

Abstract
Calcium is an essential intracellular messenger and serves critical cellular functions in both excitable and non-excitable cells. Most of the physiological functions in these cells are uniquely regulated by changes in cytosolic Ca2+ levels ([Ca2+]i), which are achieved via various mechanisms. One of these mechanism(s) is activated by the release of Ca2+ from the endoplasmic reticulum (ER), followed by Ca2+ influx across the plasma membrane (PM). Activation of PM Ca2+ channels is essential for not only refilling of the ER Ca2+ stores, but is also critical for maintaining [Ca2+]i that regulates biological functions, such as neurosecretion, sensation, long term potentiation, synaptic plasticity, gene regulation, as well as cellular growth and differentiation. Alterations in Ca2+ homeostasis have been suggested in the onset/progression of neurological diseases, such as Parkinson's, Alzheimer's, bipolar disorder, and Huntington's diseases. Available data on transient receptor potential conical (TRPC) protein indicate that these proteins initiate Ca2+ entry pathways and are essential in maintaining cytosolic, ER, and mitochondrial Ca2+ levels. A number of biological functions have been assigned to these TRPC proteins. Silencing of TRPC1 and TRPC3 has been shown to inhibit neuronal proliferation and loss of TRPC1 is implicated in neurodegeneration. Thus, TRPC channels not only contribute towards normal physiological processes, but are also implicated in several human pathological conditions. Overall, it is suggested that these channels could be used as potential therapeutic targets for many of these neurological diseases. Thus, in this review we have focused on the functional implication of TRPC channels in neuronal cells along with the elucidation of their role in neurodegeneration.
Introduction
Intracellular Ca2+ concentration plays a vital role in regulating many fundamental cellular processes such as gene regulation, muscle contraction, neurosecretion, integration of electrical signaling, neuronal excitability, synaptic plasticity, cell proliferation, and apoptosis [1-5]. In neuronal cells, Ca2+ homeostasis is very tightly regulated and disturbances in Ca2+ homeostasis have been implicated in diseases such as Parkinson's, Alzheimer's, and Huntington's [6-8]. It is not at all surprising that disturbances in Ca2+ signaling pathways underlie neuronal loss, since many factors involved in neuronal function are dependent on Ca2+ signaling [9]. However, the cellular mechanism(s) underlying neurodegeneration, due to alterations in Ca2+ homeostasis, remains to be elucidated. Accumulating evidence suggest that both excessive elevation of intracellular Ca2+ as well as loss of [Ca2+]i is crucial for neurodegeneration [10,11]. Increased [Ca2+]i leads to inappropriate activation of Ca2+-dependent processes that are normally inactive or operate at low Ca2+ levels, thereby causing metabolic derangements that ultimately lead to neuronal death [12-14]. Increase of intracellular Ca2+ load is mediated via two closely related mechanisms: excessive release of Ca2+ from the endoplasmic reticulum (ER) stores, followed by Ca2+ influx through the plasma membrane (PM) channels, a process referred to as store-operated Ca2+ entry (SOCE) [15]. Although different Ca2+ entry mechanism(s) have been identified, SOCE is the major mechanism for Ca2+ influx upon store depletion that can increase cytosolic Ca2+ [16].
In contrast, under physiological conditions Ca2+ entry through SOCE channels is essential for regulating divergent processes ranging from, exocytosis, gene transcription, cell proliferation, and differentiation [17]. Furthermore, Ca2+ influx followed by ER store depletion accomplishes several critical cellular functions. First, this Ca2+ influx replenishes the ER Ca2+ stores, thereby maintaining its ability to release Ca2+ upon subsequent stimuli. Moreover, this is also critical since Ca2+ concentrations within the ER must be maintained at sufficient levels in order for the organelle to carry out many of its fundamental functions [18,19]. Second, since ER has limited Ca2+ capability, Ca2+ influx via SOCE is essential for increasing [Ca2+]i to have a physiological response. Thus, chronic depletion of ER Ca2+, as would occur in the absence of SOCE, could not only influence ER-dependent processes such as protein folding and trafficking, but could also inhibit cellular functions that are dependent on Ca2+. Furthermore, loss of Ca2+ homeostasis, due to improper SOCE, could lead to ER stress responses, and even apoptosis [18-20]. Thus, overall a set point for Ca2+ concentrations is critical to maintain normal physiological response and alterations in this Ca2+ set point could tilt the balance, thereby resulting in pathological conditions as discussed in this review.
Store Operated Calcium Entry, Its Mechanism of Regulation, and Channels Involved
SOCE (also known as capacitative Ca2+ entry) was initially proposed as a mechanism for Ca2+ influx, which is regulated by the ER Ca2+ pool, rather than by the Ca2+ signals generated upon agonist stimulation (15). The first step that leads to an increase in [Ca2+]i via this mechanism is always the binding of a hormone or a growth factor to its receptor that is localized at the PM. Activation of this receptor regulates breakdown of phosphatidylinositol-4,5-bisphosphate (PIP2) into inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG) (Fig. 1). Binding of IP3 releases Ca2+ from the intracellular stores (ER or its specialized counterpart in muscle, the sarcoplasmic reticulum), which controls Ca2+ influx across the PM via the SOCE mechanism [15, 21]. Alternatively, Ca2+ influx can also be initiated by second messengers such as cyclic nucleotides and DAG analogues [21,22]. Although, SOCE was identified 2 decades ago, neither the ion channel(s) that regulate Ca2+ entry upon store depletion nor the mechanics of how information from the internal Ca2+ store is relayed to the PM channels, was not identified until recently. Two major families of proteins, TRPC and ORAI channels have been identified in various cell types that have been shown to be critical for SOCE [23,24]. Although, the highly selective Icrac currents have been shown to be mediated via the ORAI channels [25-28], their importance in neuronal cells has not yet been elucidated. Importantly, it has also been shown that in some cells, ORAI and transient receptor potential conical (TRPC) channels form multimers [29-31]; however, it is not known if similar association is also present in neuronal cells and if so, what is the functional implication of this interaction? Nevertheless, identification of these two proteins has opened new and exciting areas in ion channel research, which are critical for the understanding of several biological processes; importantly, it is anticipated that alterations in these proteins could contribute to various pathological conditions. To understand the mechanism for SOCE regulation/activation, classical RNAi screening was performed, which identified Stromal Interaction Molecule (STIM) proteins as the molecular link between ER Ca2+ store depletion and SOCE activation [32,33]. Recent studies have shown that both TRPC and ORAI proteins interact with STIM1, which is important for the gating of the channel [23, 24, 29, 34]. Although, there is no doubt that ORAI and STIM1 proteins are critical for SOCE and could have an extraordinarily important role in regulating basic cellular functions, we have focused mainly on TRPC channels since no data is currently available that could identify the role of ORAI or STIM1 in neuronal cells. Nevertheless, since SOCE is present in neuronal cells, it could be anticipated that alterations in ORAI or STIM1 proteins could lead to pathological conditions.
Fig. (1). Activation and functional regulation of TRPC channels.
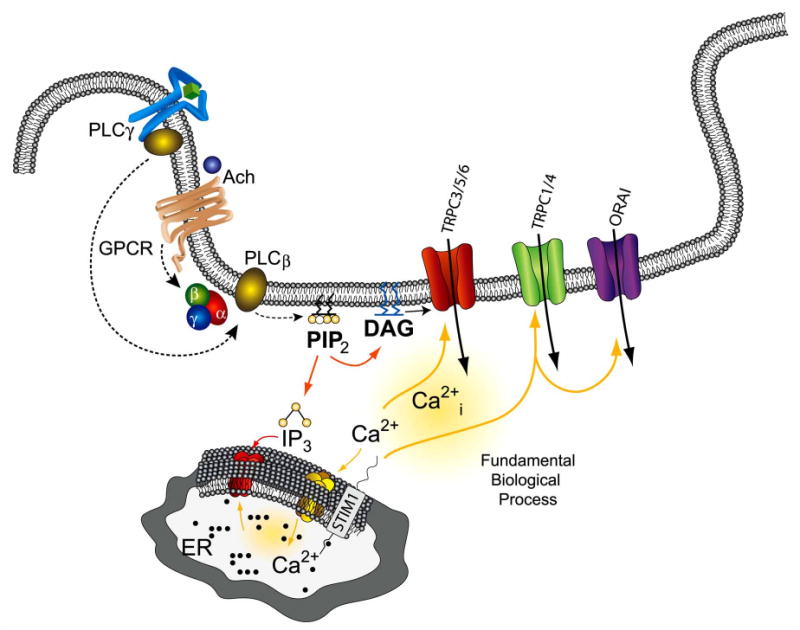
Binding of an agonist (Ach) to the G protein coupled receptor stimulates G protein, which activates PLCβ, causing the hydrolysis of phosphatidylinositol (4,5) bisphosphate (PIP2) to inositol (1,4,5)-triphosphate (IP3) and diacylglycerol (DAG). IP3 binds to IP3R, a ligand gated ion channel, which leads to the release of Ca2+ from the internal ER stores. Depletion of Ca2+ from the internal stores in turn allows STIM1 to aggregate, followed by the activation of the TRPC1, 4 or ORAI Ca2+ channels in the plasma membrane, which allows Ca2+ to enter the cell. Importantly, these signaling molecules are associated within lipid raft domains, which provide the platform for protein-protein interactions and stimulate activation of TRPC channels.
It is convincing from the current data that members of the TRPC family contribute to the elevation of [Ca2+]i, however their mode of activation as well as their biophysical properties are very distinct (Table 1). TRPC1 and TRPC4 have been shown to be activated by store depletion per se, whereas activation of TRPC3, 5, 6 and 7 has been shown to be mediated by receptor activation [35, 36] (Fig. 1). Alternatively, DAG analogues have also been shown to directly activate TRPC3 and TRPC6 channels, indicating that lipid mediators might also activate TRPC channels [37]. However, regardless, as how these TRPC channels are regulated, they all contribute towards Ca2+ entry. Importantly, a number of biological functions have already been assigned to various TRPC proteins that include mechanosensing activity, axon guidance, neurotransmitter release, neuronal proliferation/development, cell survival, axon guidance, neuronal morphogenesis, and synaptogenesis [38]. Thus, SOCE influences many aspects of neuronal biology and contributes towards normal physiology, and alterations in Ca2+ signaling could lead to the onset/progression of several pathological conditions of human health. This is intriguing, since these channels could now be used as potential therapeutic targets for several neurodegenerative diseases.
Table 1. TRPC Channel Properties and Proposed Neuronal Functions.
| Subfamily | Ion Channel Properties (Modified from Ref. [145]) | Proposed Physiological Role |
|---|---|---|
| TRPC1 | Non selective, 16-24 pS current conductance, inward rectifying with reverse potential of + 40mV | Proliferation, differentiation [38] Neuroprotection [50-52] Axon guidance [134] |
| TRPC3 | Non selective, but PCa/PNa ratio is 1.6, 42pS current conductance, slightly inward rectifying with positive current at membrane potentials above +5 mV | Proliferation, differentiation [38] Neurotransmitter release [125,126] Axon guidance [134] |
| TRPC4 | Non selective, but PCa/PNa ratio is 1.1, inward rectifying with positive current at membrane potentials above +10 mV current conductance 30- 42pS | Neurotransmitter release [146] Axonal regeneration [133] |
| TRPC5 | Partially selective with PCa/PNa ratio of 9, current conductance 47-66pS, voltage dependence decrease below −50 mV | Growth cone development [134,136] Innate fear response [137] Neurotransmitter release [126] |
| TRPC6 | Partially selective with PCa/PNa ratio of 5, current conductance 28-37pS) is also partially voltage dependent | Dentritic morphology [144] |
Diverse Function of TRPC in Neurons
Increased Ca2+ entry is implicated in both cell survival and in cell death processes [39]. Thus, it could be anticipated that different actions of Ca2+ in different cells must be dependent on its cellular concentration as well as on its location [40]. Ca2+ exerts a biphasic effect on cellular growth, a modest increase in [Ca2+]i, could promote cell proliferation, whereas relatively high [Ca2+]i would lead to increased mitochondrial Ca2+ and accounts for the release of pro-apoptotic factors resulting in cell death [39-41]. An interesting theme that is now emerging is the role of TRPC channels in cell growth, differentiation, and proliferation. TRPC1 and other members of its family, have been associated with cell proliferation and the level of expression of different TRPC proteins has been linked with the proliferative capacity in different cell lineages [38,41; Table 1]. TRPC1, 3 and 6 proteins are more highly expressed in embryonic CNS than in adult, which implicates these TRPC's in early development [42]. Furthermore, Ca2+ entry through TRPC channel plays a critical role in basic fibroblast growth factor (bFGF)-induced cortical neural stem cell (NSC) proliferation [43]. All TRPCs, except TRPC7, were present in E3 telencephalon and TRPC1-4 and 6, but not TRPC5, were expressed in NSC populations. Interestingly, TRPC1, 2 and TRPC4 showed higher expression, whereas TRPC6 expression was decreased in NSC populations, suggesting significant roles for different TRPC channels during differentiation [43]. Importantly, blocking of TRPC's channel or silencing of TRPC1 alone attenuated bFGF-induced intracellular Ca2+ elevation and NSC's proliferation [43]. In addition, Ca2+ influx through TRPC1 and TRPC3 controlled the switch between proliferation and differentiation in immortalized hippocampal neuronal cells, H19-7 [44]. On the other hand TRPC3 has been shown to be associated with neuronal growth, which is regulated by either BDNF receptor stimulation [45] or Epo-induced cell proliferation and differentiation [46]. Based on these results, TRPC proteins might play a critical role in neuronal survival, proliferation, and differentiation. Although in many cases the survival and proliferation induced by TRPCs are shown to be dependent on Ca2+ entry, it remains to be seen if TRPCs also regulate other proteins, a function that will be independent of their ability to regulate Ca2+ influx, since most TRPC have been shown to form large protein-protein complexes.
Selective degeneration of hippocampal neurons is a common feature in several neurodegenerative conditions including Alzheimer's disease, epilepsy and stroke. Accumulating evidence suggest that glutamate, an excitatory neurotransmitter intimately involved in learning and memory, is also involved in neurodegeneration [47,48]. Recently, Narayanan et al. reported that glutamate activates cell death by massive increases in [Ca2+]i via the TRPC1 channel [49]. TRPC1 expression was increased after glutamate treatment and 2APB, a blocker of TRPC channels, significantly reduces cell death in hippocampal organotypic slice cultures. Moreover, knockdown of TRPC1 significantly improved cell survival upon exposure with glutamate. Taken together, these results suggest the causative role of TRPC1 in glutamate-induced cell death in hippocampus. In contrast, TRPC1 has also been shown to protect neurons from several extracellular stimuli [50-52]. Thus, TRPC channels could act as a “double- edged sword” since TRPC channels physiologically regulate neuronal development, whereas excessive Ca2+ influx especially upon glutamate treatment can induce neuronal degeneration. Furthermore, cellular toxicity is not simply a function of increased [Ca2+]i. For example, treatments with AMPA or KCl can increase [Ca2+]i up to 1–2 μM in cortical neurons, without causing toxicity [53]. Equally high Ca2+ loads are toxic when entering via the NMDA channels, but not when entering via the voltage-dependent Ca2+ channels [53], suggesting that the source of increased [Ca2+]i or the Ca2+ channel(s) themselves are critical in deciding the fate of the neuron.
TRPC Channels in Oxidative Stress
Overproduction of reactive oxygen species (ROS) and reactive nitrogen species (RNS) have been suggested to play crucial roles in the pathogenesis of several chronic neurological disorders including Alzheimer's disease, Parkinson's disease, schizophrenia or bipolar disorder and also stroke [54-57]. Several physiological pathways are responsible for the production of ROS including, respiration (generated by mitochondrial electron transport chain) and activation of the arachidonic acid cascade [58]. ROS could activate cell death processes directly by the oxidation of proteins, lipids and/or nucleic acids or could act as initiators or second messengers in the cell death process [59-61]. The production of ROS or free radicals is tightly controlled by cellular antioxidants and oxidative stress is usually associated with the imbalance between oxidants and antioxidants [60,62,63]. Importantly, during normal ageing, oxidants are increased that alter membrane permeability and modify the function of various cellular proteins and lipids [64,65]. This modification further leads to a wide variety of cellular damages, which are believed to be an early event in neurodegeneration [66,67]. Also, oxidative stress could result in cellular defects including a defect in ER Ca2+ uptake and Ca2+ efflux, thereby increasing [Ca2+]i and Ca2+ influx [68]. This increase in [Ca2+]i induces an exacerbation of oxidative stress [69], and activates several Ca2+-dependent enzymes including, calpain and the endonuclease pathways, which ultimately cause cytoskeleton alterations and cell death [70-72].
To date, four TRP family members have been identified that are influenced by oxidative stress, TRPC3, TRPC4, TRPM2 and TRPM7. It is likely that TRPC3 and TRPC4 are directly activated in response to oxidative stress [73,74]. The causative role of TRPC3 protein in oxidative stress was first documented by Balzer and colleagues [75]. The oxidant tert.-butylhydroperoxide (tBHP) completely depolarized endothelial cells by the activation of a TRP-related cationic current and expression of a dominant negative N-terminal splice variant of TRPC3. These results suggest that the TRPC protein determines endothelial redox sensitivity and suppression of TRPC could be an approach to the treatment of oxidative stress-induced vascular dysfunction. Moreover, overexpression of TRPC3 or TRPC4 in HEK293T cells showed an increase in basal membrane conductance upon tBHP treatment, which is mainly due to the influx of Na+ [76]. This suggest that TRPC3 and TRPC4 form redox sensitive cation channels during oxidative stress and could participate in Na+ loading and membrane depolarization. Furthermore, Rosker et al. reported that extracellular Na+ regulates TRPC3 channel-mediated Ca2+ influx in HEK293 cells [77]. Agonist stimulated Ca2+ entry through TRPC3 channels was greatly reduced when the extracellular Na2+ concentration was decreased to 5mM. Moreover, an inhibitor of the Na+/Ca2+ exchanger, KB-R9743, strongly suppressed TRPC3 mediated Ca2+ influx, but not TRPC3- mediated Na+ currents. This suggests that Ca2+ entry in TRPC3-expressing cells involves Na+/Ca2+ exchange in a reversed-mode. Activation of TRPC3-mediated cationic current, by oxidative stress, could also be achieved by tyrosine phosphorylation and and activation of phospholipase C [78]. The phospholipase C inhibitor, U73122, abolished oxidative stress induced-TRPC3 cationic current, which support the conclusion that phospholipase C is necessary for ROS-induced TRPC3 activation. ROS activates tyrosine kinase receptors (RTKs) by direct oxidation of specific cysteine residues or by the formation of specific intermolecular disulfide bonds with other molecules present within the complex [79-81]. Also inhibition of tyrosine kinase, specifically Src kinases, abolishes TRPC3 activation [82], suggesting that tyrosine kinase activation plays a crucial role in redox activation of TRPC3.
Lipid rafts also play a crucial role in the initiation of oxidative stress by causing aggregation and crosslinking of kinases, which combines ROS-generating oxidation machinery with ROS-sensitive cysteine residues required for ROS generation [80, 81]. Interestingly, almost all TRPC channels are present in lipid raft domains (LRDs) and it is believed that disruption of lipid rafts in the plasma membrane could have an impact on the function of the TRPC channels [83, 84] Studies from our laboratory and others have shown that mammalian TRPC proteins are assembled in a multiprotein complex that includes key proteins involved in Ca2+ signaling and that LRDs serve as a platform for this molecular assembly. Also, TRPC1 and TRPC3 signaling complexes are associated with caveolar LRDs in the plasma membrane [83]. Caveolin 1, a substrate for nonreceptor tyrosine kinase, has a crucial role in the localization of TRPC channels at the plasma membrane [83]. Under oxidative stress, c-Src kinase phosphorylates caveolin 1 at tyrosine 14 which affects downstream signaling targets [85, 86], that in turn could serve as a crucial step for TRPC channel localization and molecular assembly. Therefore, future studies are necessary to test whether disruption of caveolar LRDs affects TRPC function and signaling mechanisms in response to oxidative stress.
Moreover, Crouzin et al. reported that pretreatment with α-tocopherol mediates long lasting neuroprotective effect against Fe2+-induced oxidative damage by suppressing Ca2+ entry through TRP-like channels in cultured hippocampal neurons [87]. Treatment with FeSO4 induces hippocampal neuronal death by increasing the intracellular Ca2+ levels. Treating hippocampal neurons with Trolox significantly prevented intracellular Ca2+ elevation and cell death; suggesting that intracellular Ca2+ elevation by FeSO4 is due to oxidative stress mediated by Fe2+ ions. FeSO4 fails to increase intracellular Ca2+ level in Ca2+ free medium; this indicates that Ca2+ entry exclusively from the extracellular medium contributes towards Fe2+ mediated [Ca2+]i elevation. TRP-like nonspecific Ca2+ ion channel blockers, Ruthenium Red, La3+, and Gd3+, prevented Fe2+ -induced oxidative stress and toxicity. Interestingly, pretreatment with α-tocopherol prevents [Ca2+]i elevation induced by Fe2+ and suppress SOCE after metabotropic glutamate receptor stimulation. Due to the lack of specific pharmacological inhibitors, these authors fail failed to identify the specific TRPC channels responsible for Fe2+ induced Ca2+ influx. Although, this study supports the toxic role of TRPC-like channels in oxidative damage, further studies are necessary to identify the specific TRPC-like channels that are involved in neuronal death induced by oxidative stress. Identification of these targets would be essential to the discovery of specific therapeutic agents that are protective against oxidative stress-induced neurodegeneration. Furthermore, ORAI and ORAI3, which contributes towards SOCE, has been also shown to be activated by arachidonic acid and since arachidonic acid is critical for ROS production, these channels could also contribute towards oxidative stress.
TRPC Channels in NO Synthesis
Nitric oxide (NO) is an important signaling molecule that plays pivotal roles in many physiological processes such as vascular remodeling, platelet activation, and synaptic plasticity [88]. NO production is dependent on Ca2+ entry and since TRPC channels allow Ca2+ influx, they could contribute to the production of NO, which subsequently would stimulate downstream targets. In striatal neurons the TRPC channel blocker SKF-96365 or addition of 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA-AM) significantly decreased NO dependent gene regulation [89]. Additionally, NO also activates protein kinase G (PKG), which is known to specifically phosphorylate TRPC3 at two specific positions (Thr-11 and Ser-263). Phosphorylation of TRPC3 at these two sites inactivates TRPC3 channel and thus PKG likely functions to fine tune [Ca2+]i through the action of NO [90]. This decrease in [Ca2+]i could protect the cells from the detrimental effects of excessive [Ca2+]i and/or NO, since increased [Ca2+]i is important for NO production. It might be nice here to note the dynamic regulatory feedback looping that seems to exist in this system with the implications of tight regulation of Ca. Importantly, PKG phosphorylation sites, Thr-11 and Ser-263, are also conserved in other members of the TRPC3/6/7 subfamily, suggesting that PKG could also phosphorylate TRPC6 and TRPC7; however, it remains to be investigated if indeed TRPC6 and TRPC7 are phosphorylated by PKG and if they exhibit protection similar to that observed by TRPC3. Importantly, TRPC5 has been shown to be activated via cysteine S-nitrosylation and TRPC5 has been particularly shown to be sensitive to NO [91]. In neuronal cells, production of amyloid β stimulates NO production from the microglial cells, which could induce Ca2+ influx via the TRPC5 channel which might contribute to neuronal cell death [92]. Since as described above NO has both inhibitory and activating roles in regulating different TRPC channels, it NO might regulate process that are dependent on the expression of a particular TRPC channels. Some processes could be neurotoxic (activation of TRPC5 might induce cell death cascades) whereas other processes might be neuroprotective (activation of PKG and other downstream targets inhibit TRPC3, thereby protecting these cells).
TRPC Channels in CCL2 Signaling Against Tat Neurotoxicity
HIV infection leads to a variety of progressive neurological disorders, including HIV-associated dementia (HAD) [93,94]. HAD is characterized by deterioration of cognitive and motor functions and behavioral changes [95,96]. A recent study reported on a role for CCL2 (Chemokine (C-C motif) ligand 2, also known as monocyte chemoattractant protein-1) in neuroprotection against toxicity mediated by Trans-Activator of Trancription (Tat) in rat midbrain neurons [52]. Ca2+ influx through TRPC channel requires CCL2-mediated neuroprotection. CCL2 is a known activator of phospholipase C that results in IP3-receptor mediated Ca2+ release of the internal stores and simultaneously activate Ca2+ influx through TRPC channels. This in turn could lead to the activation of extracellular signal-regulated kinase (ERK) followed by the activation of cAMP response element-binding protein (CREB). Blocking of TRPC channels or specific knockdown of TRPC1 and TRPC5 resulted in the suppression of CCL2-induced neuroprotection, which was attributed to the prevention of CCL2-mediated intracellular Ca2+ elevation and ERK/CREB phosphorylation [52]. These results suggest that TRPC1 and TRPC5 could contribute towards neuroprotection against HIV.
TRPC Channels in Parkinson's Disease
Parkinson's disease (PD) is the second most common neurodegenerative disorder, which involves progressive degeneration of dopaminergic neurons present in the substantia nigra pars compacta (SNpc) region. Neurotoxins, such as 1-methyl, 4-phenyl pyridinium ion (MPP+), cause selective nigral dopaminergic lesions and Parkinsonian syndromes [97]. A host of pathogenic factors have been suggested to be responsible for the degeneration of dopaminergic neurons in the SNpc. These factors include the generation of free radicals, impairment of mitochondrial function, disturbances in Ca2+ homeostasis, and apoptosis of dopaminergic neurons [98, 99]. Since so many factors might be responsibe for PD, a common factor could be involved in the pathogenesis of PD. Ca2+, being a common link in all these processes raises the possibilities that Ca2+ channel function might be a common mechanism. Additionally, the role of Ca2+ in mediating apoptosis raises the possibility that Ca2+ channels may have a salutary effect on conditions such as PD in which apoptosis is the ultimate mode of cell death. Increase in intracellular Ca2+ concentration (primarily by Ca2+ entry) has been shown to be necessary to stimulate the release of dopamine (DA) in dopaminergic neurons of the SNpc [100-102], indicating the importance of Ca2+ in PD.
Decrease of Ca2+ in the endoplasmic reticulum (ER) is known to induce ER stress, which can activate cell death cascades [18-20]. Moreover, since Ca2+ influx is critical for refilling of the ER stores, which will prevent ER stress, SOCE is likely critical for neuronal function and survival. Moreover, TRPC1 and TRPC5 are highly expressed in SNpc region [102-104] (Table 2). Therefore, the significance of TRPC's in dopaminergic neuronal function and survival merits examination. Recent data suggest that TRPC1 functions as plasma membrane Ca2+ channels in dopaminergic neurons and the expression/localization of TRPC1 was inhibited/altered by compounds that induce PD-like symptoms. Importantly, overexpression of TRPC1 reduced neurotoxicity induced by MPP+, whereas protection of dopaminergic cells against MPP+ was significantly decreased upon expression of antisense TRPC1 cDNA or by the addition of a nonspecific TRPC channel blockers [50]. Surprisingly, activation of TRPC1 by thapsigargin or carbachol decreased MPP+ mediated neurotoxicity, which was partially dependent on external Ca2+ a finding that indicates that some of the protection exhibited by TRPC1 might be contributed by its Ca2+ influx ability [50]. TRPC1 overexpression also inhibited cytochrome c release from the mitochondria, an initial step in apoptosis. Interpretations of these data suggest that reduction in the cell surface expression of TRPC1 following MPP+ treatment may be involved in dopaminergic neurodegeneration. Furthermore, TRPC1 might inhibit degenerative apoptotic signaling to provide neuroprotection against PD-inducing agents [50-51]. Similar results were also obtained when dopaminergic cells (SH-SY5Y) were treated with an endogenous neurotoxin, salsolinol, which is also involved in the pathogenesis of PD [51]. Beside these interesting findings, no data are currently available to elaborate the current understanding of the pathophysiologies exerted by impaired Ca2+ influx upon loss of TRPC1 in dopaminergic neurodegeneration. Thus, it is important to characterize TRPC1 channel activity to better define the molecular pathways involved in the onset and progression of PD. It is hypothesized that conditions such as reduced activity or mutations in TRPC1 gene could engender a predisposition towards PD. Also, other TRPC channels that are expressed in substantia nigra could also be involved in excitotoxicity. Ultimately, treatments aimed towards modulating TRPC channel activity/expression could be helpful in the treatment of PD.
Table 2. Differential Expression of Human TRPC mRNA in CNS.
| CNS | Expression of Various Levels [147] |
|---|---|
| Amygdala | TRPC5>TRPC1>TRPC4>TRPC6>TRPC3 |
| Caudate nucleus | TRPC6>TRPC1>TRPC3>TRPC5>TRPC4 |
| Cerebellum | TRPC5>TRPC1>TRPC3>TRPC6>TRPC4 |
| Hippocampus | TRPC5>TRPC1>TRPC4>TRPC6>TRPC3 |
| Putamen | TRPC1>TRPC5>TRPC3>TRPC6>TRPC4 |
| Striatum | TRPC6>TRPC1>TRPC3>TRPC5>TRPC4 |
| Substantia Nigra | TRPC1>TRPC5>TRPC6>TRPC4>TRPC3 |
TRPC Channels in Alzheimer's Disease
Cytosolic Ca2+ concentration plays a critical role in cell survival in aging brain. Thus, altered Ca2+ homeostasis might account for a number of age-related brain dysfunctions including Alzheimer's disease. The momentous role of Ca2+ in Alzheimer's disease (AD) was first postulated by Khachaturian in 1987 [105]. In recent years, accumulating evidence suggest that altered Ca2+ homeostasis is one of the main factors responsible for neuronal death in AD [106-108]. Changes in membrane structure not only alters the function of Ca2+ channels including extrusion pumps and transporters, but also changes the Ca2+ binding proteins and proteins involved in ER Ca2+ handling, which could lead to the disruption of Ca2+ homeostasis and augment the vulnerability of neurons to excitotoxicity [107,108]. Inherited mutation in genes encoding for presenilin I and presenilin 2 (PS1 and PS2) account for up to 40% of the early onset cases of familial Alzheimer's disease (FAD) [109]. PS1, 2 are expressed in neurons throughout the brain where they appear to be localized largely to the ER in cell bodies and dendrites [110, 111]. Mutations in PS1 increase the production of amyloid peptides (Aβ 40/42) by γ-secretase cleavage of amyloid precursor protein. Aβ is believed to be a crucial mediator of neuronal degeneration and impaired cognitive function in AD [112,113]. Aβ interaction with the plasma membrane results in elevated [Ca2+]i and increased susceptibility of neurons to apoptosis [114, 115]. Also, mutant PS1 deregulates intracellular Ca2+ homeostasis by increasing the intracellular pools of Ca2+ and decreasing SOCE [116]; however, no information is currently available on the expression and function of TRPC channels in AD patients.
The mechanism(s) by which presenilin might increase ER Ca2+ pools has been suggested by Tu et al. [117]. These researchers showed that unprocessed presenilin holo-proteins form constitutive ER Ca2+ leak channels that were independent of γ-secretase activity. Several in vitro and in vivo studies further demonstrated that exaggerated ER Ca2+ signaling resulting from mutations in presenilins, leads to sensitization of PS-FAD neurons to Aβ and excitotoxicity through a Ca2+-dependent mechanism [114, 118]. Recently, Bojarski et al., reported that loss of PS1 or both PS1 and PS2 resulted in enhanced SOCE, while PS1 FAD mutations exert the opposite effect on Ca2+ influx by altering the cellular level of STIM proteins [119]. Additionally, Lessard et al [120] demonstrated that PS2 controls TRPC6-mediated Ca2+ entry in HEK293T cells. In their study, the dominant-negative mutant of PS2, PS2 (D263A), enhances Angiotensin (AngII)-induced Ca2+ entry without affecting agonist induced Ca2+ release whereas wild type PS2 and FAD-linked PS2 mutants, PS2(N141I) & PS2(M239V), failed to affect the agonist-induced Ca2+ entry and release in HEK293T cells, suggesting that the endogenous PS2 act as an inhibitor of Ca2+ entry. They further revealed that the co-expression of TRPC6 with wild-type PS2 or FAD-linked PS2 mutants completely abolished AngII-induced Ca2+ entry through TRPC6 but not AngII-induced Ca2+ release. Co-expression of PS2 or the FAD-linked PS2 mutants were able to activate TRPC6 by 1-oleoyl-2-acetyl-sn-glycerol (OAG) suggesting that the negative regulation of TRPC6 by PS2 is not due to the cleavage of TRPC6 by PS2. Also treatment with Aβ peptides (1-40 and 1-42) fails to amend carbachol (CCh)-induced- Ca2+ release and Ca2+ entry in TRPC6 overexpressing cells, suggesting that the inhibitory effect of PS2 and the FAD-linked PS2 mutants in TRPC6 are independent of Aβ peptide generation.
TRPC in Neurotransmitter Release and Cerebellar Ataxia
Ca2+ plays an important role in regulating a variety of neuronal processes, which includes excitability, neurotransmitter release, synaptic plasticity, and gene transcription. Like other cells, neurons also use both extracellular and intracellular sources of Ca2+ [22,107]. Although the role of voltage gated channels in the release of neurotransmitter is well established, other ion channels also directly contribute to neurosecretory processes [121, 122]. TRPC3, a non- selective Ca2+ channel invariably allows Na+ to enter the cell, which could partially depolarize the cells and activate the voltage-dependent Ca2+ channels. This combined Ca2+ influx would further contribute to the increase of [Ca2+]i and the spatio-temporally controlled elevations of [Ca2+]i in defined cell compartments. In addition, neurtrophic factors such as BDNF and NGF have also been shown to modulate neurotransmitter release [123, 124]. TRPC3 has been shown to be activated by both BDNF and NGF [45, 125], implicating TRPC3 in enhancements of neurotransmitter efflux. Mounting evidence also implicates intracellular Ca2+ stores in the regulation of neuronal transmitter release [121, 122]. Both TRPC3, and TRPC5 interact with the SNARE complex proteins, suggesting their potential role in the release of neurotransmitter [126]. Pre-synaptic introduction of IP3, cADPR and NAADP, which can activate TRPC channels, have also been demonstrated to stimulate neurotransmitter release at cholinergic synapses [127]. Thus, although much is known about the voltage dependent Ca2+ influx pathways, there is less information on the mechanisms exerted by TRPC channels. TRPC channels might function alone or in conjunction with these mechanisms in “fine-tuning” synaptic activity through direct actions at the pre-synaptic terminals.
The hereditary ataxias have been characterized by the degeneration of the cerebellum and its associated connections, which leads to the loss of balance and coordination. Purkinje neurons represent the sole output cell of the cerebellar cortex and play an important role in the normal function of the cerebellum such as fine-tuning movement and posture [128, 129]. The molecular mechanism(s) that triggers the loss of Purkinje cells in cerebellar ataxias remain incompletely understood and more than 50 different inherited forms of cerebellar ataxia have been identified so far [130]. In a recent study, Lim et al. hypothesized that if mutations in different genes lead to death of Purkinje neurons in all ataxias, then these mutations should be part of a common biological pathway that is critical for the survival of these cerebellar neurons [131]. Recently, Becker et al. reported that a point mutation in TRPC3, threonine to alanine amino acid change (T635A) in the highly conserved S4/S5 linker region, causes abnormal Purkinje cell degeneration and cerebellar ataxia in moonwalker (Mwk) mice [132]. Dentritic development was severely affected in mutant Purkinje cells, whereas TRPC3 mRNA or protein levels were the same in both wild-type and mutant cerebellum; the expression of TRPC1 was not altered in the cerebellum. Taken together, these results suggest that the Mwk mutation results in improper TRPC3 channel activity, which could be due to altered TRPC3 phosphorylation or its gating that could promote aberrant channel opening thereby leading to cell death.
TRPC4 in Axonal Regeneration
Recently, the role of TRPC4 in axonal regeneration in adult rat dorsal root ganglia (DRG) was reported by [133]. The expression of TRPC4 increases whereas TRPC1, 3, 6 and 7 remained unaltered after nerve injury induced by either sciatic nerve transection or intra-ganglionic microinjection of dibutyryl cAMP. The TRPC4 transcript peaked at day 2 but gradually decreased by day 7 and remained low on day 14. TRPC4 protein level increased at day 4 and 7, but was reduced relative to control levels by day 14 post nerve injury. In addition, TRPC4 transcript in ND7/23 and NDC cells, hybrid cell lines derived from neonatal DRG and neuroblastoma, increased upon NGF and dibutyryl cAMP -induced differentiation [133]. Suppression of TRPC4 by a specific small interfering RNA significantly reduced the length of neuritis in cultured DRG neurons [133]. Sh-RNA mediated knockdown of TRPC4 in differentiated ND7/23 cells resulted in the reduction of neurite length, which was rescued by TRPC4 overexpression [133]. Taken together, these results suggest that TRPC4 contributes to axonal regeneration after nerve injury. If these findings have generality, TRPC4 could be an important molecular target for potential regeneration therapies in patients suffering from traumatic brain injury.
TRPC in Growth Cone Development
Development of the nervous system requires tightly controlled and directed neuronal outgrowth. Although the underlying molecular mechanism(s) for neuronal growth are not fully appreciated, the importance of Ca2+- and Na+-permeable TRPC channels is apparent. The finding that TRPC channels have such roles is consistent with recent reports, which indicate that growth cone extension and turning is dependent on Ca2+ [134]. One of the TRPC channels involved in growth cone development is TRPC5. TRPC5 is predominantly expressed in the brain where it can form heterotetrameric complexes with TRPC1 and TRPC4 channels. TRPC5 channels have been shown to have an inhibitory role on neurite extension [135,136]. TRPC5 present in growth cones and early synapses determines the growth cone morphology and motility by complexing with growth cone enriched protein Stathmin 2 [136]. That stathmin 2 is required for packing TRPC5 onto vesicular packets intended for insertion into growth cones is suggested since TRPC5 is largely absent in growth cones of rat hippocampal neurons transfected with palmitoylation-defective stathmin 2 (Stmn2mut). Neurites length and filopodia, finger like projections characteristic of growth cones, were longer when compared with control in Stmn2mut and dominant-negative TRPC5 transfected cells [136]. Growth cone filopodium and neurite length was inversely correlated with stathmin and TRPC5 expression. Moreover, it has been shown that the neuronal Ca2+ sensor-1 (NCS-1) is essential for neurite growth. NCS-1 directly binds to the C-terminus of TRPC5, suggesting that TRPC5 could sequester NCS-1 partnership and have a role in inhibiting neuronal development. Based on these results TRPC5 -mediated Ca2+ entry could be regarded as an important determinant of axonal growth and growth cone morphology [136]. However, recent reports have also shown that other TRPC proteins such as TRPC1, TRPC3, and TRPC6 might also be involved in growth cone turning upon chemotropic stimulation, expanding the general role of TRPC channels in neurite extension and growth cone development. Moreover, since organ development is followed by the development of nervous system, TRPC channels might also be essential for organ development, especially the development of exocrine glands.
TRPC5 and Response to Fear
Recently, Riccio et al. reported on the important role of TRPC5 in amygdala function and fear-related behavior [137]. A high level of TRPC5-mRNA was present in the CA1, CA2, and CA3 regions of the hippocampus and dentate gyrus, areas that regulate fear-related behaviors through projections to the amygdala. Regions of auditory cortex, somatosensory cortex, the parietalinsular cortex and the perirhinal cortex also contain TRPC5 mRNA. Interestingly, TRPC5 knockout mice exhibited less anxious behaviors (decrease innate fear), in response to innately aversive stimuli, than wild type mice. Moreover, synaptic strength at afferent inputs to the amygdala is decreased in P10-P13 TRPC5 null mice. The deletion of TRPC5 showed no detectable effect on the firing properties of lateral nucleus of the amygdala. Moreover the basal synaptic transmissions in afferent inputs to the lateral amygdale in 4- to 5-week-old TRPC5 knockout mice were also not altered. In contrast, ablation of the TRPC5 gene affected mGluR- and/or cholecystokinin2 (CCK2) receptor mediated synaptic response. Taken together, these findings support that TRPC5 has an essential function in innate fear [137].
TRPC in Dendritic Morphology
Ca2+ and its downstream signaling cascades control the development of dendrites. Elevation in intracellular Ca2+ concentration could lead to changes in dendritic morphology [138,139]. Dendritic abnormalities are commonly associated with conditions resulting in mental retardation, such as Down syndrome [140] and Fragile × syndrome [141]. Several transcription factors are involved in the Ca2+ mediated changes in dentritic morphology [142, 143]. cAMP-response element binding protein (CREB) is one of the transcription factors that is involved in dentritic morphology and is known to be regulated by TRPC3 and TRPC6 [144]. TRPC6 is highly expressed in all regions of rat hippocampus (postnatal day 14) and colocalizes with Microtubule-associated protein 2 (MAP2), a known neuronal marker in proximal dentrites of CA1 pyramidal neurons. Knockdown of TRPC6 in hippocampal neurons significantly decreased total dentritic length, whereas introduction of human TRPC6 promoted dentritic growth through the calmodulin dependent protein kinase IV (CaMKIV)-CREB pathway. Also chelating extracellular Ca2+ by EGTA or blocking of Ca2+ influx by Cd2+ prevented TRPC6-induced dentritic growth. Moreover, an increase in both basal and apical dentritic length and phosphorylation of CaMKIV and CREB was also increased in TRPC6 transgenic mice [144]. Taken together, these findings support the idea that Ca2+ influx through TRPC6 is vital for dentritic growth.
Future Challenges
Tremendous advances have been made in the past ten years in exploring the implication of TRPC channels especially with regard to neuronal survival, differentiation, and several other essential functions as discussed in this review. Although several unanswered question remained to be addressed, it has been hypothesized that Ca2+ influx via the TRPC channels and also perhaps by ORAI channels, have a significant role in the onset/progression of many neurodegenerative diseases. Thus, targeting Ca2+ entry (both inhibition as well as activation) through these channels could be critical for maintaining normal physiological functions. Recent research indicates that Ca2+ is critical for Huntington disease; however the significance of TRPC channels in this disease has not yet been identified. Furthermore, recent findings have implicated STIM1 as the regulator for SOCE making it interesting to see if STIM1 function/expression is altered in neurological diseases. Although the current data with regard to the function of TRPC in neuronal cells is overwhelming, significant information including that from human samples is still missing. Nevertheless it is anticipated that in the coming years more clinically-oriented research will be performed, which will eventually define the role of TRPC proteins in several of these diseases. A significant challenge is that specific activators and inhibitors for a specific TRPCs have not yet been identified.
Acknowledgments
We duly acknowledge the grant support from the National Science foundation (0548733) and the national Institutes of Health (DE017102, 5P20RR017699).
Abbreviations
- Aβ
Aamyloid peptide beta
- AngII
Angiotensin
- BAPTA-A
1,2-Bis(o-aminophenoxy)ethane-N,N,N′,N′-tetra acetic
- CCL2
Chemokine (C-C motif) ligand 2, also known as monocyte chemoattractant protein-1
- CREB
cAMP response element-binding protein
- DA
dopamine
- DAG
Diacylglycerol
- DRG
Dorsal root ganglia
- ER
Endoplasmic reticulum
- FAD
Familial Alzheimer's disease
- HAD
HIV-associated dementia
- MPP
1-Methyl, 4-phenyl pyridinium
- NCS-1
Neuronal Ca2+ sensor-1
- ORAI
Gene/protein
- PD
Parkinson's disease
- PKG
Protein kinase G
- PM
Plasma membrane
- PS
Presenilin
- ROS
Reactive oxygen species
- SOCE
Store operated calcium entry
- STIM
Stromal interaction molecule
- SNpc
Substantia nigra pars compacta
- TRPC
Transient receptor potential conical
References
- 1.Marty A. The physiological role of calcium-dependent channels. Trends Neurosci. 1989;12:420–424. doi: 10.1016/0166-2236(89)90090-8. [DOI] [PubMed] [Google Scholar]
- 2.Malenka RC, Kauer JA, Perkel DJ, Nicoll RA. The impact of postsynaptic calcium on synaptic transmission--its role in long-term potentiation. Trends Neurosci. 1989;12:444–450. doi: 10.1016/0166-2236(89)90094-5. [DOI] [PubMed] [Google Scholar]
- 3.Llinas RR. The intrinsic electrophysiological properties of mammalian neurons: insights into central nervous system function. Science. 1988;242:1654–1664. doi: 10.1126/science.3059497. [DOI] [PubMed] [Google Scholar]
- 4.McCormack JG, Halestrap AP, Denton RM. Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiol Rev. 1990;70:391–425. doi: 10.1152/physrev.1990.70.2.391. [DOI] [PubMed] [Google Scholar]
- 5.Orrenius S, Zhivotovsky B, Nicotera P. Regulation of cell death: the calcium-apoptosis link. Nat Rev Mol Cell Biol. 2003;4:552–565. doi: 10.1038/nrm1150. [DOI] [PubMed] [Google Scholar]
- 6.Bading H, Ginty DD, Greenberg ME. Regulation of gene expression in hippocampal neurons by distinct calcium signaling pathways. Science. 1993;260:181–186. doi: 10.1126/science.8097060. [DOI] [PubMed] [Google Scholar]
- 7.Bezprozvanny I. Calcium signaling and neurodegenerative diseases. Trends Mol Med. 2009;15:89–100. doi: 10.1016/j.molmed.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marambaud P, Dreses-Werringloer U, Vingtdeux V. Calcium signaling in neurodegeneration. Mol Neurodegener. 2009;4:20–34. doi: 10.1186/1750-1326-4-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aarts MM, Tymianski M. TRPM7 and ischemic CNS injury. Neuroscientist. 2005;11:116–123. doi: 10.1177/1073858404272966. [DOI] [PubMed] [Google Scholar]
- 10.Berridge MJ, Bootman MD, Lipp P. Calcium--a life and death signal. Nature. 1998;395:645–648. doi: 10.1038/27094. [DOI] [PubMed] [Google Scholar]
- 11.Dubinsky JM. Intracellular calcium levels during the period of delayed excitotoxicity. J Neurosci. 1993;13:623–631. doi: 10.1523/JNEUROSCI.13-02-00623.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Herman B, Gores GJ, Nieminen AL, Kawanishi T, Harman A, Lemasters JJ. Calcium and pH in anoxic and toxic injury. Crit Rev Toxicol. 1990;21:127–148. doi: 10.3109/10408449009089876. [DOI] [PubMed] [Google Scholar]
- 13.Choi DW. Calcium-mediated neurotoxicity: relationship to specific channel types and role in ischemic damage. Trends Neurosci. 1988;11:465–469. doi: 10.1016/0166-2236(88)90200-7. [DOI] [PubMed] [Google Scholar]
- 14.Arundine M, Tymianski M. Molecular mechanisms of calcium-dependent neurodegeneration in excitotoxicity. Cell Calcium. 2003;34:325–337. doi: 10.1016/s0143-4160(03)00141-6. [DOI] [PubMed] [Google Scholar]
- 15.Putney JW., Jr A model for receptor-regulated calcium entry. Cell Calcium. 1986;7:1–12. doi: 10.1016/0143-4160(86)90026-6. [DOI] [PubMed] [Google Scholar]
- 16.Parekh AB, Putney JW., Jr Store-operated calcium channels. Physiol Rev. 2005;85:757–810. doi: 10.1152/physrev.00057.2003. [DOI] [PubMed] [Google Scholar]
- 17.Parekh AB, Penner R. Store depletion and calcium influx. Physiol Rev. 1997;77:901–930. doi: 10.1152/physrev.1997.77.4.901. [DOI] [PubMed] [Google Scholar]
- 18.Sattler R, Charlton MP, Hafner M, Tymianski M. Distinct influx pathways, not calcium load, determine neuronal vulnerability to calcium neurotoxicity. J Neurochem. 1998;71:2349–2364. doi: 10.1046/j.1471-4159.1998.71062349.x. [DOI] [PubMed] [Google Scholar]
- 19.Paschen W, Doutheil J, Gissel C, Treiman M. Depletion of neuronal endoplasmic reticulum calcium stores by thapsigargin: effect on protein synthesis. J Neurochem. 1996;67:1735–1743. doi: 10.1046/j.1471-4159.1996.67041735.x. [DOI] [PubMed] [Google Scholar]
- 20.Yoshida I, Monji A, Tashiro K, Nakamura K, Inoue R, Kanba S. Depletion of intracellular Ca2+ store itself may be a major factor in thapsigargin-induced ER stress and apoptosis in PC12 cells. Neurochem Int. 2006;48:696–702. doi: 10.1016/j.neuint.2005.12.012. [DOI] [PubMed] [Google Scholar]
- 21.Putney JW. Physiological mechanisms of TRPC activation. Pflugers Arch. 2005;451:29–34. doi: 10.1007/s00424-005-1416-4. [DOI] [PubMed] [Google Scholar]
- 22.Putney JW., Jr Capacitative calcium entry in the nervous system. Cell Calcium. 2003;34:339–344. doi: 10.1016/s0143-4160(03)00143-x. [DOI] [PubMed] [Google Scholar]
- 23.Salido GM, Sage SO, Rosado JA. TRPC channels and store-operated Ca2+ entry. Biochim Biophys Acta. 2009;1793:223–230. doi: 10.1016/j.bbamcr.2008.11.001. [DOI] [PubMed] [Google Scholar]
- 24.Cahalan MD. STIMulating store-operated Ca2+ entry. Nat Cell Biol. 2009;11:669–677. doi: 10.1038/ncb0609-669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feske S, Gwack Y, Prakriya M, Srikanth S, Puppel SH, Tanasa B, Hogan PG, Lewis RS, Daly M, Rao A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature. 2006;441:179–185. doi: 10.1038/nature04702. [DOI] [PubMed] [Google Scholar]
- 26.Vig M, Peinelt C, Beck A, Koomoa DL, Rabah D, Koblan-Huberson M, Kraft S, Turner H, Fleig A, Penner R, Kinet JP. CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science. 2006;312:1220–1223. doi: 10.1126/science.1127883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Prakriya M, Feske S, Gwack Y, Srikanth S, Rao A, Hogan PG. Orai1 is an essential pore subunit of the CRAC channel. Nature. 2006;443:230–233. doi: 10.1038/nature05122. [DOI] [PubMed] [Google Scholar]
- 28.Zhang SL, Yeromin AV, Zhang XH, Yu Y, Safrina O, Penna A, Roos J, Stauderman KA, Cahalan MD. Genome-wide RNAi screen of Ca2+ influx identifies genes that regulate Ca2+ release-activated Ca2+ channel activity. Proc Natl Acad Sci USA. 2006;103:9357–9362. doi: 10.1073/pnas.0603161103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ong HL, Cheng KT, Liu X, Bandyopadhyay BC, Paria BC, Soboloff J, Pani B, Gwack Y, Srikanth S, Singh BB, Gill DL, Ambudkar IS. Dynamic assembly of TRPC1-STIM1-Orai1 ternary complex is involved in store-operated calcium influx. Evidence for similarities in store-operated and calcium release-activated calcium channel components. J Biol Chem. 2007;282:9105–9116. doi: 10.1074/jbc.M608942200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liao Y, Erxleben C, Abramowitz J, Flockerzi V, Zhu MX, Armstrong DL, Birnbaumer L. Functional interactions among Orai1, TRPCs, and STIM1 suggest a STIM-regulated heteromeric Orai/TRPC model for SOCE/Icrac channels. Proc Natl Acad Sci USA. 2008;105:2895–2900. doi: 10.1073/pnas.0712288105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liao Y, Plummer NW, George MD, Abramowitz J, Zhu MX, Birnbaumer L. A role for Orai in TRPC-mediated Ca2+ entry suggests that a TRPC:Orai complex may mediate store and receptor operated Ca2+ entry. Proc Natl Acad Sci USA. 2009;106:3202–3206. doi: 10.1073/pnas.0813346106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liou J, Kim ML, Heo WD, Jones JT, Myers JW, Ferrell JE, Jr, Meyer T. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr Biol. 2005;15:1235–1241. doi: 10.1016/j.cub.2005.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roos J, DiGregorio PJ, Yeromin AV, Ohlsen K, Lioudyno M, Zhang S, Safrina O, Kozak JA, Wagner SL, Cahalan MD, Velicelebi G, Stauderman KA. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J Cell Biol. 2005;169:435–445. doi: 10.1083/jcb.200502019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zeng W, Yuan JP, Kim MS, Choi YJ, Huang GN, Worley PF, Muallem S. STIM1 gates TRPC channels, but not Orai1, by electrostatic interaction. Mol Cell. 2008;32:439–448. doi: 10.1016/j.molcel.2008.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Birnbaumer L. The TRPC class of ion channels: a critical review of their roles in slow, sustained increases in intracellular Ca2+ concentrations. Annu Rev Pharmacol Toxicol. 2009;49:395–426. doi: 10.1146/annurev.pharmtox.48.113006.094928. [DOI] [PubMed] [Google Scholar]
- 36.Clapham DE. Calcium signaling. Cell. 2007;131:1047–1058. doi: 10.1016/j.cell.2007.11.028. [DOI] [PubMed] [Google Scholar]
- 37.Beech DJ, Bahnasi YM, Dedman AM, Al-Shawaf E. TRPC channel lipid specificity and mechanisms of lipid regulation. Cell Calcium. 2009;45:583–588. doi: 10.1016/j.ceca.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tai Y, Feng S, Du W, Wang Y. Functional roles of TRPC channels in the developing brain. Pflugers Arch. 2009;458:283–289. doi: 10.1007/s00424-008-0618-y. [DOI] [PubMed] [Google Scholar]
- 39.Orrenius S, Nicotera P. The calcium ion and cell death. J Neural Transm Suppl. 1994;43:1–11. [PubMed] [Google Scholar]
- 40.Paschen W. Role of calcium in neuronal cell injury: which subcellular compartment is involved? Brain Res Bull. 2000;53:409–413. doi: 10.1016/s0361-9230(00)00369-5. [DOI] [PubMed] [Google Scholar]
- 41.Beech DJ. TRPC1: store-operated channel and more. Pflugers Arch. 2005;451:53–60. doi: 10.1007/s00424-005-1441-3. [DOI] [PubMed] [Google Scholar]
- 42.Strubing C, Krapivinsky G, Krapivinsky L, Clapham DE. Formation of novel TRPC channels by complex subunit interactions in embryonic brain. J Biol Chem. 2003;278:39014–39019. doi: 10.1074/jbc.M306705200. [DOI] [PubMed] [Google Scholar]
- 43.Fiorio Pla A, Maric D, Brazer SC, Giacobini P, Liu X, Chang YH, Ambudkar IS, Barker JL. Canonical transient receptor potential 1 plays a role in basic fibroblast growth factor (bFGF)/FGF receptor-1-induced Ca2+ entry and embryonic rat neural stem cell proliferation. J Neurosci. 2005;25:2687–2701. doi: 10.1523/JNEUROSCI.0951-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu X, Zagranichnaya TK, Gurda GT, Eves EM, Villereal ML. A TRPC1/TRPC3-mediated increase in store-operated calcium entry is required for differentiation of H19-7 hippocampal neuronal cells. J Biol Chem. 2004;279:43392–43402. doi: 10.1074/jbc.M408959200. [DOI] [PubMed] [Google Scholar]
- 45.Li HS, Xu XZ, Montell C. Activation of a TRPC3-dependent cation current through the neurotrophin BDNF. Neuron. 1999;24:261–273. doi: 10.1016/s0896-6273(00)80838-7. [DOI] [PubMed] [Google Scholar]
- 46.Tong Q, Hirschler-Laszkiewicz I, Zhang W, Conrad K, Neagley DW, Barber DL, Cheung JY, Miller BA. TRPC3 is the erythropoietin-regulated calcium channel in human erythroid cells. J Biol Chem. 2008;283:10385–10395. doi: 10.1074/jbc.M710231200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mattson MP, Guthrie PB, Kater SB. Intrinsic factors in the selective vulnerability of hippocampal pyramidal neurons. Prog Clin Biol Res. 1989;317:333–351. [PubMed] [Google Scholar]
- 48.Arundine M, Tymianski M. Molecular mechanisms of glutamate-dependent neurodegeneration in ischemia and traumatic brain injury. Cell Mol Life Sci. 2004;61:657–668. doi: 10.1007/s00018-003-3319-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Narayanan KL, Irmady K, Subramaniam S, Unsicker K, von Bohlen und Halbach O. Evidence that TRPC1 is involved in hippocampal glutamate-induced cell death. Neurosci Lett. 2008;446:117–122. doi: 10.1016/j.neulet.2008.09.034. [DOI] [PubMed] [Google Scholar]
- 50.Bollimuntha S, Singh BB, Shavali S, Sharma SK, Ebadi M. TRPC1-mediated inhibition of 1-methyl-4-phenylpyridinium ion neurotoxicity in human SH-SY5Y neuroblastoma cells. J Biol Chem. 2005;280:2132–2140. doi: 10.1074/jbc.M407384200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bollimuntha S, Ebadi M, Singh BB. TRPC1 protects human SH-SY5Y cells against salsolinol-induced cytotoxicity by inhibiting apoptosis. Brain Res. 2006;1099:141–149. doi: 10.1016/j.brainres.2006.04.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yao H, Peng F, Dhillon N, Callen S, Bokhari S, Stehno-Bittel L, Ahmad SO, Wang JQ, Buch S. Involvement of TRPC channels in CCL2-mediated neuroprotection against tat toxicity. J Neurosci. 2009;29:1657–1669. doi: 10.1523/JNEUROSCI.2781-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen Q, Surmeier DJ, Reiner A. NMDA and non-NMDA receptor-mediated excitotoxicity are potentiated in cultured striatal neurons by prior chronic depolarization. Exp Neurol. 1999;159:283–296. doi: 10.1006/exnr.1999.7135. [DOI] [PubMed] [Google Scholar]
- 54.Behl C. Amyloid beta-protein toxicity and oxidative stress in Alzheimer's disease. Cell Tissue Res. 1997;290:471–480. doi: 10.1007/s004410050955. [DOI] [PubMed] [Google Scholar]
- 55.Dexter DT, Holley AE, Flitter WD, Slater TF, Wells FR, Daniel SE, Lees AJ, Jenner P, Marsden CD. Increased levels of lipid hydroperoxides in the parkinsonian substantia nigra: an HPLC and ESR study. Mov Disord. 1994;9:92–97. doi: 10.1002/mds.870090115. [DOI] [PubMed] [Google Scholar]
- 56.Kuloglu M, Ustundag B, Atmaca M, Canatan H, Tezcan AE, Cinkilinc N. Lipid peroxidation and antioxidant enzyme levels in patients with schizophrenia and bipolar disorder. Cell Biochem Funct. 2002;20:171–175. doi: 10.1002/cbf.940. [DOI] [PubMed] [Google Scholar]
- 57.Hou ST, MacManus JP. Molecular mechanisms of cerebral ischemia-induced neuronal death. Int Rev Cytol. 2002;221:93–148. doi: 10.1016/s0074-7696(02)21011-6. [DOI] [PubMed] [Google Scholar]
- 58.Sies H. Oxidative stress: oxidants and antioxidants. Exp Physiol. 1997;82:291–295. doi: 10.1113/expphysiol.1997.sp004024. [DOI] [PubMed] [Google Scholar]
- 59.Nakamura T, Lipton SA. Cell death: protein misfolding and neurodegenerative diseases. Apoptosis. 2009;14:455–468. doi: 10.1007/s10495-008-0301-y. [DOI] [PubMed] [Google Scholar]
- 60.Chandra J, Samali A, Orrenius S. Triggering and modulation of apoptosis by oxidative stress. Free Radic Biol Med. 2000;29:323–333. doi: 10.1016/s0891-5849(00)00302-6. [DOI] [PubMed] [Google Scholar]
- 61.Gopalakrishna R, Jaken S. Protein kinase C signaling and oxidative stress. Free Radic Biol Med. 2000;28:1349–1361. doi: 10.1016/s0891-5849(00)00221-5. [DOI] [PubMed] [Google Scholar]
- 62.Berlett BS, Stadtman ER. Protein oxidation in aging, disease, and oxidative stress. J Biol Chem. 1997;272:20313–20316. doi: 10.1074/jbc.272.33.20313. [DOI] [PubMed] [Google Scholar]
- 63.Bast A, Haenen GR, Doelman C. Oxidants and antioxidants: state of the art. J Am J Med. 1991;91:2S–13S. doi: 10.1016/0002-9343(91)90278-6. [DOI] [PubMed] [Google Scholar]
- 64.Nunomura A, Perry G, Aliev G, Hirai K, Takeda A, Balraj EK, Jones PK, Ghanbari H, Wataya T, Shimohama S, Chiba S, Atwood CS, Petersen RB, Smith MA. Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol. 2001;60:759–767. doi: 10.1093/jnen/60.8.759. [DOI] [PubMed] [Google Scholar]
- 65.Cutler RG, Kelly J, Storie K, Pedersen WA, Tammara A, Hatanpaa K, Troncoso JC, Mattson MP. Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer's disease. Proc Natl Acad Sci USA. 2004;101:2070–2075. doi: 10.1073/pnas.0305799101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Moreira PI, Smith MA, Zhu X, Nunomura A, Castellani RJ, Perry G. Oxidative stress and neurodegeneration. Ann N Y Acad Sci. 2005;1043:545–552. doi: 10.1196/annals.1333.062. [DOI] [PubMed] [Google Scholar]
- 67.Perry G, Nunomura A, Hirai K, Takeda A, Aliev G, Smith MA. Oxidative damage in Alzheimer's disease: the metabolic dimension. Int J Dev Neurosci. 2000;18:417–421. doi: 10.1016/s0736-5748(00)00006-x. [DOI] [PubMed] [Google Scholar]
- 68.Orrenius S, Zhivotovsky B, Nicotera P. Regulation of cell death: the calcium-apoptosis link. Nat Rev Mol Cell Biol. 2003;4:552–565. doi: 10.1038/nrm1150. [DOI] [PubMed] [Google Scholar]
- 69.Orrenius S, McConkey DJ, Bellomo G, Nicotera P. Role of Ca2+ in toxic cell killing. Trends Pharmacol Sci. 1989;10:281–285. doi: 10.1016/0165-6147(89)90029-1. [DOI] [PubMed] [Google Scholar]
- 70.Ermak G, Davies KJ. Calcium and oxidative stress: from cell signaling to cell death. Mol Immunol. 2002;38:713–721. doi: 10.1016/s0161-5890(01)00108-0. [DOI] [PubMed] [Google Scholar]
- 71.Herson PS, Lee K, Pinnock RD, Hughes J, Ashford ML. Hydrogen peroxide induces intracellular calcium overload by activation of a non-selective cation channel in an insulin-secreting cell line. J Biol Chem. 1999;274:833–841. doi: 10.1074/jbc.274.2.833. [DOI] [PubMed] [Google Scholar]
- 72.Nicotera P, Bellomo G, Orrenius S. Calcium-mediated mechanisms in chemically induced cell death. Annu Rev Pharmacol Toxicol. 1992;32:449–470. doi: 10.1146/annurev.pa.32.040192.002313. [DOI] [PubMed] [Google Scholar]
- 73.Aarts MM, Tymianski M. TRPMs and neuronal cell death. Pflugers Arch. 2005;451:243–249. doi: 10.1007/s00424-005-1439-x. [DOI] [PubMed] [Google Scholar]
- 74.Miller BA. The role of TRP channels in oxidative stress-induced cell death. J Membr Biol. 2006;209:31–41. doi: 10.1007/s00232-005-0839-3. [DOI] [PubMed] [Google Scholar]
- 75.Balzer M, Lintschinger B, Groschner K. Evidence for a role of Trp proteins in the oxidative stress-induced membrane conductances of porcine aortic endothelial cells. Cardiovasc Res. 1999;42:543–549. doi: 10.1016/s0008-6363(99)00025-5. [DOI] [PubMed] [Google Scholar]
- 76.Poteser M, Graziani A, Rosker C, Eder P, Derler I, Kahr H, Zhu MX, Romanin C, Groschner K. TRPC3 and TRPC4 associate to form a redox-sensitive cation channel: evidence for expression of native TRPC3-TRPC4 heteromeric channels in endothelial cells. J Biol Chem. 2006;281:13588–13595. doi: 10.1074/jbc.M512205200. [DOI] [PubMed] [Google Scholar]
- 77.Rosker C, Graziani A, Lukas M, Eder P, Zhu MX, Romanin C, Groschner K. Ca2+ signaling by TRPC3 involves Na(+) entry and local coupling to the Na+/Ca2+ exchanger. J Biol Chem. 2004;279:13696–13704. doi: 10.1074/jbc.M308108200. [DOI] [PubMed] [Google Scholar]
- 78.Groschner K, Rosker C, Lukas M. Role of TRP channels in oxidative stress. Novartis Found Symp. 2004;258:222–230. [PubMed] [Google Scholar]
- 79.Giannoni E, Buricchi F, Raugei G, Ramponi G, Chiarugi P. Intracellular reactive oxygen species activate Src tyrosine kinase during cell adhesion and anchorage-dependent cell growth. Mol Cell Biol. 2005;25:6391–6403. doi: 10.1128/MCB.25.15.6391-6403.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nakashima I, Kato M, Akhand AA, Suzuki H, Takeda K, Hossain K, Kawamoto Y. Redox-linked signal transduction pathways for protein tyrosine kinase activation. Antioxid Redox Signal. 2002;4:517–531. doi: 10.1089/15230860260196326. [DOI] [PubMed] [Google Scholar]
- 81.Rhee SG, Yang KS, Kang SW, Woo HA, Chang TS. Controlled elimination of intracellular H(2)O(2): regulation of peroxiredoxin, catalase, and glutathione peroxidase via post-translational modification. Antioxid Redox Signal. 2005;7:619–626. doi: 10.1089/ars.2005.7.619. [DOI] [PubMed] [Google Scholar]
- 82.Vazquez G, Wedel BJ, Kawasaki BT, Bird GS, Putney JW., Jr Obligatory role of Src kinase in the signaling mechanism for TRPC3 cation channels. J Biol Chem. 2004;279:40521–40528. doi: 10.1074/jbc.M405280200. [DOI] [PubMed] [Google Scholar]
- 83.Ambudkar IS, Brazer SC, Liu X, Lockwich T, Singh B. Plasma membrane localization of TRPC channels: role of caveolar lipid rafts. Novartis Found Symp. 2004;258:63–70. [PubMed] [Google Scholar]
- 84.Pani B, Singh BB. Lipid rafts/caveolae as microdomains of calcium signaling. Cell Calcium. 2009;45:625–633. doi: 10.1016/j.ceca.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Parat MO, Fox PL. Oxidative stress, caveolae and caveolin-1. Subcell Biochem. 2004;37:425–441. doi: 10.1007/978-1-4757-5806-1_13. [DOI] [PubMed] [Google Scholar]
- 86.Sanguinetti AR, Mastick CC. c-Abl is required for oxidative stress-induced phosphorylation of caveolin-1 on tyrosine 14. Cell Signal. 2003;15:289–298. doi: 10.1016/s0898-6568(02)00090-6. [DOI] [PubMed] [Google Scholar]
- 87.Crouzin N, de Jesus Ferreira MC, Cohen-Solal C, Aimar RF, Vignes M, Guiramand J. Alpha-tocopherol-mediated long-lasting protection against oxidative damage involves an attenuation of calcium entry through TRP-like channels in cultured hippocampal neurons. Free Radic Biol Med. 2007;42:1326–1337. doi: 10.1016/j.freeradbiomed.2007.01.032. [DOI] [PubMed] [Google Scholar]
- 88.Tiruppathi C, Minshall RD, Paria BC, Vogel SM, Malik AB. Role of Ca2+ signaling in the regulation of endothelial permeability. Vascul Pharmacol. 2002;39:173–185. doi: 10.1016/s1537-1891(03)00007-7. [DOI] [PubMed] [Google Scholar]
- 89.Gokce O, Runne H, Kuhn A, Luthi-Carter R. Short-term striatal gene expression responses to brain-derived neurotrophic factor are dependent on MEK and ERK activation. PLoS One. 2009;4:e5292. doi: 10.1371/journal.pone.0005292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yao X. TRPC, cGMP-dependent protein kinases and cytosolic Ca2+ Handb Exp Pharmacol. 2007;179:527–540. doi: 10.1007/978-3-540-34891-7_31. [DOI] [PubMed] [Google Scholar]
- 91.Chen J, Crossland RF, Noorani MM, Marrelli SP. Am J Physiol Heart Circ Physiol. 2009;297:H417. doi: 10.1152/ajpheart.01130.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yamamoto S, Wajima T, Hara Y, Nishida M, Mori Y. Transient receptor potential channels in Alzheimer's disease. Biochim Biophys Acta. 2007;1772:958–967. doi: 10.1016/j.bbadis.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 93.Chakrabarti L, Hurtrel M, Maire MA, Vazeux R, Dormont D, Montagnier L, Hurtrel B. Early viral replication in the brain of SIV-infected rhesus monkeys. Am J Pathol. 1991;139:1273–1280. [PMC free article] [PubMed] [Google Scholar]
- 94.Davis LE, Hjelle BL, Miller VE, Palmer DL, Llewellyn AL, Merlin TL, Young SA, Mills RG, Wachsman W, Wiley CA. Early viral brain invasion in iatrogenic human immunodeficiency virus infection. Neurology. 1992;42:1736–1739. doi: 10.1212/wnl.42.9.1736. [DOI] [PubMed] [Google Scholar]
- 95.Navia BA, Jordan BD, Price RW. The AIDS dementia complex: I. Clinical features. Ann Neurol. 1986;19:517–524. doi: 10.1002/ana.410190602. [DOI] [PubMed] [Google Scholar]
- 96.Price RW, Brew B, Sidtis J, Rosenblum M, Scheck AC, Cleary P. The brain in AIDS: central nervous system HIV-1 infection and AIDS dementia complex. Science. 1988;239:586–592. doi: 10.1126/science.3277272. [DOI] [PubMed] [Google Scholar]
- 97.Javitch JA, D'Amato RJ, Strittmatter SM, Snyder SH. Parkinsonism-inducing neurotoxin, N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine: uptake of the metabolite N-methyl-4-phenylpyridine by dopamine neurons explains selective toxicity. Proc Natl Acad Sci USA. 1985;82:2173–2177. doi: 10.1073/pnas.82.7.2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lotharius J, Dugan LL, O'Malley KL. Distinct mechanisms underlie neurotoxin-mediated cell death in cultured dopaminergic neurons. J Neurosci. 1999;19:1284–1293. doi: 10.1523/JNEUROSCI.19-04-01284.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hoffman AF, Gerhardt GA. Differences in pharmacological properties of dopamine release between the substantia nigra and striatum: an in vivo electrochemical study. J Pharmacol Exp Ther. 1999;289:455–463. [PubMed] [Google Scholar]
- 100.Chen BT, Rice ME. Novel Ca2+ dependence and time course of somatodendritic dopamine release: substantia nigra versus striatum. J Neurosci. 2001;21:7841–7847. doi: 10.1523/JNEUROSCI.21-19-07841.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Patel JC, Witkovsky P, Avshalumov MV, Rice ME. Mobilization of calcium from intracellular stores facilitates somatodendritic dopamine release. J Neurosci. 2009;29:6568–6579. doi: 10.1523/JNEUROSCI.0181-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tozzi A, Bengtson CP, Longone P, Carignani C, Fusco FR, Bernardi G, Mercuri NB. Involvement of transient receptor potential-like channels in responses to mGluR-I activation in midbrain dopamine neurons. Eur J Neurosci. 2003;18:2133–2145. doi: 10.1046/j.1460-9568.2003.02936.x. [DOI] [PubMed] [Google Scholar]
- 103.Kim SJ, Kim YS, Yuan JP, Petralia RS, Worley PF, Linden DJ. Activation of the TRPC1 cation channel by metabotropic glutamate receptor mGluR1. Nature. 2003;426:285–291. doi: 10.1038/nature02162. [DOI] [PubMed] [Google Scholar]
- 104.Martorana A, Giampa C, DeMarch Z, Viscomi MT, Patassini S, Sancesario G, Bernardi G, Fusco FR. Distribution of TRPC1 receptors in dendrites of rat substantia nigra: a confocal and electron microscopy study. Eur J Neurosci. 2006;24:732–738. doi: 10.1111/j.1460-9568.2006.04932.x. [DOI] [PubMed] [Google Scholar]
- 105.Khachaturian ZS. Hypothesis on the regulation of cytosol calcium concentration and the aging brain. Neurobiol Aging. 1987;8:345–346. doi: 10.1016/0197-4580(87)90073-x. [DOI] [PubMed] [Google Scholar]
- 106.Small DH. Dysregulation of calcium homeostasis in Alzheimer's disease. Neurochem Res. 2009;34:1824–1829. doi: 10.1007/s11064-009-9960-5. [DOI] [PubMed] [Google Scholar]
- 107.Michaelis ML, Foster CT, Jayawickreme C. Regulation of calcium levels in brain tissue from adult and aged rats. Mech Ageing Dev. 1992;62:291–306. doi: 10.1016/0047-6374(92)90114-s. [DOI] [PubMed] [Google Scholar]
- 108.Arispe N, Rojas E, Pollard HB. Alzheimer disease amyloid beta protein forms calcium channels in bilayer membranes: blockade by tromethamine and aluminum. Proc Natl Acad Sci USA. 1993;90:567–571. doi: 10.1073/pnas.90.2.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Katzman R. Editorial: The prevalence and malignancy of Alzheimer disease: a major killer. Arch Neurol. 1976;33:217–218. doi: 10.1001/archneur.1976.00500040001001. [DOI] [PubMed] [Google Scholar]
- 110.De Strooper B, Beullens M, Contreras B, Levesque L, Craessaerts K, Cordell B, Moechars D, Bollen M, Fraser P, George-Hyslop PS, Van Leuven F. Phosphorylation, subcellular localization, and membrane orientation of the Alzheimer's disease-associated presenilins. J Biol Chem. 1997;272:3590–3598. doi: 10.1074/jbc.272.6.3590. [DOI] [PubMed] [Google Scholar]
- 111.Cook DG, Sung JC, Golde TE, Felsenstein KM, Wojczyk BS, Tanzi RE, Trojanowski JQ, Lee VM, Doms RW. Expression and analysis of presenilin 1 in a human neuronal system: localization in cell bodies and dendrites. Proc Natl Acad Sci USA. 1996;93:9223–9228. doi: 10.1073/pnas.93.17.9223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N, Bird TD, Hardy J, Hutton M, Kukull W, Larson E, Levy-Lahad E, Viitanen M, Peskind E, Poorkaj P, Schellenberg G, Tanzi R, Wasco W, Lannfelt L, Selkoe D, Younkin S. Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer's disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer's disease. Nat Med. 1996;2:864–870. doi: 10.1038/nm0896-864. [DOI] [PubMed] [Google Scholar]
- 113.Haass C. Presenilins: genes for life and death. Neuron. 1997;18:687–690. doi: 10.1016/s0896-6273(00)80309-8. [DOI] [PubMed] [Google Scholar]
- 114.Green KN, LaFerla FM. Linking calcium to Abeta and Alzheimer's disease. Neuron. 2008;59:190–194. doi: 10.1016/j.neuron.2008.07.013. [DOI] [PubMed] [Google Scholar]
- 115.Guo Q, Sopher BL, Furukawa K, Pham DG, Robinson N, Martin GM, Mattson MP. Alzheimer's presenilin mutation sensitizes neural cells to apoptosis induced by trophic factor withdrawal and amyloid beta-peptide: involvement of calcium and oxyradicals. J Neurosci. 1997;17:4212–4222. doi: 10.1523/JNEUROSCI.17-11-04212.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Yoo AS, Cheng I, Chung S, Grenfell TZ, Lee H, Pack-Chung E, Handler M, Shen J, Xia W, Tesco G, Saunders AJ, Ding K, Frosch MP, Tanzi RE, Kim TW. Presenilin-mediated modulation of capacitative calcium entry. Neuron. 2000;27:561–572. doi: 10.1016/s0896-6273(00)00066-0. [DOI] [PubMed] [Google Scholar]
- 117.Tu H, Nelson O, Bezprozvanny A, Wang Z, Lee SF, Hao YH, Serneels L, De Strooper B, Yu G, Bezprozvanny I. Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer's disease-linked mutations. Cell. 2006;126:981–993. doi: 10.1016/j.cell.2006.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Bezprozvanny I, Mattson MP. Neuronal calcium mishandling and the pathogenesis of Alzheimer's disease. Trends Neurosci. 2008;31:454–463. doi: 10.1016/j.tins.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Bojarski L, Pomorski P, Szybinska A, Drab M, Skibinska-Kijek A, Gruszczynska-Biegala J, Kuznicki J. Presenilin-dependent expression of STIM proteins and dysregulation of capacitative Ca2+ entry in familial Alzheimer's disease. Biochim Biophys Acta. 2009;1793:1050–1057. doi: 10.1016/j.bbamcr.2008.11.008. [DOI] [PubMed] [Google Scholar]
- 120.Lessard CB, Lussier MP, Cayouette S, Bourque G, Boulay G. The overexpression of presenilin2 and Alzheimer's-disease-linked presenilin2 variants influences TRPC6-enhanced Ca2+ entry into HEK293 cells. Cell Signal. 2005;17:437–445. doi: 10.1016/j.cellsig.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 121.Petersen OH, Cancela JM. New Ca2+-releasing messengers: are they important in the nervous system? Trends Neurosci. 1999;22:488–495. doi: 10.1016/s0166-2236(99)01456-3. [DOI] [PubMed] [Google Scholar]
- 122.Waterman SA. Voltage-gated calcium channels in autonomic neuroeffector transmission. Prog Neurobiol. 2000;60:181–210. doi: 10.1016/s0301-0082(99)00025-8. [DOI] [PubMed] [Google Scholar]
- 123.Thoenen H. Neurotrophins and neuronal plasticity. Science. 1995;270:593–598. doi: 10.1126/science.270.5236.593. [DOI] [PubMed] [Google Scholar]
- 124.Li YX, Zhang Y, Lester HA, Schuman EM, Davidson N. Enhancement of neurotransmitter release induced by brain-derived neurotrophic factor in cultured hippocampal neurons. J Neurosci. 1998;18:10231–10240. doi: 10.1523/JNEUROSCI.18-24-10231.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Tyler WJ, Perrett SP, Pozzo-Miller LD. The role of neurotrophins in neurotransmitter release. Neuroscientist. 2002;8:524–531. doi: 10.1177/1073858402238511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Singh BB, Lockwich TP, Bandyopadhyay BC, Liu X, Bollimuntha S, Brazer SC, Combs C, Das S, Leenders AG, Sheng ZH, Knepper MA, Ambudkar SV, Ambudkar IS. VAMP2-dependent exocytosis regulates plasma membrane insertion of TRPC3 channels and contributes to agonist-stimulated Ca2+ influx. Mol Cell. 2004;15:635–646. doi: 10.1016/j.molcel.2004.07.010. [DOI] [PubMed] [Google Scholar]
- 127.Chameau P, Van de Vrede Y, Fossier P, Baux G. Ryanodine-, IP3- and NAADP-dependent calcium stores control acetylcholine release. Pflugers Arch. 2001;443:289–296. doi: 10.1007/s004240100691. [DOI] [PubMed] [Google Scholar]
- 128.Cheramy A, Barbeito L, Godeheu G, Glowinski J. Riluzole inhibits the release of glutamate in the caudate nucleus of the cat in vivo. Neurosci Lett. 1992;147:209–212. doi: 10.1016/0304-3940(92)90597-z. [DOI] [PubMed] [Google Scholar]
- 129.Glickstein M. Mossy-fibre sensory input to the cerebellum. Prog Brain Res. 1997;114:251–259. doi: 10.1016/s0079-6123(08)63368-3. [DOI] [PubMed] [Google Scholar]
- 130.Matilla-Duenas A. The highly heterogeneous spinocerebellar ataxias: from genes to targets for therapeutic intervention. Cerebellum. 2008;7:97–100. doi: 10.1007/s12311-008-0020-5. [DOI] [PubMed] [Google Scholar]
- 131.Lim J, Hao T, Shaw C, Patel AJ, Szabo G, Rual JF, Fisk CJ, Li N, Smolyar A, Hill DE, Barabasi AL, Vidal M, Zoghbi HY. A protein-protein interaction network for human inherited ataxias and disorders of Purkinje cell degeneration. Cell. 2006;125:801–814. doi: 10.1016/j.cell.2006.03.032. [DOI] [PubMed] [Google Scholar]
- 132.Becker EB, Oliver PL, Glitsch MD, Banks GT, Achilli F, Hardy A, Nolan PM, Fisher EM, Davies KE. A point mutation in TRPC3 causes abnormal Purkinje cell development and cerebellar ataxia in moonwalker mice. Proc Natl Acad Sci USA. 2009;106:6706–6711. doi: 10.1073/pnas.0810599106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wu D, Huang W, Richardson PM, Priestley JV, Liu M. TRPC4 in rat dorsal root ganglion neurons is increased after nerve injury and is necessary for neurite outgrowth. J Biol Chem. 2008;283:416–426. doi: 10.1074/jbc.M703177200. [DOI] [PubMed] [Google Scholar]
- 134.Wang GX, Poo MM. Requirement of TRPC channels in netrin-1-induced chemotropic turning of nerve growth cones. Nature. 2005;434:898–904. doi: 10.1038/nature03478. [DOI] [PubMed] [Google Scholar]
- 135.Bezzerides VJ, Ramsey IS, Kotecha S, Greka A, Clapham DE. Rapid vesicular translocation and insertion of TRP channels. Nat Cell Biol. 2004;6:709–720. doi: 10.1038/ncb1150. [DOI] [PubMed] [Google Scholar]
- 136.Greka A, Navarro B, Oancea E, Duggan A, Clapham DE. TRPC5 is a regulator of hippocampal neurite length and growth cone morphology. Nat Neurosci. 2003;6:837–845. doi: 10.1038/nn1092. [DOI] [PubMed] [Google Scholar]
- 137.Riccio A, Li Y, Moon J, Kim KS, Smith KS, Rudolph U, Gapon S, Yao GL, Tsvetkov E, Rodig SJ, Van't Veer A, Meloni EG, Carlezon WA, Jr, Bolshakov VY, Clapham DE. Essential role for TRPC5 in amygdala function and fear-related behavior. Cell. 2009;137:761–772. doi: 10.1016/j.cell.2009.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Redmond L, Ghosh A. Regulation of dendritic development by calcium signaling. Cell Calcium. 2005;37:411–416. doi: 10.1016/j.ceca.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 139.Wu GY, Cline HT. Stabilization of dendritic arbor structure in vivo by CaMKII. Science. 1998;279:222–226. doi: 10.1126/science.279.5348.222. [DOI] [PubMed] [Google Scholar]
- 140.Becker LE, Armstrong DL, Chan F. Dendritic atrophy in children with Down's syndrome. Ann Neurol. 1986;20:520–526. doi: 10.1002/ana.410200413. [DOI] [PubMed] [Google Scholar]
- 141.Dierssen M, Ramakers GJ. Dendritic pathology in mental retardation: from molecular genetics to neurobiology. Genes Brain Behav. 2006;5(Suppl 2):48–60. doi: 10.1111/j.1601-183X.2006.00224.x. [DOI] [PubMed] [Google Scholar]
- 142.Aizawa H, Hu SC, Bobb K, Balakrishnan K, Ince G, Gurevich I, Cowan M, Ghosh A. Dendrite development regulated by CREST, a calcium-regulated transcriptional activator. Science. 2004;303:197–202. doi: 10.1126/science.1089845. [DOI] [PubMed] [Google Scholar]
- 143.Gaudilliere B, Konishi Y, de la Iglesia N, Yao G, Bonni A. A CaMKII-NeuroD signaling pathway specifies dendritic morphogenesis. Neuron. 2004;41:229–241. doi: 10.1016/s0896-6273(03)00841-9. [DOI] [PubMed] [Google Scholar]
- 144.Tai Y, Feng S, Ge R, Du W, Zhang X, He Z, Wang Y. TRPC6 channels promote dendritic growth via the CaMKIV-CREB pathway. J Cell Sci. 2008;121:2301–2307. doi: 10.1242/jcs.026906. [DOI] [PubMed] [Google Scholar]
- 145.Owsianik G, Talavera K, Voets T, Nilius B. Permeation and selectivity of TRP channels. Annu Rev Physiol. 2006;68:685–717. doi: 10.1146/annurev.physiol.68.040204.101406. [DOI] [PubMed] [Google Scholar]
- 146.Munsch T, Freichel M, Flockerzi V, Pape HC. Contribution of transient receptor potential channels to the control of GABA release from dendrites. Proc Natl Acad Sci USA. 2003;100:16065–16070. doi: 10.1073/pnas.2535311100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Riccio A, Medhurst AD, Mattei C, Kelsell RE, Calver AR, Randall AD, Benham CD, Pangalos MN. mRNA distribution analysis of human TRPC family in CNS and peripheral tissues. Brain Res Mol Brain Res. 2002;109:95–104. doi: 10.1016/s0169-328x(02)00527-2. [DOI] [PubMed] [Google Scholar]