiello L, Laghi L, Ramamirtham P, et al. JC virus DNA sequences are frequently present in the human upper and lower gastrointestinal tract. Gastroenterology. 2000;119:1228–35. doi: 10.1053/gast.2000.19269. [DOI] [PubMed] [Google Scholar]
- 10.Padgett BL, Walker DL, ZuRhein GM, Eckroade RJ, Dessel BH. Cultivation of papova-like virus from human brain with progressive multifocal leucoencephalopathy. Lancet. 1971;1:1257–60. doi: 10.1016/s0140-6736(71)91777-6. [DOI] [PubMed] [Google Scholar]
- 11.Reiss K, Khalili K. Viruses and cancer: lessons from the human polyomavirus, JCV. Oncogene. 2003;22:6517–23. doi: 10.1038/sj.onc.1206959. [DOI] [PubMed] [Google Scholar]
- 12.White MK, Gordon J, Reiss K, et al. Human polyomaviruses and brain tumors. Brain Res Brain Res Rev. 2005;50:69–85. doi: 10.1016/j.brainresrev.2005.04.007. [DOI] [PubMed] [Google Scholar]
- 13.Dyson N, Bernards R, Friend SH, et al. Large T antigens of many polyomaviruses are able to form complexes with the retinoblastoma protein. J Virol. 1990;64:1353–6. doi: 10.1128/jvi.64.3.1353-1356.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krynska B, Gordon J, Otte J, et al. Role of cell cycle regulators in tumor formation in transgenic mice expressing the human neurotropic virus, JCV, early protein. J Cell Biochem. 1997;67:223–30. doi: 10.1002/(sici)1097-4644(19971101)67:2<223::aid-jcb7>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 15.Enam S, Del VL, Lara C, et al. Association of human polyomavirus JCV with colon cancer: evidence for interaction of viral T-antigen and β-catenin. Cancer Res. 2002;62:7093–101. [PubMed] [Google Scholar]
- 16.Ricciardiello L, Baglioni M, Giovannini C, et al. Induction of chromosomal instability in colonic cells by the human polyomavirus JC virus. Cancer Res. 2003;63:7256–62. [PubMed] [Google Scholar]
- 17.Goel A, Li MS, Nagasaka T, et al. Association of JC virus T-antigen expression with the methylator phenotype in sporadic colorectal cancers. Gastroenterology. 2006;130:1950–61. doi: 10.1053/j.gastro.2006.02.061. [DOI] [PubMed] [Google Scholar]
- 18.Verdonk RC, Haagsma EB, Jonker MR, et al. Effects of different immunosuppressive regimens on regulatoryT-cells in noninflamed colon of liver transplant recipients. Inflamm Bowel Dis. 2007;13:703–9. doi: 10.1002/ibd.20087. [DOI] [PubMed] [Google Scholar]
- 19.Koornstra JJ, Rijcken FE, De JS, Hollema H, de Vries EG, Kleibeuker JH. Assessment of apoptosis by M30 immunoreactivity and the correlation with morphologic criteria in normal colorectal mucosa, adenomas and carcinomas. Histopathology. 2004;44:9–17. doi: 10.1111/j.1365-2559.2004.01739.x. [DOI] [PubMed] [Google Scholar]
- 20.Hamilton SR, Vogelstein B, Kudo S. World Health Organization classification of tumours: pathology and genetics of tumours of the digestive system. Lyon: IARC; 2000. [Google Scholar]
- 21.Jung WT, Li MS, Goel A, Boland CR. JC virusT-antigen expression in sporadic adenomatous polyps of the colon. Cancer. 2008;112:1028–36. doi: 10.1002/cncr.23266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vallejo GH, Romero CJ, de Vicente JC. Incidence and risk factors for cancer after liver transplantation. Crit Rev Oncol Hematol. 2005;56:87–99. doi: 10.1016/j.critrevonc.2004.12.011. [DOI] [PubMed] [Google Scholar]
- 23.Higashi H, Yanaga K, Marsh JW, Tzakis A, Kakizoe S, Starzl TE. Development of colon cancer after liver transplantation for primary sclerosing cholangitis associated with ulcerative colitis. Hepatology. 1990;11:477–80. doi: 10.1002/hep.1840110320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Loftus EV, Jr, Aguilar HI, Sandborn WJ, et al. Risk of colorectal neoplasia in patients with primary sclerosing cholangitis and ulcerative colitis following orthotopic liver transplantation. Hepatology. 1998;27:685–90. doi: 10.1002/hep.510270308. [DOI] [PubMed] [Google Scholar]
- 25.Silva MA, Jambulingam PS, Mirza DF. Colorectal cancer after orthotopic liver transplantation. Crit Rev Oncol Hematol. 2005;56:147–53. doi: 10.1016/j.critrevonc.2004.12.013. [DOI] [PubMed] [Google Scholar]
- 26.Vera A, Gunson BK, Ussatoff V, et al. Colorectal cancer in patients with inflammatory bowel disease after liver transplantation for primary sclerosing cholangitis. Transplantation. 2003;75:1983–8. doi: 10.1097/01.TP.0000058744.34965.38. [DOI] [PubMed] [Google Scholar]
- 27.Amital A, Shitrit D, Raviv Y, et al. Development of malignancy following lung transplantation. Transplantation. 2006;81:547–51. doi: 10.1097/01.tp.0000195774.26382.34. [DOI] [PubMed] [Google Scholar]
- 28.Nagele H, Bahlo M, Klapdor R, Rodiger W. Tumor marker determination after orthotopic heart transplantation. J Heart Lung Transplant. 1999;18:957–62. doi: 10.1016/s1053-2498(99)00069-8. [DOI] [PubMed] [Google Scholar]
- 29.Kasiske BL, Snyder JJ, Gilbertson DT, Wang C. Cancer after kidney transplantation in the United States. Am J Transplant. 2004;4:905–13. doi: 10.1111/j.1600-6143.2004.00450.x. [DOI] [PubMed] [Google Scholar]
- 30.Rodriguez-Larrain JM, Ziebert JJ, Kfoury AG, Kuwada S, Taylor DO, Renlund DG. Incidence of adenomatous colorectal polyps in cardiac transplant recipients. Transplantation. 1997;64:528–30. doi: 10.1097/00007890-199708150-00025. [DOI] [PubMed] [Google Scholar]
- 31.Saidi RF, Dudrick PS, Goldman MH. Colorectal cancer after renal transplantation. Transplant Proc. 2003;35:1410–2. doi: 10.1016/s0041-1345(03)00478-0. [DOI] [PubMed] [Google Scholar]
- 32.Stewart T, Henderson R, Grayson H, Opelz G. Reduced incidence of rectal cancer, compared to gastric and colonic cancer, in a population of 73,076 men and women chronically immunosuppressed. Clin Cancer Res. 1997;3:51–5. [PubMed] [Google Scholar]
- 33.Pina-Oviedo S, Urbanska K, Radhakrishnan S, et al. Effects of JC virus infection on anti-apoptotic protein survivin in progressive multifocal leukoencephalopathy. Am J Pathol. 2007;170:1291–304. doi: 10.2353/ajpath.2007.060689. [DOI] [PMC free article] [PubMed] [Google Scholar]