

Abstract
A causal relationship between diet-induced hyperhomocysteinemia (HHcy) and accelerated atherosclerosis has been established in apolipoprotein E-deficient (apoE−/−) mice. However, it is not known whether the proatherogenic effect of HHcy in apoE−/− mice is independent of hyperlipidemia and/or deficiency of apoE. In this study, a comprehensive dietary approach using C57BL/6J mice was used to investigate whether HHcy is an independent risk factor for accelerated atherosclerosis or dependent on additional dietary factors that increase plasma lipids and/or inflammation. C57BL/6J mice at 4 wk of age were divided into 6 dietary groups: chow diet (C), chow diet + methionine (C+M), western-type diet (W), western-type diet + methionine (W+M), atherogenic diet (A), or atherogenic diet + methionine (A+M). After 2, 10, 20, or 40 wk on the diets, mice were sacrificed, and the levels of total plasma homocysteine, cysteine, and glutathione, as well as total plasma cholesterol and triglycerides were analyzed. Aortic root sections were examined for atherosclerotic lesions. HHcy was induced in all groups supplemented with methionine, compared to diet-matched control groups. Plasma total cholesterol was significantly increased in mice fed the W or A diet. However, the W diet increased LDL/IDL and HDL levels, while the A diet significantly elevated plasma VLDL and LDL/IDL levels without increasing HDL. No differences in plasma total cholesterol levels or lipid profiles were observed between methionine-supplemented groups and the diet-matched control groups. Early atherosclerotic lesions containing macrophage foam cells were only observed in mice fed the A or A + M diet. Furthermore, lesion size was significantly larger in the A + M group compared to the A group at 10 and 20 wk; however, mature lesions were never observed even after 40 wk on these diets. The presence of lymphocytes, increased hyaluronan staining, and the expression of endoplasmic reticulum (ER) stress markers were also increased in atherosclerotic lesions from the A + M group. Taken together, these results suggest that HHcy does not independently cause atherosclerosis in C57BL/6J mice even in the presence of increased total plasma lipids induced by the W diet. However, HHcy can accelerate atherosclerotic lesion development under dietary conditions that increase plasma VLDL levels and/or inflammation.
Keywords: homocysteine, inflammation, VLDL, atherogenesis, dietary intervention
Clinical and epidemiological studies have established that elevated plasma total homocysteine (tHcy) is an independent risk factor for cardiovascular disease and stroke (1, 2). Furthermore, accelerated atherosclerosis has been demonstrated in apolipoprotein E-deficient (apoE−/−) mice with dietary hyperhomocysteinemia (HHcy) (3–5) and in cystathionine β-synthase (CBS)/apoE−/− double-knockout mice (6). These findings suggest that HHcy accelerates atherogenesis in a hyperlipidemic mouse model that develops spontaneous atherosclerosis. However, a causal relationship between atherogenesis and HHcy has not been established in other species, including rats, rabbits, pigs, and primates (7–10).
Patients with inborn errors of methionine metabolism due to deficiencies in CBS or 5,10-methylenetetrahydrofolate reductase (MTHFR) present with severe HHcy and have a 50% chance of developing a major vascular event by the age of 30 yr if untreated (11, 12). However, the vascular complications associated with these patients frequently result from arterial and venous thrombosis, while pathological studies rarely show typical lipid-rich atherosclerotic lesions (13). This was recapitulated by Wilcken (14), who reported that the underlying process in children before the application of effective therapy is in the nature of a “nonlipid” model of arteriosclerosis. Although clinical investigations have shown that increased intima/media thickness (IMT) of carotid and femoral arteries is associated with elevated tHcy by B-mode ultrasound (15-17), there is no evidence that the increased IMT represents a typical atherosclerotic plaque, other than the fibrosis alone. Furthermore, these human studies have not demonstrated that HHcy accelerates atheroma formation, such as larger lipid core, greater number of inflammatory cells, and/or higher incidences of plaque rupture. On the contrary, a recent postmortem study on men with coronary disease demonstrated that increased tHcy is associated with the presence of lipid-poor, fibrous plaques (18). On the basis of these findings, it may be questioned whether 1) vascular changes observed in homocystinuric patients with severe HHcy have anything to do with atherogenesis; 2) the atherogenic effect of HHcy observed in atherosclerosis-prone apoE−/− mice is dependent on other factors, such as elevated plasma lipids, deficiency in apoE, or enhanced proinflammatory mediators; or 3) HHcy can induce and/or promote atherosclerosis in wild-type mouse models, similar to that observed in apoE−/− mice.
In the present study, C57BL/6J mice were treated with various dietary regimens to examine the causal effects of HHcy on atherogenesis. Our findings suggest that HHcy does not directly cause atherogenesis in C57BL/6J mice fed control or high-fat diets. However, HHcy only accelerated atherosclerotic lesion development in mice fed an atherogenic diet, a diet known to significantly increase VLDL levels. Given that elevated VLDL levels are a risk factor for cardiovascular disease and induce a proinflammatory response, our findings imply that homocysteine does not directly accelerate atherogenesis but can, under certain dietary conditions that alter plasma lipid levels and/or inflammation, contribute to atherosclerotic lesion development.
Materials and Methods
Mice and diets
Two hundred fourteen female C57BL/6J mice at 4 wk of age were obtained from Charles River (Quebec, QC, Canada). Mice were fed a normal chow diet without (C, n=36) or with 0.5% (w/v) l-methionine (Sigma-Aldrich, St. Louis, MO, USA) supplementation in the drinking water (C+M, n=36), or a western-type diet (0.15% cholesterol and 21% fat) without (W, n=36) or with 0.5% l-methionine supplementation in the drinking water (W+M, n=36), or an atherogenic diet (1.25% cholesterol, 15% fat, and 0.5% cholic acid) without (A, n=36) or with 0.5% l-methionine (A+M, n=36) supplementation in the drinking water. All mice were provided food and water ad libitum during the course of the study. Twelve mice in each group were sacrificed after 2, 10, and 20 wk on different diets for the measurement of plasma tHcy, total cholesterol (TC), triglycerides (TGs), lipid profiles, and the examination of atherosclerotic lesions. An additional 24 C57BL/6J mice were sacrificed after 40 wk on an atherogenic diet without or with 0.5% l-methionine supplementation in the drinking water to investigate the long-term effects of both atherogenic diet and HHcy on the progression of atherosclerosis. All procedures were approved by the McMaster University Animal Research Ethics Board.
Plasma tHcy and lipids
Plasma tHcy, cysteine, and glutathione levels were determined by the monobromobimane method of Jacobsen et al. (19) using high-performance liquid chromatography with fluorescence detection. Plasma TC and TG levels were assayed enzymatically using reagents from Sigma-Aldrich (Oakville, ON, Canada). Plasma lipoproteins were separated by automated gel filtration chromatography on an AKTA fast-protein liquid chromatography (FPLC) system (GE Healthcare Bio-Sciences, Inc., Baie d'Urfé, QC, Canada), and cholesterol concentrations were determined enzymatically (ThermoDMA) on an autoanalyzer (Molecular Devices Corp., Sunnyvale, CA, USA) (20).
Tissue sample preparations and histology
Mice were sacrificed, the maximum amount of blood was taken from the right ventricle, and the intact circulation was flushed with 1× PBS (3). Half of the mice in each group were perfusion fixed using 10% neutral buffered formalin, as described previously (5). The heart (including the aortic root) was removed and cut transversely as described by Paigen et al. (21) and embedded in paraffin. Four-micrometer cross sections of the aortic root were collected on glass slides for the measurement of lesion size [hematoxylin and eosin (H&E) staining] and IMT (Orcein staining; ref. 5) and for immunohistochemical (IHC) staining. Cryostat sections from the aortic root of the other half of mice in each group were stained with Oil red O (ORO) for the measurement of lipid-rich lesion size (22).
Immunohistochemistry
IHC analysis was performed using Vectastain ABC Systems (Vector Laboratories, Burlingame, CA, USA), as described previously (23). Antibodies directed against phospho-IκB-α (Cell Signaling, Danvers, MA, USA), KDEL (StressGen Biotechnologies, Victoria, BC, Canada), phospho-PERK (Cell Signaling), lymphocytes (CD3; Santa Cruz Technology, Santa Cruz, CA, USA), smooth muscle cells (SMCs) (α-actin; Stress-Gen Biotechnologies or Sigma), or macrophages (Mac-3; PharMingen, San Diego, CA, USA) were assessed in atherosclerotic lesions using IHC staining. Images were captured using a charge-coupled device (CCD) color video camera (Sony, Tokyo, Japan), and images were analyzed using Paint-Shop Pro 7 (Jasc Software Inc., Eden Prairie, MN, USA) and Northern Exposure (Empix Inc., Mississauga, ON, Canada). The mean lesion size and the intensity of IHC staining were measured, and the average value in each group was calculated as described previously (23).
Hyaluronan (HA) deposition
Affinity histochemistry was used to stain aortic root sections for HA, as described previously (24, 25). Briefly, sections were deparaffinized, rehydrated, and incubated in PBS. Nonspecific binding was blocked with PBS containing 3% fetal bovine serum (FBS). Samples were then incubated with 5 μg/ml biotinylated HA-binding protein (Seikagaku America, Falmouth, MA, USA) in PBS containing 3% FBS. After extensive washing, the sections were incubated with 2 μg/ml fluoresceinated avidin (Kirkegaard and Perry Laboratories, Gaithers-burg, MD, USA). The samples were then extensively washed and mounted with mounting medium containing 4′,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories, Burlin-game, CA, USA) to counterstain the nuclei. Samples were examined with a confocal microscope equipped for fluorescence. Biotinylated HA-binding protein was omitted when staining the samples serving as negative controls.
Statistical analyses
Results were expressed as mean ± se. Unpaired Student's t test was used to assess the difference between the experimental group and the control group. Values of P < 0.05 were considered significant, if not otherwise stated.
Results
Effects of diets on body weight
The initial mean body weight of 4-wk-old mice from all groups ranged from 17.5 to 18.3 g and did not differ significantly among groups. However, mean body weight of mice fed the W or W + M diet increased significantly at 2, 10, and 20 wk, compared to mice fed the C or C + M diet (Fig. 1). In contrast, mean body weight was significantly lower in the A and A + M groups after 20 wk, compared to mice fed the C or C + M diet. For all dietary groups, methionine supplementation in the drinking water had no effect on mean body weight over the 20-wk time period when compared to the appropriate controls.
Figure 1.
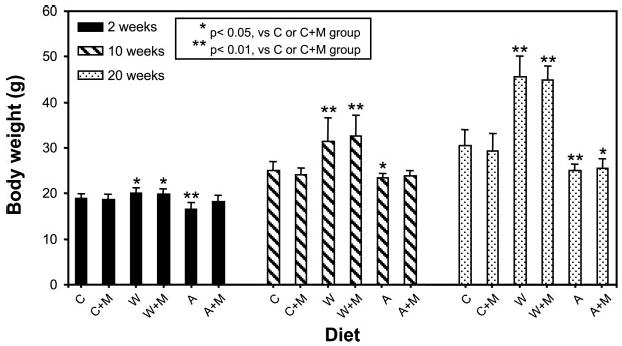
Body weight of C57BL/6J mice fed different diets for 2, 10, and 20 wk. C57BL/6J mice at 4 wk of age were fed different diets without or with 0.5% l-methionine (M) in the drinking water for 2, 10, or 20 wk. The body weight of the mice in the W and W + M groups increased significantly over all time periods, compared to the C and C + M groups. The A and A + M groups showed a significant decrease in weight gain at 20 wk, compared to the C and C + M groups. *P < 0.05; **P < 0.01. Methionine supplementation had no significant effect on body weight in the dietary groups tested.
Effects of diets on plasma tHcy and lipid levels
Plasma tHcy levels were significantly elevated in all dietary groups supplemented with methionine in the drinking water, compared to the diet-matched controls (Table 1). However, methionine supplementation had no significant effect on plasma total cysteine or glutathione levels over all time periods examined (Table 1). Plasma TC was significantly increased in mice fed the W, W + M, A, or A + M diet, compared to the mice fed the C or C + M diet (Table 2). However, no difference in plasma TC levels was found between the methionine-supplemented groups and diet-matched controls. There was also some evidence of increased plasma total TG in the W diet group, but a decrease in the A diet group, compared to the C diet group (Table 2). Methionine supplementation did not change plasma TG levels, compared with the diet-matched groups (Table 2). The analysis of plasma lipoprotein cholesterol profiles from mice fed diets for 20 wk showed that the W diet increased both LDL/IDL and HDL levels, compared to the C diet group (Fig. 2). The A diet significantly increased VLDL and LDL/IDL levels, without altering HDL levels, compared to the C diet group (Fig. 2). No significant difference in lipoprotein profile was found between methionine-supplemented and diet-matched groups.
TABLE 1. Plasma tHcy, cysteine, and glutathione levels in C57BL/6J mice fed different diets, with or without methionine supplementation, for 2, 10, or 20 wk.
| tHcy (μmol/L) | Cysteine (μmol/L) | Glutathione (μmol/L) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Diet group | 2 wk | 10 wk | 20 wk | 2 wk | 10 wk | 20 wk | 2 wk | 10 wk | 20 wk |
| C | 3.82 ± 0.79 | 2.94 ± 0.76 | 2.48 ± 0.55 | 153.42 ± 31.06 | 181.57 ± 50.06 | 182.02 ± 29.75 | 15.55 ± 4.28 | 15.71 ± 4.27 | 15.54 ± 2.21 |
| C + M | 10.12 ± 3.64* | 7.99 ± 4.95* | 6.02 ± 5.84* | 158.01 ± 33.3 | 186.94 ± 44.55 | 154.07 ± 26.6 | 18.43 ± 3.51 | 16.32 ± 5.32 | 13.55 ± 2.62 |
| W | 5.97 ± 1.81 | 3.56 ± 1.16 | 2.68 ± 0.95 | 167.6 ± 31.18 | 170.85 ± 29.7 | 170.13 ± 45.19 | 22.87 ± 5.14 | 19.18 ± 4.31 | 24.48 ± 10.04 |
| W + M | 26.35 ± 18.7* | 11.60 ± 7.73* | 6.59 ± 5.04* | 161.5 ± 37.11 | 165.53 ± 62.24 | 156.58 ± 51.52 | 20.04 ± 2.47 | 24.47 ± 11.46 | 21.14 ± 7.78 |
| A | 2.41 ± 0.55 | 1.51 ± 0.66 | 1.61 ± 0.46 | 191.33 ± 37.11 | 184.94 ± 36.25 | 149.58 ± 23.59 | 18.31 ± 5.84 | 28.89 ± 9.91 | 20.25 ± 5.61 |
| A + M | 8.51 ± 4.95* | 11.77 ± 12.58* | 3.54 ± 2.32* | 196.77 ± 26.86 | 187.0 ± 38.38 | 127.99 ± 27.98 | 17.43 ± 4.81 | 28.73 ± 6.66 | 17.43 ± 5.01 |
Data are presented as mean ± se; n = 12 mice/group.
P < 0.01 vs. diet-matched control group.
TABLE 2. Plasma TC and TG levels in C57BL/6 mice fed different diets, with or without methionine supplementation, for 2, 10, or 20 wk.
| TC (mM/L) | TG (mM/L) | |||||
|---|---|---|---|---|---|---|
| Diet group | 2 wk | 10 wk | 20 wk | 2 wk | 10 wk | 20 wk |
| C | 1.68 ± 0.09 | 3.72 ± 0.15 | 2.07 ± 0.10 | 1.21 ± 0.1 | 0.94 ± 0.11 | 0.86 ± 0.04 |
| C + M | 1.51 ± 0.07 | 3.72 ± 0.25 | 2.09 ± 0.11 | 0.89 ± 0.12 | 0.97 ± 0.12 | 1.04 ± 0.12 |
| W | 2.54 ± 0.28** | 5.26 ± 0.32** | 6.13 ± 0.45** | 1.86 ± 0.17* | 1.49 ± 0.22 | 1.31 ± 0.22 |
| W + M | 2.54 ± 0.07†† | 5.57 ± 0.32†† | 5.80 ± 0.67†† | 1.60 ± 0.37 | 1.99 ± 0.30†† | 1.71 ± 0.35 |
| A | 4.87 ± 0.84** | 8.24 ± 0.29** | 6.25 ± 0.72** | 0.65 ± 0.03** | 0.89 ± 0.07 | 0.49 ± 0.04** |
| A + M | 4.14 ± 0.23†† | 8.40 ± 0.38†† | 5.78 ± 0.60†† | 0.54 ± 0.07†† | 1.03 ± 0.11 | 0.60 ± 0.16†† |
Data are presented as mean ± se; n = 12 mice/group.
P < 0.05,
P < 0.01 vs. C diet group;
P < 0.01 vs. C + M diet group.
Figure 2.

Plasma lipoprotein profiles of C57BL/6J mice fed different diets, with or without methionine supplementation, for 20 wk. Plasma lipoproteins were size fractionated by FPLC. Chromatograms shown are from C57BL/6J mice fed different diets for 20 wk. Data are presented as micrograms per fraction of TC for each dietary group (n=5 mice/group).
Effect of HHcy on atherosclerotic lesion size in C57BL/6J mice fed the A or A + M diet
Paraffin sections from the aortic root of C57BL/6J mice were stained with H&E to assess lesion growth and gross cellular morphology (Fig. 3). Early atherosclerotic lesions containing fatty streaks and macrophage foam cells were consistently observed in the aortic root of C57BL/6J mice fed the A or A + M diet for at least 10 wk (Figs. 3 and 4A). The accumulation of lipid, as determined by ORO staining, was also increased in the vessel wall of mice fed the A or A + M diet (Fig. 4A). In contrast, early atherosclerotic lesions were not detected in mice fed the C, C + M, W, or W + M diet over the 20-wk time period (Fig. 3). Mean lesion size in the aortic root of mice fed the A + M diet was significantly larger at both 10 and 20 wk, compared to mice fed the A diet alone (Fig. 4B). However, the early lesions observed in the mice at 20 wk failed to develop into mature atherosclerotic lesions consisting of a smooth muscle/fibrous cap and lipid-rich necrotic core with cellular debris, even after 40 wk on the A or A + M diet (data not shown). Furthermore, there was no significant difference in IMT of the aorta between the methionine-supplemented groups and the diet-matched controls (data not shown).
Figure 3.

Morphology of the aortic root from C57BL/6J mice fed different diets, with or without methionine supplementation, for 20 wk. H&E staining of representative sections from the aortic root of C57BL/6J mice fed the C, C + M, W, W + M, A, or A + M diet for 20 wk. Arrows in sections from the A and A + M diet groups show the presence of early atherosclerotic lesions. No evidence of early atherosclerotic lesions was observed in mice fed the C, C + M, W, or W + M diet for 20 wk. Original view, ×40.
Figure 4.

Atherosclerotic lesion development in C57BL/6J mice fed the A or A + M diet for 10 or 20 wk. A) Atherosclerotic lesion morphology was assessed by H&E and ORO staining. Arrows indicate early atherosclerotic lesions. B) Atherosclerotic lesion size in mice fed the A or A + M diet for 10 or 20 wk (n=8 lesions assessed per group). Original view, ×100.
Identification of macrophages and lymphocytes in early atherosclerotic lesions from C57BL/6J mice fed the A or A + M diet
Lesional macrophages were identified by immunostaining using an anti-Mac-3 antibody, whereas lymphocytes were identified with an anti-CD-3 antibody (Fig. 5). In the early atherosclerotic lesions from mice fed the A or A + M diet, Mac-3 immunostaining revealed the presence of intimal lipid-rich macrophage foam cells. The intensity of staining and distribution of Mac-3-positive macrophages were increased in the atherosclerotic lesions from mice fed the A + M diet for 20 wk, compared to mice fed the A diet. Consistent with these findings, the intensity of staining and distribution of CD-3-positive lymphocytes were also increased in the atherosclerotic lesions from mice fed the A + M diet. In addition, the distribution of macrophages and lymphocytes seemed to overlap in the early lesions from mice fed the A or A + M diet.
Figure 5.

Identification of T-lymphocytes, macrophages, and endoplasmic reticulum stress in the atherosclerotic lesions from C57BL/6J mice Representative aortic root sections from C57BL/6J mice fed the A or A + M diet for 20 wk were immunostained for CD3 (T-lymphocytes), Mac-3 (macrophages), or p-PERK (phospho-PERK). Immunostaining for CD3, Mac-3, and p-PERK were increased in the atherosclerotic lesions from mice fed the A + M diet. Arrows in the H&E panel indicate a large population of lesion-resident inflammatory cells. Original view, ×100.
Previous studies have demonstrated that UPR activation occurs at all stages of atherosclerotic lesion development (26) and is increased in apoE−/− mice fed a high-methionine diet (23). To assess whether UPR activation occurs in the early lesions from C57BL/6J mice fed the A or A + M diet, aortic root sections were immunostained with an antiphospho-PERK antibody. Consistent with previous findings (26), phospho-PERK imunostaining colocalized with lesion-resident macrophages, as well as a subset of lymphocytes (Fig. 5). Although phospho-PERK staining was markedly increased in lymphocyte-rich lesions from mice fed the A + M diets, it was not uniform in all macrophages, a finding similar to that observed in apoE−/− mice (26).
Not all atherosclerotic lesions found in mice fed the A or A + M diet were rich in lymphocytes. Therefore, the morphology and composition of these lymphocyte-poor lesions were also assessed using IHC staining for KDEL (UPR marker), phospho-IκB-α (proinflammatory marker), and SMC α-actin (Fig. 6). Positive immunostaining for all of these markers were observed in lesions from mice fed the A or A + M diet for 20 wk, a result similar to that observed in apoE−/− mice (26).
Figure 6.

Expression of GRP78/94, phospho-IκB-α, and SMC α-actin in early atherosclerotic lesions from C57BL/6J mice. Representative aortic root sections from C57BL/6J mice fed the A or A + M diet for 20 wk were immunostained for GRP78/94 (KDEL), phospho-IκB-α (p-IκB-α) and SMC α-actin. Original view, ×100.
HA deposition in atherosclerotic lesions from C57BL/6J mice fed the A or A + M diet
Previous studies have demonstrated that HHcy enhances the activation of NF-κB in the atherosclerotic lesions from apoE−/− mice (4, 23). Given the marked increase in lymphocytes and macrophages in the early lesions from mice fed the A + M diet, aortic root sections were stained for HA, a linear glycosaminoglycan having proinflammatory properties (24, 25). Marked HA deposition was observed in the early atherosclerotic lesions from mice fed either the A or A + M diet. The intensity of HA staining also correlated with atherosclerotic lesion size, with HA staining being increased in the lesions from mice fed the A + M diet, compared to mice fed the A diet. In contrast, HA accumulation did not achieve the levels observed in mice fed the A or A + M diet in mice fed the C, C + M, W, or W + M diet for 20 wk (Fig. 7).
Figure 7.

Effect of different diets on HA deposition in the aortic root of C57BL/6J mice. Representative aortic root sections from C57BL/6J mice fed different diets with or without 0.5% methionine in the drinking water for 20 wk were immunostained for HA. Sections were counterstained with DAPI to reveal nuclei. Arrows indicate lesion-resident HA immunostaining in the aortic root of the A and A + M groups. HA immunostaining is absent in the aortic root sections from the C, C + M, W, and W + M groups. Original views, ×40; ×100 (inset details; right panels).
Discussion
The aim of this study was to utilize a comprehensive dietary approach in C57BL/6J mice to investigate whether HHcy is an independent risk factor for accelerated atherosclerosis or is dependent on additional dietary factors that contribute to atherogenesis. The results of this study indicate that HHcy does not independently cause atherosclerosis in C57BL/6J mice. This is based on several lines of evidence. Although methionine supplementation in the drinking water significantly increased plasma tHcy levels, there was no evidence of atherosclerotic lesion development in mice fed a chow diet. Similar to these findings, there was no evidence of atherosclerotic lesions in C57BL/6J mice fed a western-type diet, even though mean total body weight and plasma total lipid levels were significantly elevated. However, methionine supplementation was shown to accelerate lesion development in mice fed an atherogenic diet. Thus, our data suggest that diet-induced HHcy per se does not cause atherosclerosis in mice. However, given that the atherogenic diet significantly increases plasma VLDL levels (21, 27), a known cardiovascular risk factor having proinflammatory properties (28–31), our findings suggest that HHcy can accelerate atherosclerotic lesion development in mice under dietary conditions that elevate plasma VLDL. This is similar to previous studies in rabbits showing that HHcy exacerbates coronary artery atherosclerotic lesions only in the presence of dietary conditions that increase plasma levels of lipids such as cholesterol (32).
Although methionine supplementation caused a significant increase in plasma tHcy in all diet groups, it had no effect on mean body weight, plasma total lipid levels, or plasma profiles. These findings imply that diet-induced HHcy does not contribute to atherosclerotic lesion development by increasing weight gain or altering plasma lipid levels in C57BL/6J mice. However, previous reports have demonstrated that diet-induced HHcy causes sterol regulatory element-binding protein (SREBP) activation, the major transcriptional switch that induces intracellular cholesterol and TG biosynthesis, in the livers of apoE−/− mice (33, 34). This effect on SREBP activation is through a cellular mechanism involving the ability of elevated intracellular homocysteine levels to induce ER stress, thereby leading to SREBP cleavage (33, 35). The observation that diet-induced HHcy does not significantly affect plasma total lipid levels suggests that the effects of homocysteine may be intracellular and localized to tissues or cell types that are sensitive to increases in intracellular homocysteine. This may, in part, explain why diet-induced HHcy does not lead to an overall effect on plasma total lipid levels, despite the ability of homocysteine to activate SREBP and its downstream gene targets (33, 35).
We observed a decrease in plasma tHcy levels in mice fed methionine-enriched diets by 20 wk, compared to 2 and 10 wk. A similar age-dependent decline in plasma tHcy levels was observed in apoE−/− mice fed methionine-enriched control or high-fat diets (36). Previous studies in rats (37) also showed that plasma levels of total, free, and protein-bound thiols, including homocysteine, decreased with age. These observations suggest that the age-dependent decline in plasma tHcy could be related to the induction of genes involved in intracellular homocysteine metabolism and/or export. In support of this concept, previous reports using cultured endothelial cells showed that exogenously added homocysteine can increase the expression of methylenetetrahydrofolate dehydrogenase and methyl-enetetrahydrofolate cyclohydrolase, enzymes which are involved in homocysteine metabolism (38). These enzymes were also induced in the brains of MTHFR−/− mice having 10-fold higher levels of plasma tHcy (39).
In contrast to the W diet, the A diet contains 0.5% cholic acid, an abundant bile acid produced in the liver from cholesterol. Several studies have investigated the mechanisms by which dietary cholic acid promotes atherosclerosis. Cholic acid increases plasma levels of apolipoprotein B-containing VLDL particles in mice by a posttranscriptional mechanism (40). It is well established that elevated levels of plasma VLDL are considered to be an independent risk factor for the progression of atherosclerosis (41, 42). VLDL has also been shown to potentiate the inflammatory processes associated with atherosclerosis by 1) enhancing the expression of tumor necrosis factor α and interleukin-1β expression in macrophages (29, 30), and 2) modulating the cytokine secretion profile of lymphocytes to a predominantly proinflammatory response (28). Previous studies have also demonstrated that cholic acid can activate genes associated with hepatic fibrosis, while the cholesterol component of the atherogenic diet was found to induce genes responsible for the acute inflammatory response (31). In contrast to VLDL, cholic acid lowers plasma levels of HDL in mice and humans by a transcriptional mechanism (43). This would subsequently alter the VLDL/HDL ratio to a more proatherogenic condition.
In the present study, phospho-IκB-α immunostaining and T-lymphocytes were observed in the lesions of mice fed the A or A + M diet and were increased in the A + M diet group. Our findings imply that dietary conditions that increase VLDL/cholesterol levels are likely the driving force responsible for the accelerated atherosclerosis in mice fed A diets. Furthermore, our findings demonstrate that HHcy accelerates atherosclerotic lesion development under dietary conditions that increase VLDL/cholesterol and induce a proinflammatory response. Thus, given its known proinflammatory characteristics (2, 4, 23), HHcy may also contribute to the proinflammatory response in C57BL/6J mice fed the A diet, thereby acting in concert with VLDL to accelerate atherosclerotic lesion development. Al-though anti-inflammatories such as curcumin (44), resveratrol (45), indomethacin (46) or statins (47–49) reduce atherosclerosis in apoE−/− mice, further experiments are necessary to determine whether treatment with anti-inflammatories can reduce atherogenesis in C57BL/6J mice fed the A or A + M diet.
Increased HA staining was observed in mice fed the A or A + M diet. HA is a glycosaminoglycan containing repeating disaccharide units composed of glucuronic acid and N-acetylglucosamine (25) and accumulates in numerous inflammatory conditions, including atherosclerosis. HA-receptor interactions are associated with the differentiation of monocytes, the uptake of oxidized LDL by monocytes, the activation of macrophages and T-cells, and the migration of leukocytes, suggesting that HA has a dynamic role in the inflammatory process. An important property of HA is its ability to mediate the interactions of immune and nonimmune cells. Activated leukocytes can bind to endothelial cell HA, and nonactivated leukocytes can bind to stressed SMCs via a unique structural form of HA. Interestingly, overexpression of HA by aortic SMCs in apoE−/− mice accelerates atherosclerosis (50). Together, these findings indicate that HA production in response to inflammatory stimuli may have an active role in regulating leukocyte adhesion and retention within inflamed tissues. Interactions of leukocytes with HA produced by SMCs may be important for modulating and perpetuating the inflammatory response in atherosclerotic lesions. The observation that HHcy increases HA staining in atherosclerotic lesions from mice fed the atherogenic diet provides further support that HHcy contributes to atherogenesis through inflammatory-mediated processes.
Recent in vitro studies have shown that bile acids and salts can activate transcription factors that respond to several cellular stress pathways, including DNA damage, ER stress (GRP78, GADD153), and oxidative stress (NF-κB, HSP70) (51). In the present study, the expression of phospho-PERK and GRP78/94 were observed in the early lesions from mice fed the A or A + M diet. Furthermore, phospho-PERK was increased in atherosclerotic lesions in mice fed the A + M diet, compared to the A diet group. Whether this can be attributed solely to the effects of cholic acid is unknown. However, these studies show for the first time that ER stress/UPR activation can be induced by the A diet and may play a role in the development of atherosclerosis in C57BL/6J mice.
The lack of atherosclerotic lesions observed in C57BL/6J mice fed the C or W diet supplemented with methionine is reminiscent of findings in children with homocystinuria in which large and medium-sized arteries showed a lack of lipid accumulation (52). Other morphological changes in the arteries consisted of swelling and hyperplasia of endothelial cells, fibrous intimal thickenings, disruption of the internal elastic membrane, and perivascular fibrosis. Recently, Rubba et al. (13) updated the existing knowledge on vascular complications of homocystinuria from the autopsy of 12 cases of premature cardiovascular death and found that both arterial and venous thrombosis are frequent and occur at an early age but apparently often without concomitant atherosclerosis. Therefore, it is unlikely that typical atherosclerotic lesions precede thrombus formation in young homocystinuric patients.
In summary, diet-induced HHcy does not increase atherosclerotic lesion development in C57BL/6J mice fed either chow or high-fat diets. However, diet-induced HHcy increased atherosclerotic lesion development in mice fed an atherogenic diet. These studies imply that homocysteine does not directly accelerate atherogenesis but can, under dietary conditions that increase plasma VLDL levels and/or cause inflammation, contribute to atherosclerotic lesion development.
Acknowledgments
This work was supported in part by research grants to R.C.A from the Heart and Stroke Foundation of Ontario (T-6146, NA-6204), the Canadian Institutes of Health Research (MOP-74477, MOP-67116) and the Ontario Research and Development Challenge Fund. Support was also provided by the Heart, Lung and Blood Institute of the U.S. National Institutes of Health (HL52234 to D.W.J.). R.C.A. is a Career Investigator of the Heart and Stroke Foundation of Ontario (CI-5959) and holds the Amgen Canada Research Chair in Nephrology.
References
- 1.Boushey CJ, Beresford SA, Omenn GS, Motulsky AG. A quantitative assessment of plasma homocysteine as a risk factor for vascular disease. Probable benefits of increasing folic acid intakes. JAMA. 1995;274:1049–1057. doi: 10.1001/jama.1995.03530130055028. [DOI] [PubMed] [Google Scholar]
- 2.Eikelboom JW, Lonn E, Genest J, Jr, Hankey G, Yusuf S. Homocyst(e)ine and cardiovascular disease: a critical review of the epidemiologic evidence. Ann Intern Med. 1999;131:363–375. doi: 10.7326/0003-4819-131-5-199909070-00008. [DOI] [PubMed] [Google Scholar]
- 3.Zhou J, Moller J, Danielsen CC, Bentzon J, Ravn HB, Austin RC, Falk E. Dietary supplementation with methionine and homocysteine promotes early atherosclerosis but not plaque rupture in ApoE-deficient mice. Arterioscler Thromb Vasc Biol. 2001;21:1470–1476. doi: 10.1161/hq0901.096582. [DOI] [PubMed] [Google Scholar]
- 4.Hofmann MA, Lalla E, Lu Y, Gleason MR, Wolf BM, Tanji N, Ferran LJ, Jr, Kohl B, Rao V, Kisiel W, Stern DM, Schmidt AM. Hyperhomocysteinemia enhances vascular inflammation and accelerates atherosclerosis in a murine model. J Clin Invest. 2001;107:675–683. doi: 10.1172/JCI10588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhou J, Moller J, Ritskes-Hoitinga M, Larsen ML, Austin RC, Falk E. Effects of vitamin supplementation and hyperhomocysteinemia on atherosclerosis in apoE-deficient mice. Atherosclerosis. 2003;168:255–262. doi: 10.1016/s0021-9150(03)00138-2. [DOI] [PubMed] [Google Scholar]
- 6.Wang H, Jiang X, Yang F, Gaubatz JW, Ma L, Magera MJ, Yang X, Berger PB, Durante W, Pownall HJ, Schafer AI. Hyperhomocysteinemia accelerates atherosclerosis in cystathionine beta-synthase and apolipoprotein E double knock-out mice with and without dietary perturbation. Blood. 2003;101:3901–3907. doi: 10.1182/blood-2002-08-2606. [DOI] [PubMed] [Google Scholar]
- 7.Matthias D, Becker CH, Riezler R, Kindling PH. Homocysteine induced arterosclerosis-like alterations of the aorta in normotensive and hypertensive rats following application of high doses of methionine. Atherosclerosis. 1996;122:201–216. doi: 10.1016/0021-9150(95)05740-4. [DOI] [PubMed] [Google Scholar]
- 8.Donahue S, Sturman JA, Gaull G. Arterisclerosis due to homocyst(e)inemia. Failure to reproduce the model in weanling rabbits. Am J Pathol. 1974;77:167–174. [PMC free article] [PubMed] [Google Scholar]
- 9.Reddy GSR, Wilcken DEL. Experimental homocysteinemia in pigs: comparison with studies in sixteen homocystinuric patients. Metabolism. 1982;31:778–783. doi: 10.1016/0026-0495(82)90075-0. [DOI] [PubMed] [Google Scholar]
- 10.Harker LA, Ross R, Slichter SJ, Scott CR. Homocystine-induced arterosclerosis. The role of endothelial cell injury and platelet response in its genesis. J Clin Invest. 1976;58:731–741. doi: 10.1172/JCI108520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mudd SH, Skovby F, Levy HL, Pettigrew KD, Wilcken B, Pyeritz RE, Andria G, Boers GHJ, Bromberg IL, Cerone R, Fowler B, Grobe H, Schmidt H, Schweitzer L. The natural history of homocystinuria due to cystathionine-β-synthase deficiency. Am J Hum Genet. 1985;37:1–31. [PMC free article] [PubMed] [Google Scholar]
- 12.Rosenblatt DS, Fenton WA. Inherited disorders of folate and cobalamin transport and metabolism. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW, Vogelstein B, editors. The Metabolic & Molecular Bases of Inherited Disease. 8th. McGraw Hill; New York: 2001. pp. 3897–3933. [Google Scholar]
- 13.Rubba P, Minno GD, Andria G. Vascular complications of homocystinuria: incidence, clinical pattern and treatment. In: Robinson K, editor. Homocysteine and Vascular Disease. Kluwer Academic Publishers; Dordrecht, The Netherlands: 2000. pp. 117–134. [Google Scholar]
- 14.Wilcken DEL. Historical aspects of the relationship between homocysteine and vascular disease. In: Robinson K, editor. Homocysteine and Vascular Disease. Kluwer Academic Publishers; Dordrecht, The Netherlands: 2000. pp. 5–14. [Google Scholar]
- 15.Malinow MR. Plasma homocyst(e)ine and arterial occlusive diseases: a mini-review. Clin Chem. 1995;41:173–176. [PubMed] [Google Scholar]
- 16.McQuillan BM, Beilby JP, Nidorf M, Thompson PL, Hung J. Hyperhomocysteinemia but not the C677T mutation of methylenetetrahydrofolate reductase is an independent risk determinant of carotid wall thickening. The Perth Carotid Ultrasound Disease Assessment Study (CUDAS) Circulation. 1999;99:2383–2388. doi: 10.1161/01.cir.99.18.2383. [DOI] [PubMed] [Google Scholar]
- 17.Smilde TJ, van den Berkmortel FW, Boers GH, Wollersheim H, de Boo T, van Langen H, Stalenhoef AF. Carotid and femoral artery wall thickness and stiffness in patients at risk for cardiovascular disease, with special emphasis on hyperhomocysteinemia. Arterioscler Thromb Vasc Biol. 1998;18:1958–1963. doi: 10.1161/01.atv.18.12.1958. [DOI] [PubMed] [Google Scholar]
- 18.Burke AP, Fonseca V, Kolodgie F, Zieske A, Fink L, Virmani R. Increased serum homocysteine and sudden death resulting from coronary atherosclerosis with fibrous plaques. Arterioscler Thromb Vasc Biol. 2002;22:1936–1941. doi: 10.1161/01.atv.0000035405.16217.86. [DOI] [PubMed] [Google Scholar]
- 19.Jacobsen DW, Gatautis VJ, Green R, Robinson K, Savon SR, Secic M, Ji J, Otto JM, Taylor LM., Jr Rapid HPLC determination of total homocysteine and other thiols in serum and plasma: sex differences and correlation with cobalamin and folate levels in normal subjects. Clin Chem. 1994;40:873–881. [PubMed] [Google Scholar]
- 20.Rigotti A, Trigatti BL, Penman M, Rayburn H, Herz J, Krieger M. A targeted mutation in the murine gene encoding the high-density lipoprotein (HDL) receptor scavenger receptor class B type I reveals its key role in HDL metabolism. Proc Natl Acad Sci U S A. 1997;94:12610–12615. doi: 10.1073/pnas.94.23.12610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Paigen B, Morrow A, Holmes PA, Mitchell D, Williams RA. Quantitative assessment of atherosclerotic lesions in mice. Atherosclerosis. 1987;68:231–240. doi: 10.1016/0021-9150(87)90202-4. [DOI] [PubMed] [Google Scholar]
- 22.Dixon JL, Shen S, Vuchetich JP, Wysocka E, Sun GY, Sturek M. Increased atherosclerosis in diabetic dyslipidemic swine: protection by atorvastatin involves decreased VLDL triglycerides but minimal effects on the lipoprotein profile. J Lipid Res. 2002;43:1618–1629. doi: 10.1194/jlr.m200134-jlr200. [DOI] [PubMed] [Google Scholar]
- 23.Zhou J, Werstuck GH, Lhoták S, de Koning ABL, Sood SK, Hossain GS, Møller J, Ritskes-Hoitinga M, Falk E, Dayal S, Lentz SR, Austin RC. Association of multiple cellular stress pathways with accelerated atherosclerosis in hyperhomocysteinemic apolipoprotein E-deficient mice. Circulation. 2004;110:207–213. doi: 10.1161/01.CIR.0000134487.51510.97. [DOI] [PubMed] [Google Scholar]
- 24.Majors AK, Austin RC, de la Motte CA, Pyeritz RE, Hascall VC, Kessler SP, Sen G, Strong SA. Endoplasmic reticulum stress induces hyaluronan deposition and leukocyte adhesion. J Biol Chem. 2003;278:47223–47231. doi: 10.1074/jbc.M304871200. [DOI] [PubMed] [Google Scholar]
- 25.Hascall VC, Majors AK, De La Motte CA, Evanko SP, Wang A, Drazba JA, Strong SA, Wight TN. Intracellular hyaluronan: a new frontier for inflammation? Biochim Biophys Acta. 2004;1673:3–12. doi: 10.1016/j.bbagen.2004.02.013. [DOI] [PubMed] [Google Scholar]
- 26.Zhou J, Lhotak S, Hilditch BA, Austin RC. Activation of the unfolded protein response occurs at all stages of atherosclerotic lesion development in apolipoprotein E-deficient mice. Circulation. 2005;111:1814–1821. doi: 10.1161/01.CIR.0000160864.31351.C1. [DOI] [PubMed] [Google Scholar]
- 27.Paigen B, Morrow A, Brandon C, Mitchell D, Holmes P. Variation in susceptibility to atherosclerosis among inbred strains of mice. Atherosclerosis. 1985;57:65–73. doi: 10.1016/0021-9150(85)90138-8. [DOI] [PubMed] [Google Scholar]
- 28.Sampedro MC, Motrán C, Gruppi A, Kivatinitz SC. VLDL modulates the cytokine secretion profile to a proinflmmatory pattern. Biochem Biophys Res Commun. 2001;285:393–399. doi: 10.1006/bbrc.2001.5202. [DOI] [PubMed] [Google Scholar]
- 29.Stollenwerk MM, Lindholm MW, Pörn-Ares MI, Larsson A, Nilsson J, Ares MP. Very low-density lipoprotein induces interleukin-1β expression in macrophages. Biochem Biophys Res Commun. 2005;335:603–608. doi: 10.1016/j.bbrc.2005.07.123. [DOI] [PubMed] [Google Scholar]
- 30.Stollenwerk MM, Schiopu A, Fredrikson GN, Dichtl W, Nilsson J, Ares MP. Very low density lipoprotein potentiates tumor necrosis factor-alpha expression in macrophages. Atherosclerosis. 2005;179:247–254. doi: 10.1016/j.atherosclerosis.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 31.Vergnes L, Phan J, Strauss M, Tafuri S, Reue K. Cholesterol and cholate components of an atherogenic diet induce distinct changes of hepatic inflammatory gene expression. J Biol Chem. 2003;278:42774–42784. doi: 10.1074/jbc.M306022200. [DOI] [PubMed] [Google Scholar]
- 32.Zulli A, Hare DL, Buxton BF, Black MJ. High dietary methionine plus cholesterol exacerbates atherosclerosis formation in the left main coronary artery of rabbits. Atherosclerosis. 2004;176:83–89. doi: 10.1016/j.atherosclerosis.2004.05.013. [DOI] [PubMed] [Google Scholar]
- 33.Werstuck GH, Lentz SR, Dayal S, Hossain GS, Sood SK, Shi YY, Zhou J, Maeda N, Krisans SK, Malinow MR, Austin RC. Homocysteine-induced endoplasmic reticulum stress causes dysregulation of the cholesterol and triglyceride biosynthetic pathways. J Clin Invest. 2001;107:1263–1273. doi: 10.1172/JCI11596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ji C, Kaplowitz N. Betaine decreases hyperhomocysteinemia, endoplasmic reticulum stress, and liver injury in alcohol-fed mice. Gastroenterol. 2003;124:1488–1499. doi: 10.1016/s0016-5085(03)00276-2. [DOI] [PubMed] [Google Scholar]
- 35.Colgan SM, Tang D, Werstuck GH, Austin RC. Endoplasmic reticulum stress causes the activation of sterol regulatory element binding protein-2. Int J Biochem Cell Biol. 2007;39:1843–1851. doi: 10.1016/j.biocel.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 36.Wilson KM, McCaw RB, Leo L, Arning E, Lhoták S, Bottiglieri T, Austin RC, Lentz SR. Prothrombotic effects of hyperhomocysteinemia and hypercholesterolemia in apoE-deficient mice. Arterioscler Thromb Vasc Biol. 2007;27:233–240. doi: 10.1161/01.ATV.0000251607.96118.af. [DOI] [PubMed] [Google Scholar]
- 37.Iciek M, Chwatko G, Lorenc-Koci E, Bald E, Wlodek L. Plasma levels of total, free and protein bound thiols as well as sulfane sulfur in different age groups of rats. Acta Biochim Pol. 2004;51:815–824. [PubMed] [Google Scholar]
- 38.Kokame K, Kato H, Miyata T. Homocysteine-respondent genes in vascular endothelial cells identified by differential display analysis. BRP78/BiP and novel genes. J Biol Chem. 1996;271:29659–29665. doi: 10.1074/jbc.271.47.29659. [DOI] [PubMed] [Google Scholar]
- 39.Chen Z, Ge B, Hudson TJ, Rozen R. Microarray analysis of brain RNA in mice with methylenetetrahydrofolate reductase deficiency and hyperhomocysteinemia. Brain Res Gene Expr Patterns. 2002;1:89–93. doi: 10.1016/s1567-133x(01)00018-7. [DOI] [PubMed] [Google Scholar]
- 40.Srivastava RA, Averna M, Srivastava N, Pape ME. Dietary cholate increases plasma levels of apolipoprotein B in mice by posttranscriptional mechanisms. Int J Biochem Cell Biol. 2001;33:1215–1226. doi: 10.1016/s1357-2725(01)00080-2. [DOI] [PubMed] [Google Scholar]
- 41.Tatami R, Mabuchi H, Ueda K, Ueda R, Haba T, Kametani T, Ito S, Koizumi J, Ohta M, Miyamoto S, Nakayama A, Kanaya H, Oiwake H, Genda A, Takeda R. Intermediate-density lipoprotein and cholesterol-rich very low density lipoprotein in angiographically determined coronary artery disease. Circulation. 1981;64:1174–1184. doi: 10.1161/01.cir.64.6.1174. [DOI] [PubMed] [Google Scholar]
- 42.Mack WJ, Krauss RM, Hodis HN. Lipoprotein subclasses in the Monitored Atherosclerosis Regression Study (MARS). Treatment effects and relation to coronary angiographic progression. Arterioscler Thromb Vasc Biol. 1996;16:697–704. doi: 10.1161/01.atv.16.5.697. [DOI] [PubMed] [Google Scholar]
- 43.Srivastava RA, Srivastava N, Averna M. Dietary cholic acid lowers plasma levels of mouse and human apolipoprotein A-I primarily via a transcriptional mechanism. Eur J Biochem. 2000;267:4272–4280. doi: 10.1046/j.1432-1033.2000.01473.x. [DOI] [PubMed] [Google Scholar]
- 44.Olszanecki R, Jawień J, Gajda M, Mateuszuk L, Gebska A, Korabiowska M, Chłopicki S, Korbut R. Effect of curcumin on atherosclerosis in apoE/LDLR-double knockout mice. J Physiol Pharmacol. 2005;56:627–635. [PubMed] [Google Scholar]
- 45.Norata GD, Marchesi P, Passamonti S, Pirillo A, Violi F, Catapano AL. Anti-inflammatory and anti-atherogenic effects of cathecin, caffeic acid and trans-resveratrol in apolipoprotein E deficient mice. Atherosclerosis. 2007;191:265–271. doi: 10.1016/j.atherosclerosis.2006.05.047. [DOI] [PubMed] [Google Scholar]
- 46.Burleigh ME, Babaev VR, Patel MB, Crews BC, Remmel RP, Morrow JD, Oates JA, Marnett LJ, Fazio S, Linton MF. Inhibition of cyclooxygenase with indomethacin phenethylamide reduces atherosclerosis in apoE-null mice. Biochem Pharmacol. 2005;70:334–342. doi: 10.1016/j.bcp.2005.04.044. [DOI] [PubMed] [Google Scholar]
- 47.Monetti M, Canavesi M, Camera M, Parente R, Paoletti R, Tremoli E, Corsini A, Bellosta S. Rosuvastatin displays anti-atherothrombotic and anti-inflammatory properties in apoE-deficient mice. Pharmacol Res. 2007;55:441–449. doi: 10.1016/j.phrs.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 48.Scalia R, Gooszen ME, Jones SP, Hoffmeyer M, Rimmer DM, Trocha SD, Huang PL, Smith MB, Lefer AM, Lefer DJ. Simvastatin exerts both anti-inflammatory and cardioprotective effects in apolipoprotein E-deficient mice. Circulation. 2001;103:2598–2603. doi: 10.1161/01.cir.103.21.2598. [DOI] [PubMed] [Google Scholar]
- 49.Kleeman R, Princen HM, Emeis JJ, Jukema JW, Fontijn RD, Horrevoets AJ, Kooistra T, Haveke LM. Rosuvastatin reduces atherosclerosis development beyond and independent of its plasma cholesterol-lowering effect in APO*3-Leiden transgenic mice: evidence for anti-inflammatory effects of rosuvastatin. Circulation. 2003;108:1368–1374. doi: 10.1161/01.CIR.0000086460.55494.AF. [DOI] [PubMed] [Google Scholar]
- 50.Chai S, Chai Q, Danielsen CC, Hjorth P, Nyengaard JR, Ledet T, Yamaguchi Y, Rasmussen LM, Wogensen L. Overexpression of hyaluronan in the tunica media promotes the development of atherosclerosis. Circ Res. 2005;96:583–591. doi: 10.1161/01.RES.0000158963.37132.8b. [DOI] [PubMed] [Google Scholar]
- 51.Bernstein H, Payne CM, Bernstein C, Schneider J, Beard SE, Crowley CL. Activation of the promoters of genes associated with DNA damage, oxidative stress, ER stress and protein malfolding by the bile salt, deoxycholate. Toxicol Lett. 1999;108:37–46. doi: 10.1016/s0378-4274(99)00113-7. [DOI] [PubMed] [Google Scholar]
- 52.McCully KS. Vascular pathology of homocysteinemia: implications for the pathogenesis of arteriosclerosis. Am J Pathol. 1969;56:111–128. [PMC free article] [PubMed] [Google Scholar]