

Abstract
In the heart, adenosine binds to pharmacologically distinct G‐protein‐coupled receptors (A1‐R, A2A‐R, and A3‐R). While the role of A1‐and A3‐Rs in the heart has been clarifed, the effect of genetically manipulating the A2A‐R has not been defned. Thus, we created mice overexpressing a cardiac‐restricted A2A‐R transgene. Mice with both low (Lo) and high (Hi) levels of A2A‐R overexpression demonstrated an increase in cardiac contractility at 12 weeks. These changes were associated with a signifcantly higher systolic but not diastolic [Ca2+]i, higher maximal contraction amplitudes, and a signifcantly enhanced sarcoplasmic reticulum Ca2+ uptake activity. At 20 weeks, the effects of A2A‐R overexpression on cardiac contractility diminished. The positive effects elicited by A2A‐R overexpression differ from the heart failure phenotype we observed with A1‐R overexpresson. Interestingly, coexpression of A2A‐R TGHi, but not A2A‐R TGLo, enhanced survival, prevented the development of left ventricular dysfunction and heart failure, and improved Ca2+ handling in mice overexpressing the A1‐R. These results suggest that adenosine‐mediated signaling in the heart requires a balance between A1‐ and A2A‐Rs—a fnding that may have important implications for the ongoing clinical evaluation of adenosine receptor subtype‐specifc agonists and antagonists for the treatment of cardiovascular diseases.
Keywords: adenosine receptors, Ca2+ transients, cardiac myocytes, heart failure, transgenic mice
Introduction
The ubiquitous purine nucleotide, adenosine, regulates a variety of cardiac pathways. including growth and differentiation, angiogenesis, coronary blood fow, cardiac conduction, and heart rate, as well as mediating the cardioprotective process of ischemic preconditioning. These biologic actions of adenosine are mediated by a family of G‐protein‐coupled receptors found on the sarcolemmal surface of cardiac myocytes, including the A1‐, A2A‐, a n d A 3‐ adenosine receptors (R) and the A2B‐R, which is expressed only in the cardiac vasculature. 1 In contrast to other G‐protein‐coupled receptors in the heart, activation of select adenosine receptor subtypes can result in diametrically opposite physiologic efects. For example, activation of the A1‐R or A3‐R pathways inhibits adenylyl cyclase due to coupling with the inhibitory guanine nucleotide binding protein Gi. 2 By contrast, activation of the A2A‐R enhances the production of the intracellular second messenger cAMP through Gs‐mediated activation of adenylyl cyclase. Thus, these two receptor‐mediated signaling pathways subserve very different physiologic functions, leading investigators to propose that the role of A2A‐R in the normal heart is to reduce A1‐R mediated antiadrenergic actions. 3
Studies using transgenic mouse models harboring a transgene overexpressing the A1‐R or A3‐R 4 , 5 , 6 as well as mice in which the A1‐R has been ablated 7 have helped to clarify the role of these receptor subtypes and have confirmed their importance in cardiac protection during ischemia‐reperfusion and myocardial infarction. 8 Interestingly, recent studies using transgenic models have suggested that robust activation of A1‐R and A3‐R can have adverse consequences on cardiac morphology and function, albeit in a strain‐ and dose‐specifc manner. 9 , 10 , 11
While transgenic models have helped to clarify the role of A1/A3‐R pathways in cardiac physiology, the role of A2A‐R pathway in the heart has remained controversial. For example, studies have shown disparate effects of A2A‐R activation on cardiac contractility. 12 , 13 , 14 , 15 This controversy is due in large part to the fact that subtype‐selective adenosine receptor agonists and antagonists lack receptor‐subtype selectivity and to species‐specifc responses to pharmacologic agents. 16 Therefore, to more efectively evaluate the role of A2A‐R in cardiac physiology, we created transgenic mice with cardiac‐restricted overexpression of the A2A‐R. In contrast with mice overexpressing the A1‐R, overexpression of the A2A‐R markedly enhanced cardiac function. These salutary efects of A2A‐R activation on myocardium were due at least in part to enhanced sarcoplasmic reticulum Ca2+ uptake, resulting in higher systolic but not diastolic [Ca2+]i. Furthermore, overexpression of the A2A‐R prevented the cardiodepressant efects of A1‐R activation. These results provide important insights regarding the use of adenosine receptor subtype‐selective agonists and antagonists in the treatment of patients with cardiovascular diseases.
Materials and Methods
Transgenic mouse generation
Experiments were carried out in transgenic mice with cardiac‐restricted constitutive overexpression of the human A2A‐R engineered on an FVB background as previously described. 9 , 17 All protocols were approved by the Institutional Animal Care and Use Committee of Thomas Jeferson University.
Methods: Calculating Copy Number of Inserted Transgene
To quantify the number of transgenes inserted into the genome, genomic DNA from mouse‐tail was isolated using the Qiagen DNAeasy kit. 100 ng of genomic DNA were used for Real‐time PCR using the A2A‐AR‐specifc primer set (5′‐CAG CTG AAG CAG ATG GAG AGC‐3′; 5′‐GAT GGC CAG GTA CAT GAG CCA‐3′) and actin set (5′ AGG ACC TGT ACG CCA ACA AC 3′, 5′ACA TCT GCT GGA AGG TGG AC 3′). A2A‐R‐specifc primers were conserved between mouse and human A2A‐AR and detected human and mouse A2A gene with equal efciency (data not shown).
Real time PCR was performed in a 30 μL reaction (100 ng of genomic DNA; 250 nM each primer; 1X SYBRE Green Master Mix). Each experimental sample was performed in triplicate. The ACT method was used to quantify the results, which are presented as relative fold changes to the actin gene. Each primer set was designed to assume a Tm of 60°C and 50% GC content.
Echocardiography
Echocardiographic studies were performed using an ultrasonographic system (ACUSON Sequoia C256, Siemens, Malvern, PA, USA) as described. 18 , 19 Age matching, nontransgenic mice in FVB background served as controls. Mice were anesthetized with 2.5% Avertin (10 μL/g body weight, IP, Aldrich Chemical Co) and placed in the supine position. A 14‐MHz transducer was applied to the left hemithorax. Two‐dimensional targeted M‐mode imaging was obtained from the short‐axis view at the level of the greatest lef ventricular dimension at baseline. M‐mode measurements of left ventricular end‐diastolic and end‐systolic diameter and left ventricular anterior‐ and posterior‐wall thickness were made using the leading‐edge convention of the American Society of Echocardiography. End diastole was determined at the maximal left ventricular diastolic dimension, and end systole was taken at the peak of posterior‐wall motion.
Left ventricular hemodynamics measurement
After anesthetization with 2.5% Avertin (10 μL/g body weight, IP, Aldrich Chemical Co), mice were placed in the supine position. A 1.4 F micromanometer catheter (Millar Instruments) was inserted into the left ventricle through the right carotid artery. 18 , 19 Left ventricular pressure and heart rate were then recorded.
Immunoblotting and histopathology of myocardium
Frozen ventricular tissues were homogenized on ice using a nonionic detergent‐based lysis buffer (25 mM Tris‐HCl pH 7.6, 137 mM NaCl, 10% glycerol, 1% NP40, or IGEPAL CA‐630, 10 mM NaF) freshly supplemented with 1 mM sodium pyrophosphate, 5 μg/mL leupeptin, 5 μg/mL aprotinin, 1 mM EDTA, 10 mM PMSF, and 1 mM NaVO4. After electrophoresis in SDS‐PAGE and transfer onto nitrocellulose membranes, the blots were probed with anti‐A1‐R (Affinity BioReagents), anti‐A2A‐R (Millipore), anti‐actin (Sigma), anti‐Gαi (Abcam), anti‐NCX1 (Swant), anti‐SERCA2 (Bethyl lab), anti‐pT308‐Akt (Cell Signaling), anti‐total Akt (BD Biosciences), anti‐calsequestrin (Swant) and anti‐Na+‐K+‐ATPase (Gif from Dr. R. Levenson, Pennsylvania State University) as previously described. 9 , 20 For anti‐A1‐R and anti‐A2A‐R blots, special precaution was taken to denature protein extract in the absence of reducing agent at 70°C for 5 minutes. All blots were incubated with either IRDye 700 or 800 secondary antibodies and visualized using the Odyssey Infrared Imaging System software (Li‐Cor, Lincoln, NE, USA). Histopathology staining was performed by RADIL, University of Missouri and imaged at TJU Pathology Imaging Facility.
Isolation of adult murine cardiac myocytes
Cardiac myocytes were isolated from the septum and left ventricular free wall of wild‐type and transgenic mice (male, 8–9‐week old) as recently described. 20 Briefy, mice were heparinized (1,500 U/kg ip) and anesthetized (pentobarbital sodium, 50 mg/kg ip). Excised heart was mounted on a steel cannula and retrograde perfused (100 cmH2O, 37°C) with Ca2+‐free bicarbonate bufer followed by enzymatic digestion (collagenases B and D, protease XIV). Isolated myocytes were cultured on laminin‐coated glass cover slips and the Ca2+ concentration of the buffer was progressively increased from 0.05 to 0.125 to 0.25 to 0.5 mM in three steps (10‐minute interval each). The 0.5 mM Ca2+ buffer was then aspirated and replaced with minimal essential medium (MEM, Sigma M1018) containing 1.2 mM Ca2+, 2.5% FBS, and antibiotics (1% penicillin/streptomycin). After 1 hour (5% CO2, 37°), media were replaced with FBS‐free MEM. Myocytes were used within 2–8 hours of isolation.
Myocyte shortening measurements
Myocytes adherent to cover slips were bathed in 0.6 mL of air‐ and temperature‐equilibrated (37°), HEPES‐bufered (20 mM, pH 7.4) medium 199 containing 0.6, 1.8, or 5.0 mM [Ca2+]o. Measurements of myocyte contraction (1 Hz) were performed using a charge‐coupled device video camera and edge‐detection software (Ionoptix, Milton, MA, USA) as previously described. 20 , 21 , 22
[Ca2+]i transient measurements
Myocytes were exposed to 0.67 μM of fura‐2 AM for 15 minutes at 37°C Fura‐2‐loaded myocytes were feld‐stimulated to contract (1 Hz, 37°C) in medium 199 containing 0.6, 1.8, or 5.0 mM [Ca2+]o. Fura‐2‐loaded myocytes mounted on [Ca2+]i transient measurements using a Dvorak‐Stotler chamber situated in a temperature‐controlled stage (37°C) of a Zeiss IM 35 inverted microscope were performed as previously described. 20 , 21 , 22
Immunofuorescence microscopy
Freshly isolated myocytes were fxed in 2 mM EGTA/phosphate‐buffers saline (PBS‐EGTA) containing 4% paraformaldehyde, 20 minutes at room temperature. Cells were washed with PBS‐EGTA and pelleted by centrifugation (Beckman GS6, 600 rpm, 5 minutes). Approximately 3,000 rod‐shaped myocytes suspended in 600 μL PBS‐EGTA were mounted onto polylysine‐coated slides by cytospin (400 rpm, 2 minutes). The mounted myocytes were made permeable by incubating with blocking buffer (0.01 M Tris‐HCl, pH7.5, 0.15 M NaCl, 0.1% BSA and 0.002% NaN3) containing 0.2% Triton X‐100 for 5 minutes at room temperature. Subsequently, the slides were incubated with anti‐A1‐R (Affinity BioReagents), anti‐A2A‐R (Millipore and Affinity BioReagents) and anti‐α‐actinin (Sigma) antibodies in a 37°C humidified incubator for 45 minutes. After rinsing with blocking buffer, the myocytes were incubated with Alexa‐594‐conjugated anti‐rabbit and Alexa‐488‐conjugated anti‐mouse secondary antibodies (1:2,000) and 350 nM DAPI nuclear stain. After incubation and washing, the myocyte slides were mounted and sealed with ProLong® Gold reagent (Invitrogen) and stored in the dark at 2–6°C Myocyte images were taken with a Confocal Microscope (Zeiss LSM 510 META Confocal, KCC Bioimaging facility).
Statistics
All results are expressed as means ± SE. Kaplan‐Meier survival curves were compared between groups using log‐rank tests. Two‐way analysis of variance was used to analyze the calcium transient and contraction results. Commercial sofware package were used for all statistical analysis (JMP version 4.05; SAS Institute, Cary NC, USA) or SPSS for Windows (version 11.5). Categorical differences were analyzed using the Mann‐Whitney test. In all analyses, p < 0.05 was taken to be statistically significant.
Results
Creation of transgenic mice overexpress ing the A2A‐adenosine receptor
We placed the human A2A‐ adenosine receptor (A2A‐R) cDNA under the control of a cardiac‐specifc promoter as described previously. 9 Using an anti‐A2A‐R antibody, we analyzed levels of A2A‐R expression in both wild‐type and 15 lines of transgenic mice. Based on these measurements, the transgenic lines were classified as low expression (2–5×, A2A‐TGLo) or high expression (more than 50×, A2A‐TGHi) ( Figure 1A ). Transgenic expression was confirmed at the mRNA level by real‐time polymerase chain reaction RT‐PCR (Data not shown). Consistently, RT‐PCR assay showed that the A2A‐TGLo line has seven genomic copies of the transgene, whereas, the A2A‐TGHi line has over 25 copies ( Figure 2 ). Immunofuorescence staining of permeabilized myocytes showed that the overexpressed A2A‐R receptors were mostly localized at the sarcolemma ( Figure 1B ).
Figure 1.
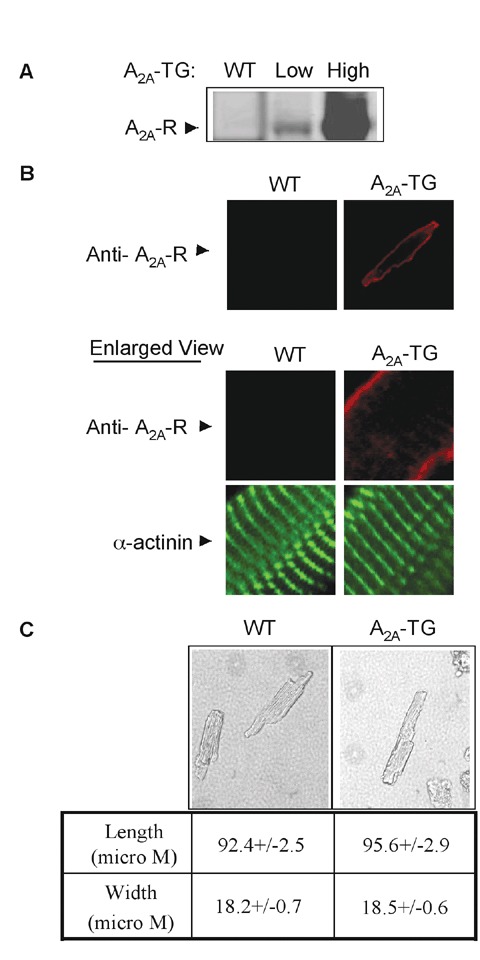
Creation of transgenic mice expressing cardiac‐specific a2a‐r. (a) A2a‐R transgenic founder lines expressing low and high levels of A2a‐R. Total ventricular protein extracts from 6‐week‐old male mice were immunoblotted with anti‐A2a‐R antibody. (B) Immunofuorescence staining of isolated myocytes showing A2a‐R and α‐actinin. 60× whole cell and enlarged images are shown. (C) Morphology of isolated myocytes and their length and width measurements. 20× images are shown. Values are means +/‐SE. n= 38–56 myocytes pooled from four to five mice.
Figure 2.

Genomic copies of inserted A2a‐R transgene. Copy number was calculated by real‐time pcr using 100 ng of genomic dna and mouse/human specifc A2a‐R primers. Data were normalized to actin and wild‐type A2a‐R gene values. Each sample was measured in triplicate and 4–6 samples were used for each genotype. Graph is shown with standard error.
A2A‐R overexpression increased cardiac contractility
As seen in Table 1 , overex pression of the A2A‐R resulted in a small but significant increase in left ventricular mass (ventricular wt/body wt) at 8–12 weeks in mice with high levels of A2A‐R overexpression but not in those with low levels of A2A‐R overexpression. Interestingly, heart rates and wall thickness were significantly increased when compared with WT mice, but they were not significantly different between A2A‐TGHi and A2A‐TGLo transgenic lines ( Table 1 ). Furthermore, measurements of single cell morphology showed no increase in either ventricular myocyte width or length ( Figure 1C ). Echocardiography demonstrated an increase in heart rate and fractional shortening as well as a signifcant decrease in left ventricular end‐systolic dimension (LVESD) in mice with both high and low levels of A2A‐R overexpression ( Table 1 ).
Table 1.
Basal physiology of high and low expression lines of A2a‐TG mice.
| WT | A2a‐TGHi | p value vs. Wt | A2a ‐TGlo | p value vs. Wt | |
|---|---|---|---|---|---|
| N = | 15 | 17 | 12 | ||
| VW/BW | 3.86 | 4.57 | <0.001 | 4.04 | NS |
| +/−0.04 | +/−0.2 | +/−0.09 | |||
| Heart Rate | 372 | 457 | <0.001 | 494 | <0.001 |
| +/−13 | +/−13 | +/−11 | |||
| LVEDD, mm | 3.28 | 3.04 | NS | 2.94 | <0.005 |
| +/−0.06 | +/−0.01 | +/−0.08 | |||
| LVESD, mm | 1.86 | 1.35 | <0.005 | 1.33 | <0.005 |
| +/−0.09 | +/−0.12 | +/−0.09 | |||
| FS,% | 43.6 | 56.6 | <0.005 | 55.05 | <0.005 |
| +/−Ί.9 | +/−2.5 | +/−2.4 | |||
| AWT | 0.70 | 0.90 | <0.005 | 0.90 | <0.005 |
| +/−0.02 | +/−0.03 | +/−0.03 | |||
| PWT | 0.73 | 0.85 | <0.005 | 0.95 | <0.005 |
| +/−0.02 | +/−0.03 | +/‐0.03 |
Organ weights and echocardiography data in mice expressing high levels (A2a‐TGHi) or low levels (A2a‐TGLo) of A2a‐R. Mice were examined at 8–12 weeks. VW/BW = ventricular weight/body weight. LVEDD and LVESD = left ventricular end‐diastolic and ‐systolic dimensions respectively. FS =% of Fractional Shortening; AWT = anterior wall thickness; PWT = posterior wall thickness.
Overexpression of the A2A‐R prevents the development of the cardiomyopathic phenotype in mice overexpressing the A1‐R
In a previous study, we demonstrated that constitutive overexpression of the A1‐R decreased cardiac contractility in FVB mice. 9 Te present finding that A2A‐R increased cardiac contractility led us to hypothesize that the adverse effects of A1‐R overexpression were due at least in part to a lack of balance in the endogenous stoichiometry of the A1‐ and A2A‐Rs. To test this hypothesis, we created double recombinants harboring both the A1‐R and A2A‐RHi transgenes (A1/A2A‐TGHi). As seen in Figure 3A , coexpression of the A1‐R and A2A‐R transgenes did not infuence the expression of either the A1‐R or A2A‐R receptors when compared with mice in which either the A1‐R‐ or A2A‐R transgenes were expressed alone. Also, immunofuorescence microscopy of isolated myocytes confirmed that both receptors colocalized to the sarcolemmal surface ( Figure 3B ). Importantly, while mice overexpressing A1‐R alone developed profound cardiac dilatation, A2A‐R coexpression prevented the profound cardiac dilatation in A1‐TG mice ( Figure 3C ). Furthermore, as seen in Figure 4 , coexpression of the A1‐R and A2A‐R transgenes significantly improved cardiac hemodynamics when compared with mice overexpressing the A1‐R. Indeed, fractional shortening, left ventricular end‐diastolic pressures, +dp/dt, and ‐dp/dt were similar in A1/A2A‐TG mice and wild‐type controls. However, heart rate remained depressed in the A1/A2A‐TG mice, albeit at a rate signifcantly higher than that seen in the A1‐TG mice. Furthermore, as seen in Figure 5A , coexpression of the A1‐R and A2A‐RHi transgenes significantly improved survival compared to mice overexpressing A1‐R alone. By contrast, coexpression of the A1‐R with the transgenic line expressing lower levels of A2A‐R (A2A‐R TGLo) had no effect on the cardiac phenotype when compared with A1‐TG mice ( Figure 5B and data not shown).
Figure 3.

Creation of transgenic mice overexpressing both A1‐R and A2A‐R. (A) A1‐R and A2A‐R expression in WT, A1‐TG, A2A‐TG and A1/A2A‐TG mice. Ventricular extracts from 8‐ week‐old male mice were probed with indicated antibodies. (B) A1‐R and A2A‐R colocalized on cardiac myocyte membrane. 60× confocal images are shown. (C) Horizontal sections of mouse hearts stained with hematoxylin‐eosin (HE). Minimum two animals (8‐week‐old male) of each genotype were stained and representative scaled photographic images (1×) are shown.
Figure 4.

A2a‐R expression improved cardiac function and hemodynamics without affecting heart rate in A1‐TG mice. (A) Percent fractional shortening of indicated mouse groups; (B) Heart rate of indicated mouse groups; (C) and (D) Hemodynamic functions of transgenic mice: maximum rate of ventricular pressure rise and decline (+ DP/DT and ‐DP/DT.) *p < 0.001 versus WT, †p < 0.001 versus A1TG. (n= 15–21, SE, male 8–12‐week‐old mice).
Figure 5.

Animal survival, calcium handling, and gene expression in A1TG and A2a‐TG mice. (a) Kaplan‐Meier survival curve for A1‐R transgenic lines coexpressing high levels of A2a‐R (A2a‐TGHi). A1 versus A1/A2a p < 0.001. (B) Kaplan‐Meier survival curve for A1‐R transgenic lines coexpressing low levels of A2a‐R (A2a‐TGlo). A1 versus A1/A2a NS. (C) Representative tracings of myocyte Ca2+ transients and contractions. Detailed calculations are shown in Table 2. (D) SERCA2 and Gαi2 expression. Ventricular extracts from 8‐week‐old male mice were probed with indicated antibodies. SERCA2 and Gαi2 signals were normalized to average intensity in WT hearts (n= 4–7) and were analyzed by one‐way analysis of variance followed by Dunnett's test. Analyzed values were mean +/‐ SE, *p < 0.05 versus WT.
Overexpression of A2A‐R enhances myocyte [Ca2+]i transport
In view of the marked increase in contractility in the A2A‐TGHi mice and their ability to prevent heart failure in the A1‐TG mice, we hypothesized that mice harboring the A2A transgene might have enhanced Ca2+ handling. To test this hypothesis, we first compared Ca2+ handling in myocytes isolated from A2A‐TGHi, A1‐TG, and wild‐type nontransgenic littermates. As seen in Figure 5C and Table 2 , when compared to WT myocytes, constitutive overexpression of A2A‐R resulted in significant higher systolic (p < 0.0001) but not diastolic [Ca2+]i (p < 0.60). Maximal contraction amplitudes (p < 0.0001), maximal shortening (p < 0.0003), and relengthening velocities (p < 0.006) were all higher in myoctyes overexpressing A2A‐R ( Table 2 ). Sarcoplasmic reticulum (SR) Ca2+ uptake activity was significantly faster (p < 0.0001).
Table 2.
Effects of adenosine receptor overexpression on [Ca2+]i transients, myocyte contraction, and calcium handling protein expression.
| WT | A2a‐TCHi | Ai‐TC | Ai‐TC A2a‐TCHi | |
|---|---|---|---|---|
| Calcium transients [Ca2+]0 mM | ||||
| Systolic [Ca2+]I, nM | ||||
| 0.6 | 167 ± 10 | 205 ± 13* | 110 ± 9* | 181 ± 14*$ |
| (20) | (28) | (20) | (12) | |
| 1.8 | 215 ± 10 | 295 ± 14* | 172 ± 12* | 271 ± 22*$ |
| (39) | (32) | (38) | (32) | |
| 5.0 | 336 ± 11 | 488 ± 18* | 304 ± 30* | 480 ± 80*$ |
| (41) | (35) | (15) | (19) | |
| Diastolic [Ca2*], nM | ||||
| 0.6 | 118 ± 7 | 108 ± 6+ | 73 ± 9* | 94 ± 13* |
| 1.8 | 111 ± 7 | 116 ± 6+ | 95 ± 7* | 86 ± 11* |
| 5.0 | 131 ± 5 | 125 ± 5+ | 113 ± 11* | 85 ± 17* |
| t1/2[Ca2+]| transient decline, ms | ||||
| 0.6 | 216 ± 13 | 175 ± 11*+ | 307 ± 32* | 231 ± 22*$ |
| 1.8 | 174 ± 7 | 124 ± 7*+ | 243 ± 13* | 143 ± 8*$ |
| 5.0 | 151 ± 5 | 100 ± 4*+ | 200 ± 8* | 133 ± 8*$ |
| Myocyte contraction [Ca2+]0 | ||||
| Maximal contraction amplitude,% cell length | ||||
| 1.8 | 6.80 ± 0.43 | 9.07 ± 0.42*+ | 4.99 ± 0.45* | 7.85 ± 0.41 *$ |
| (56) | (38) | (36) | (36) | |
| 5.0 | 10.59 ± 0.44 | 12.79 ± 0.57*+ | 7.17 ± 0.82* | 11.92 ± 0.35*$ |
| (38) | (49) | (11) | (22) | |
| Maximal shortening velocity, ce1l length/s | ||||
| 1.8 | 1.05 ± 0.07 | 1.39 ± 0.06*+ | 0.63 ± 0.06* | 1.00 ± 0.05$ |
| 5.0 | 1.58 ± 0.10 | 1.84 ± 0.09*+ | 0.90 ± 0.13* | 1.53 ± 0.07$ |
| Maximal relengthening velocity,cell length/s | ||||
| 1.8 | 0.92 ± 0.07 | 1.17 + 0.07*+ | 0.42 ± 0.04* | 0.75 ± 0.05$ |
| 5.0 | 1.29 ± 0.10 | 1.49 ± 0.09*+ | 0.68 ± 0.13 | 1.17 ± 0.05$ |
| Selected calcium handling protein expression | ||||
| SERCA2 | 1.00 ± 0.07 | 1.42 ± 0.05*$ | 0.63 ± 0.11* | 1.37 ± 0.14*$ |
| (7) | (4) | (7) | (4) | |
| Na+ pump | 1.00 ± 0.03 | 0.78 ± 0.09$ | 0.43 ± 0.11* | 0.71 ± 0.04*$ |
| (4) | (4) | (4) | (4) | |
| NCX1 | 1.00 ± 0.12 | 1.45 ± 0.15$ | 0.83 ± 0.19 | 1.32 ± 0.17$ |
| (4) | (4) | (4) | (4) | |
For calcium transient and contraction measurements, numbers in parentheses are myocytes pooled from 4–5 mice/genotype group. Mouse groups: wild‐type (WT), A2a‐TGHi, A1‐TG) and A1/A2a‐TG. Values are means ± SE. Two‐way analysis of variance was used for analysis. *p < 0.05, compared to WT; $p < 0.0001, A1‐TG vs. A1‐TG/A2a‐TGHi; + p < 0.05, A2a‐TG vs. A1‐TG/A2a‐TGHi. Protein expression was determined by immunoblotting. Numbers in parentheses are number of hearts from each mouse group. Signal band intensities on each blot were normalized to the average intensity of that protein measured in wild‐type hearts. One‐way analysis of variance followed by Dunnett's test was used to analyze the results. *p < 0.05, compared to WT; $A1‐TG vs, A2a‐TGHi or A1‐TG vs. A1/A2a‐TGHi.
By contrast, myocytes in which A1‐R was constitutively overexpressed had significantly lower systolic (p < 0.0005) and diastolic [Ca2+]i (p < 0.0007) at all [Ca2+]o examined ( Table 2 ). Altered [Ca2+]i transients in myocytes with constitutively overexpressed A1‐R resulted in decreased maximal contraction amplitudes as well as maximal shortening and relengthening velocities (p < 0.0001 for all three parameters; Table 2 ). Furthermore, the t1/2 of [Ca2+]i transient decline, an estimate of sarcoplasmic reticulum Ca2+ uptake, 23 was significantly prolonged in A1‐R TG myocytes when compared with WT (p < 0.0001) myocytes.
Co‐overexpression of A1‐ and A2A‐R ameliorates the abnormal [Ca2+]i handling seen in the A1‐R TG myocytes
When analyzing myocyte function in our dual A1‐/A2A‐R mice, we found that the overexpression of the A2A‐R significantly ameliorated the contractile abnormalities observed in A1‐R TG myocytes (p < 0.0001 for maximal contraction amplitude, maximal shortening, and relengthening velocities, Table 2 ). In addition, maximal contraction amplitude of A1/A2A–R TG myocytes was significantly higher (p < 0.015) than that observed in WT myocytes but lower (p < 0.05) than that measured in A2A‐R overexpressed myocytes ( Table 2 ). With respect to [Ca2+]i homeostasis, SR Ca2+ uptake activity (p < 0.0001) and systolic (p < 0.0001) but not diastolic [Ca2+]i (p < 0.36) was improved by co‐overexpression of A1 and A2A‐R when compared with constitutive overexpression of A1‐R alone ( Table 2 ). Diastolic [Ca2+]i was significantly lower in A1/A2A myocytes when compared to WT (p < 0.0001) or A2A‐R overexpressed (p < 0.0001) myocytes. Te t1/2 of [Ca2+]i transient decline was significantly shorter in A1/A2A myocytes when compared to WT myocytes (p < 0.009) but longer when compared to A2A‐R overexpressed myocytes (p < 0.002).
Efects of enhanced A2A‐R signaling on biochemical pathways in the heart
In order to understand the mechanisms responsible for the enhanced contractility effected by increased A2A‐R signaling, we assessed the levels of proteins involved in Ca2+ homeostasis and G‐protein‐coupled receptor signaling in the heart. As seen in Figure 5D , the amount of SERCA2 was significantly increased in hearts from A2A‐R TGHi mice when compared with wild‐type controls. The level of SERCA2 protein was also significantly elevated in A1/A2A‐TGHi mice but by contrast was significantly lower than WT controls in mice overexpressing the A1‐R. Te levels of the Na+ pump and NCX1 in the A2A‐TG mice had changing trends, but they were not statistically different from the WT mice. By contrast, mice overexpressing the A1‐R had decreased Na+ pump protein levels. It is interesting to note that co‐overexpression of A2A‐R in A1‐TG mice enhanced the expression of all three proteins involved in Ca2+ homeostasis when compared to mice expressing only A 1‐R ( Figure 5D and Table 2 ). However, the change in contractility seen in the A2A‐R TGHi mice as well as in the A2A‐R TGLo mice was not associated with an increase in steady‐state adenylyl cyclase activity (data not shown). By contrast, levels of the G‐inhibitory protein, Gαi‐2, as well as the levels of mRNA encoding Gαi‐2 were increased in all three experimental models (A2a‐TGHi, A1‐TG, a1/a2a‐tg; Figure 5D and data not shown).
Long‐term effects of enhanced A2A‐R signaling
Although overexpression of the A2A‐R markedly enhanced cardiac contractility in mice up to 12 weeks of age, long‐term overexpression was not associated with an increase in cardiac contractility. Indeed, cardiac function was identical in 20‐week‐old transgenic and nontransgenic controls as seen by measurement of fractional shortening (42 ± 2.0% WT, n = 7 vs. 40.3 ± 3.4% A2A‐TGHi, n = 13, p= NS). At 20 weeks, A2A‐TG mice remained hypertrophied, but without dilation (data not shown). However, compared to younger mice, the 20‐week‐old A2A‐TG hearts developed significant fibrosis ( Figure 6 ). Furthermore, the fibrosis progressively worsened at 30 weeks (data not shown). Also, SERCA2 levels were not elevated in 20‐week‐old A2A‐TGHi mice (1.1 ± 0.1, n= 7) when compared with wild‐type controls (1.0 + 0.1,H = 13, p= NS). Similarly, while phosphorylation (i.e., activation) of Akt was signifcantly enhanced in young mice (12 weeks old) overexpressing the A2A‐R transgene (1.03 ± 0.2 WT, n= 3 vs. 1.8 ± 0.16, A2A‐TGHi, n = 6, p < 0.01), pAkt levels were not elevated in 20‐week‐old A2A‐R TGHi mice (0.91 ± 0.12, n = 9) when compared with wild‐type controls (1.12 ± 0.15, n = 7, p= NS). However, levels of Gαi remained elevated in the hearts of the 20‐week‐old A2A‐TGHi mice (1.9 ± 0.3 A2A‐R TGHi, n= 13 vs. 1.0 ± 0.1 WT, n = 7, p < 0.01).
Figure 6.

Pico Sirius Red staining of 12‐week and 20‐week‐old A2a‐TGhi myocardium (collagen‐stained purple). Representative 10× objective images are shown. Two independent observers evaluated fbrosis based on 4–7 samples/group.
Discussion
In the present study, we have utilized cardiac‐restricted overexpression of the A2A‐R to assess for the first time the effects of A2A‐R‐signaling on cardiac morphology and function independent of the limitations imposed by experiments utilizing receptor subtype “selective” agonists and antagonists. Constitutive overexpression of the A2A‐R in young mice resulted in super normal contractility, which was associated with a modest increase in heart rate and a small but significant increase in LV mass. In myocytes isolated from hearts overexpressing the A2A‐R, systolic but not diastolic [Ca2+]i was elevated and t1/2 of [Ca2+]i transient decline was much shorter when compared with wild‐type controls and myocyte size did not change. These observations are consistent with enhanced SR Ca2+ uptake, resulting in increased SR Ca2+ content, more Ca2+ available for release per beat, and larger systolic [Ca2+]i values and twitch amplitudes in A2A‐R overexpressed myocytes. In support of this mechanism of action is the observation that SERCA2 but not Na+‐K+‐ATPase or NCX1 expression was increased in A2A‐R overexpressed myocytes. Thus, viewed in terms of excitation‐contraction coupling, the major alteration induced by A2A‐R overexpression was increased SERCA2 expression and SR Ca2+ uptake activity.
These findings are disparate from previous studies evaluating the role of adenosine receptor subtype‐specific agonists and antagonists, which suggested that A2A‐R‐mediated inotropy was accompanied by only a small increase in Ca2+ transients leading investigators to suggest that the A2A‐R‐induced contractile effects were mediated by Ca2+‐independent inotropic mechanisms. 24 Furthermore, these earlier studies also suggested that A2A‐R activation could increase shortening and the rate of maximal shortening in isolated myocytes without an effect on maximal rate of relaxation 25 while in isolated hearts, A2A‐R activation could increase left ventricular pressure and the maximal rate of pressure development (+dP/dt/max) without infuencing cardiac relaxation. 14 By contrast, in this study we demonstrate enhanced contractility as well as enhanced relaxation, these effects being associated with increased expression of SERCA2 and robust changes in Ca2+ handling. Importantly, the changes in cardiac function after A2A‐R overexpression were not due to an increase in heart rate as we were able to see enhanced contractility both invivo as well as in isolated and paced myocytes. The disparity between the present results and earlier studies may be due to the lack of “specifcity” of pharmacologic agonists and antagonists. However, we cannot exclude the possibility of species‐related differences as earlier studies were performed in rats.
Because of the contrasting effects of A1‐ and A2A‐R overexpression on cardiac function, we wondered whether abnormalities in calcium homeostasis might also be responsible for the adverse effects of A1‐R overexpression. Indeed, left ventricular myocyte contractility was severely depressed in A1‐TG myocytes. In addition, systolic [Ca2+]i in A1‐R myocytes was lower at all 3 [Ca2+]o examined, suggesting that SR Ca2+ uptake was diminished. 26 The decreased SERCA2 expression and prolonged t1/2 of [Ca2+]i transient decline in A1‐TG myocytes are consistent with deffective SR Ca2+ uptake activity, resulting in decreased SR Ca2+ content and diminished twitch amplitudes. Diastolic [Ca2+]i was also lower with A1‐R overexpression, suggesting that accelerated Ca2+ effux may further contribute to reduced SR Ca2+ content and twitch amplitude. As one of the major alterations in excitation‐contraction coupling in A2A‐TG myocytes was enhanced SERCA2 expression and SR Ca 2+ uptake, we hypothesized that enhanced A2A‐R signaling might ameliorate the negative inotropic effects of enhanced A1‐R signaling. Indeed, co‐overexpression of the A2A‐R improved cardiac contractility, decreased end‐diastolic pressure, enhanced SERCA2 expression, and markedly improved survival when compared with mice overexpressing the A1‐R. These salutary benefits were also seen at the single cell level as coexpression of the A1‐R and A2A‐R ameliorated the marked cellular abnormalities found in the A1‐TG mice. Because systolic [Ca2+]i and t1/2 of [Ca2+]i transient decline improved without a change in diastolic [Ca2+]i in the A1‐/A2A‐TG mice when compared with A1‐TG mice, it is likely that defects in SR Ca2+ uptake but not Ca2+ effux pathways induced by constitutive A1‐R overexpression were influenced by co‐overexpression of A2A‐R. However, it is also noteworthy that the ability of A2A‐R signaling to ameliorate the adverse efects of A1‐R overexpression was dose related as crossing the A1‐TG with A2A‐R TGLo mice had no effect on cardiac hemodynamics or outcomes despite an increase in contractility in the A2A‐TGLo mice.
Despite the salutary benefits of short‐term overexpression of the A2A‐R, long‐term benefits were not obvious as by 20 weeks of age, contractility decreases substantially to levels less similar to those seen in WT controls. Interestingly, this fnding was consistent with results seen after overexpression of other G‐protein‐coupled receptors. For example, overexpression of the β2‐adrenergic receptor (AR) results in left ventricular dysfiunction in older mice while mice overexpressing the β1‐AR develop signifcant LV dysfunction and early death. The markedly different phenotype in young mice overexpressing the β2‐AR and those overexpressing the β1‐AR has been attributed to the cardioprotective effects of enhanced Gαi signaling in the β2‐AR overexpressing mice. 27 The increase in Gαi we observed in mice overexpressing the A2A‐R may have provided similar beneficial effects in young mice by limiting or opposing the effects of activation of Gas. However, it is interesting to note that in contrast with β‐AR overexpression, both mice overexpressing the A2A‐R and mice overexpressing Gαs 28 did not demonstrate an increase in steady‐state adenylyl cyclase activity. Thus, the enhanced inotropy secondary to increased A2A‐R signaling in the mouse may be mediated through adenylyl cyclase dependent and independent pathways. Regardless, increased cardiac fbrosis in older A2A‐R overexpressing mice suggested a potential mechanism for diminishing contractile benefits derived from A2A‐R overexpression. Since A2A‐R, through Gs coupling, increased cAMP‐dependent transsarcolemmal calcium fux and contraction, it is highly possible that sustained contractile overdrive in A2A‐TG mice may ultimately lead to “calcium‐overload” leading to dysfunction and structural injury. However, similar to the β‐adrenergic receptor transgenic mice, 27 other pathways may also participate in the late myopathy.
Another possible explanation for the finding that early activation of A2A‐R signaling did not cause left ventricular dysfunction or pathology is the signifcant increase in Akt levels in young mice. Phosphorylation (activation) of Akt is known to have cardioprotective effects. 29 Indeed, while phosphorylation of Akt was enhanced in young mice overexpressing the A2A‐R, it was not increased in older A2A‐TG mice with “normalization” of left ventricular function. These results were consistent with our recent fnding that Akt activity was substantially decreased in mice with left ventricular dysfunction secondary to overexpression of the A1‐R. 9 Finally, our results suggest that the therapeutic usefulness of A2A‐R overexpression might be predicated on “dose” as despite an inability to reverse left ventricular dysfunction caused by A1‐R overexpression, low levels of A2A‐R overexpression significantly increased cardiac contractility without causing an increase in either ventricular or atrial hypertrophy. Nonetheless, additional studies will be required to identify the role of low levels of A2A‐R overexpression in preventing or rescuing left ventricular dysfunction in other experimental models. Furthermore, additional studies using controlled overexpression of the A2A‐R receptor may shed additional light on the important downstream signaling pathways that play a role in the development of the early and late A2A‐R phenotype.
Taken together, our results suggest that despite the fact that the A1‐R and A2A‐R signaling pathways affect different downstream events, the physiologic integrity of the heart, at least in a chronic sense, requires an ongoing balance between the activation of these two pathways. Indeed, the family of adenosine receptor subtypes is an anomaly because it is the only group of G‐protein‐coupled, seven‐transmembrane‐spanning receptors in which a single ligand can bind to multiple receptor subtypes in the same tissue and in so doing mediate opposing signaling pathways—that is, adenylylcyclase activation in the case of the A2A‐R and adenylyl cyclase inhibition viathe A1‐R. Thus, it would appear axiomatic that the chronic use of subtype selective adenosine receptor agonists or antagonists might have unexpected consequences. These fndings should be considered when designing adenosine agonists and/or antagonists for chronic use in patients with cardiovascular diseases.
Acknowledgments
This study was supported by Pennsylvania Research Formulary Fund (A.M.F., T.O.C.) and the American Heart Association SDG F64702 (T.O.C).
Disclosures: None.
References
- 1. Yang D, Zhang Y, Nguyen HG, Koupenova M, Chauhan AK, Makitalo M, Jones MR, St Hilaire C, Seldin DC, Toselli P, Lamperti E, Schreiber BM, Gavras H, Wagner DD, Ravid K. The A2B adenosine receptor protects against inflammation and excessive vascular adhesion. J Clin Invest. 2006; 116(7): 1913–1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Klinger M, Freissmuth M, Nanoff C. Adenosine rec eptors: G protein‐mediated signalling and the role of accessory proteins. Cell Signal. 2002; 14(2): 99–108. [DOI] [PubMed] [Google Scholar]
- 3. Norton GR, Woodiwiss AJ, McGinn RJ, Lorbar M, Chung ES, Honeyman TW, Fenton RA, Dobson JG Jr, Meyer TE. Adenosine A1 receptor‐mediated antiadrenergic effects are modulated by A2a receptor activation in rat heart. Am J Physiol. 1999; 276(2 Pt 2): H341–H349. [DOI] [PubMed] [Google Scholar]
- 4. Cross HR, Murphy E, Black RG, Auchampach J, Steenbergen C. Overexpression of A(3) adenosine receptors decreases heart rate, preserves energetics, and protects ischemic hearts. Am J Physiol Heart Circ Physiol. 2002; 283(4): H1562–H1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yang Z, Cerniway RJ, Byford AM, Berr SS, French BA, Matherne GP. Cardiac overexpression of A1‐adenosine receptor protects intact mice against myocardial infarction. Am J Physiol Heart Circ Physiol. 2002; 282(3): H949–H955. [DOI] [PubMed] [Google Scholar]
- 6. Morrison RR, Jones R, Byford AM, Stell AR, Peart J, Headrick JP, Matherne GP. Transgenic overexpression of cardiac A(1) adenosine receptors mimics ischemic preconditioning. Am J Physiol Heart Circ Physiol. 2000; 279(3): H1071–H1078. [DOI] [PubMed] [Google Scholar]
- 7. Reichelt ME, Willems L, Molina JG, Sun CX, Noble JC, Ashton KJ, Schnermann J, Blackburn MR, Headrick JP. Genetic deletion of the A1 adenosine receptor limits myocardial ischemic tolerance. Circ Res. 2005; 96(3): 363–367. [DOI] [PubMed] [Google Scholar]
- 8. Lankford AR, Yang JN, Rose'Meyer R, French BA, Matherne GP, Fredholm BB, Yang Z. Effect of modulating cardiac A1 adenosine receptor expression on protection with ischemic preconditioning. Am J Physiol Heart Circ Physiol. 2006; 290(4): H1469–H1473. [DOI] [PubMed] [Google Scholar]
- 9. Funakoshi H, Chan TO, Good JC, Libonati JR, Piuhola J, Chen X, MacDonnell SM, Lee LL, Herrmann DE, Zhang J, Martini J, Palmer TM, Sanbe A, Robbins J, Houser SR, Koch WJ, Feldman AM. Regulated overexpression of the A1‐adenosine receptor in mice results in adverse but reversible changes in cardiac morphology and function. Circulation. 2006; 114(21): 2240–2250. [DOI] [PubMed] [Google Scholar]
- 10. Gauthier NS, Headrick JP, Matherne GP. Myocardial function in the working mouse heart overexpressing cardiac A1 adenosine receptors. J Mol Cell Cardiol. 1998; 30(1): 187–193. [DOI] [PubMed] [Google Scholar]
- 11. Matherne GP, Linden J, Byford AM, Gauthier NS, Headrick JP. Transgenic A1 adenosine receptor overexpression increases myocardial resistance to ischemia. Proc Natl Acad Sci U S A. 1997; 94(12): 6541–6546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kilpatrick EL, Narayan P, Mentzer RM Jr, Lasley RD. Cardiac myocyte adenosine A2a receptor activation fails to alter cAMP or contractility: role of receptor localization. Am J Physiol Heart Circ Physiol. 2002; 282(3): H1035–H1040. [DOI] [PubMed] [Google Scholar]
- 13. Lasley RD, Jahania MS, Mentzer RM Jr. Benefcial effects of adenosine A(2a) agonist CGS‐21680 in infarcted and stunned porcine myocardium. Am J Physiol Heart Circ Physiol. 2001; 280(4): H1660–H1666. [DOI] [PubMed] [Google Scholar]
- 14. Monahan TS, Sawmiller DR, Fenton RA, Dobson JG Jr. Adenosine A(2a)‐receptor activation increases contractility in isolated perfused hearts. Am J Physiol Heart Circ Physiol. 2000; 279(4): H1472–H1481. [DOI] [PubMed] [Google Scholar]
- 15. Shryock J, Song Y, Wang D, Baker SP, Olsson RA, Belardinelli L. Selective A2‐adenosine receptor agonists do not alter action potential duration, twitch shortening, or cAMP accumulation in guinea pig, rat, or rabbit isolated ventricular myocytes. Circ Res. 1993; 72(1): 194–205. [DOI] [PubMed] [Google Scholar]
- 16. Headrick JP, Hack B, Ashton KJ. Acute adenosinergic cardioprotection in ischemic‐reperfused hearts. Am J Physiol Heart Circ Physiol. 2003; 285(5): H1797–H1818. [DOI] [PubMed] [Google Scholar]
- 17. Sanbe A, Gulick J, Hanks MC, Liang Q, Osinska H, Robbins J. Reengineering inducible cardiac‐specific transgenesis with an attenuated myosin heavy chain promoter. Circ Res. 2003; 92(6): 609–616. [DOI] [PubMed] [Google Scholar]
- 18. Funakoshi H, Kubota T, Kawamura N, Machida Y, Feldman AM, Tsutsui H, Shimokawa H, Takeshita A. Disruption of inducible nitric oxide synthase improves beta‐adrenergic inotropic responsiveness but not the survival of mice with cytokine‐induced cardiomyopathy. Circ Res. 2002; 90(9): 959–965. [DOI] [PubMed] [Google Scholar]
- 19. Funakoshi H, Kubota T, Machida Y, Kawamura N, Feldman AM, Tsutsui H, Shimokawa H, Takeshita A. Involvement of inducible nitric oxide synthase in cardiac dysfunction with tumor necrosis factor‐alpha. Am J Physiol Heart Circ Physiol. 2002; 282(6): H2159–H2166. [DOI] [PubMed] [Google Scholar]
- 20. Tucker AL, Song J, Zhang XQ, Wang J, Ahlers BA, Carl LL, Mounsey JP, Moorman JR, Rothblum LI, Cheung JY. Altered contractility and [Ca2+]i homeostasis in phospholemman‐deficient murine myocytes: role of Na+/Ca2 +exchange. Am J Physiol Heart Circ Physiol. 2006; 291(5): H2199–H2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Song J, Zhang XQ, Ahlers BA, Carl LL, Wang J, Rothblum LI, Stahl RC, Mounsey JP, Tucker AL, Moorman JR, Cheung JY. Serine 68 of phospholemman is critical in modulation of contractility, [Ca2+]i transients, and Na+/Ca2+ exchange in adult rat cardiac myocytes. Am J Physiol Heart Circ Physiol. 2005; 288(5): H2342–H2354. [DOI] [PubMed] [Google Scholar]
- 22. Song J, Zhang XQ, Carl LL, Qureshi A, Rothblum LI, Cheung JY. Overexpression of phospholemman alters contractility and [Ca(2+)](i) transients in adult rat myocytes. Am J Physiol Heart Circ Physiol. 2002; 283(2): H576–H583. [DOI] [PubMed] [Google Scholar]
- 23. Zhang XQ, Ng YC, Moore RL, Musch TI, Cheung JY. In situ SR function in postinfarction myocytes. J Appl Physiol. 1999; 87(6): 2143–2150. [DOI] [PubMed] [Google Scholar]
- 24. Woodiwiss AJ, Honeyman TW, Fenton RA, Dobson JG Jr. Adenosine A2a‐receptor activation enhances cardiomyocyte shortening via Ca2+‐independent and ‐dependent mechanisms. Am J Physiol. 1999; 276(5 Pt 2): H1434–H1441. [DOI] [PubMed] [Google Scholar]
- 25. Dobson JG Jr, Fenton RA. Adenosine A2 receptor function in rat ventricular myocytes. Cardiovasc Res. 1997; 34(2): 337–347. [DOI] [PubMed] [Google Scholar]
- 26. Ahlers BA, Song J, Wang J, Zhang XQ, Carl LL, Tadros GM, Rothblum LI, Cheung JY. Effects of sarcoplasmic reticulum Ca2+‐ATPase overexpression in postinfarction rat myocytes. J Appl Physiol. 2005; 98(6): 2169–2176. [DOI] [PubMed] [Google Scholar]
- 27. Foerster K, Groner F, Matthes J, Koch WJ, Birnbaumer L, Herzig S. Cardioprotection specifc for the G protein Gi2 in chronic adrenergic signaling through beta 2‐adrenoceptors. Proc Natl Acad Sci U S A. 2003; 100(24): 14475–14480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gaudin C, Ishikawa Y, Wight DC, Mahdavi V, Nadal‐Ginard B, Wagner TE, Vatner DE, Homcy CJ. Overexpression of Gs alpha protein in the hearts of transgenic mice. J Clin Invest. 1995; 95(4): 1676–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol. 2006; 7(8): 589–600. [DOI] [PubMed] [Google Scholar]