

Abstract
We report the use of the molecular signatures known as “Property-Encoded Shape Distributions” (PESD) together with standard Support Vector Machine (SVM) techniques to produce validated models that can predict the binding affinity of a large number of protein ligand complexes. This “PESD-SVM” method uses PESD signatures that encode molecular shapes and property distributions on protein and ligand surfaces as features to build SVM models that require no subjective feature selection. A simple protocol was employed for tuning the SVM models during their development, and the results were compared to SFCscore – a regression-based method that was previously shown to perform better than 14 other scoring functions. Although the PESD-SVM method is based on only two surface property maps, the overall results were comparable. For most complexes with a dominant enthalpic contribution to binding (ΔH/-TΔS > 3), a good correlation between true and predicted affinities was observed. Entropy and solvent were not considered in the present approach and further improvement in accuracy would require accounting for these components rigorously.
Introduction
Accurate prediction of protein-ligand binding affinity is a key component of computer-aided drug discovery. There are many techniques for affinity prediction1-15, with notable accuracy (1 kcal/mol) being seen with combination of molecular dynamics and free energy perturbation techniques12,16,17. In drug discovery applications, fast computation of affinity is highly desirable to enable rapid virtual screening for potency, which is currently attempted using scoring functions based on the static structures of protein-ligand complexes. In spite of the progress made over several years, the applicability of the scoring functions for affinity prediction across different proteins remains limited as demonstrated by recent benchmarking studies18. Binding affinity is a thermodynamic process that involves both enthalpic and entropic contributions to ligand pose stability. Unfortunately, accounting for entropy from a static model is difficult, and most scoring functions provide only minimal treatment (generally as a “rotor” term) for this important contribution. Ladbury and Williams19 pointed out that “specific attribution of thermodynamic parameters to the formation/breaking of particular local non-covalent interactions, to conformational or dynamic change, or to solvent reorganisation is not easy to achieve”. However, good correlation between change in buried apolar surface area on complex formation and free energy (though not necessarily with entropy) 20, and improved performance of empirical scoring functions on enrichment of the training set11 have also been previously noted. These could be contributors to the modest to good correlations between true affinity and predicted affinity observed in some protein-ligand systems. Until such time that entropic contributions to binding affinity can be accurately assessed in high-throughput virtual screening applications, the development of new generalized scoring functions needs to be coupled with an increased awareness of the applicability domains of those new scoring functions. Such an analysis appears later in this report.
Recently, we developed the “Property-Encoded Shape Distributions” (PESD) concept that enabled us to determine similarities between many functionally related binding sites by analyzing structural similarity at the level of molecular surface21. PESD signatures account for distribution of polar and apolar regions as well as electrostatic potential on the molecular surface. In this study, we investigate to what extent the encoding of surface property distributions within PESD signatures can explain observed variance in binding affinity in the absence of any explicit treatment for solvent and entropy given the observed correlation between change in buried apolar surface area and free energy. Surface property distributions have also been encoded by methods such as the MaP approach22 by Stiefl and Baumann, the autocorrelation descriptors of surfaces23 by Wagener, Sadowski and Gasteiger, Surfcats descriptors24 by Renner and Schneider, PEST descriptors by Breneman and coworkers25 and shape signatures of Zauhar and coworkers26. However, unlike others, the PESD algorithm is a novel approach that is based on a fixed number of randomly sampled point pairs on the molecular surface that does not require ray-tracing or the equal spacing of ligand or protein surface points. In the current study, PESD signatures calculated from both protein and ligand interaction surfaces are utilized as features for creating Support Vector Machine27 (SVM) models for binding affinity prediction. Therefore, the binding affinity prediction approach is proteochemometric, a term coined by Wikberg and coworkers28. Proteochemometric approaches use both the protein (usually in and around the binding site) and the ligand structural features to build predictive models11, 28-36. We chose a recently published proteochemometric method called SFCscore for comparison with the PESD-SVM method. SFCscore is an empirical scoring function that is trained on descriptors (including surface based) derived from the ligand as well as the protein component of each complex.
Following a description of our approach, we discuss the results of applying PESD-SVM models to complexes in the PDBbind37, 38 data set for affinity prediction. We next compare PESD-SVM results with those of SFCscore that was previously benchmarked against 14 other scoring functions39 for affinity prediction. Finally, we analyze the results and discuss the strengths and shortcomings of the present method.
Methods
Protein structure preparation
Protein structures obtained from the PDBbind database version 200537, 38 were appropriately protonated with the Protonate3D routine40 in MOE41 at the pH at which the complexes were crystallized42, 43. The pH values were extracted directly from the PDB44 files of the respective complexes. For structures not having a specified pH, a default pH of 7.0 was used. The electrostatics cutoff was set at 12 Å. To reduce the computational time used in preprocessing protein structures, the following “sliding scale” was used to determine whether to include specific waters during structure optimization: If the protein structure was of significantly high resolution (less than 2.2 Å resolution and an r-factor of less than 0.29) and was small enough (less than 6000 daltons), all waters were included in subsequent optimization. For all other structures, waters were included only if they were located less than 3.8 Å from the ligand.
Generation of molecular interaction surfaces
Property mapped interaction surfaces were generated using the MOE41 package. The protein interaction surface was defined as the Gauss-Connolly surface of the protein at 4.5 Å or less from any ligand atom; the ligand interaction surface was defined as the Gauss-Connolly surface of the ligand at 3 Å or less from any protein atom. A 4.5 Å cutoff (default) is typically used for defining an active site in MOE, whereas the 3 Å cutoff for the ligand was chosen to eliminate solvent exposed ligand surfaces further away from the interfacial region. Ligand and protein interaction surfaces were encoded with EP and Active LP (ALP) surface maps45. The EP map was a Ewald-type screened molecular electrostatic potential that covered a range of -35 to 35 kcal/mol 46. Potential values occurring outside the range were clamped to lie inside the range. Gasteiger-Huckel partial charges from PDBbind ligand structures were used for computing the ligand EP surface map, whereas partial charges assigned to the protein from the structure optimization step with Protonate3D were used for computing the EP map for the protein interaction surface. The ALP surface map displays different colors that represent hydrogen-bonding regions, mildly polar regions and hydrophobic regions.
PESD signature generation
The Property Encoded Shape Distributions (PESD) method was originally developed to find similarities between binding site shape and surface properties by comparing protein interaction surface PESD signatures. PESD signatures are invariant to rotation and translation, exploiting the concept of Shape Distributions47 and extending it by adding the capability of capturing three dimensional distributions of mapped properties on a molecular surface. Triangulated molecular surfaces are commonly generated by molecular graphics programs, including MOE, for visualization purposes - thus, PESD was designed to work directly from such surface files. Each vertex of a property mapped MOE-generated surface mesh is represented by its Cartesian coordinates and a 24-bit RGB color code representing the mapped property magnitude. The PESD routine samples pairs of points from random locations on the triangulated molecular surface mesh. Collections of point pairs are then binned in a two-dimensional binning grid by the distance between the point pairs as well as the property magnitudes or “color combinations” on both endpoints (Figure 1). A coarse-grained binning scheme is employed that utilizes twenty-four uniform distance bins 1 Å wide (recording distances from 0 to 24 Å). For building predictive models all 24 bins were used. For determining chi-squared distances between protein targets for assessing the domain of applicability for a model, an extended signature of 25 bins was used. The 25th bin records all distances greater than 24 Å in the signature and its inclusion enhanced the performance of the applicability domain assessment routine.
Figure 1.

(Left) Side view of EP mapped protein interaction surface of complex 1fbp. P1 and P2 are two points chosen from random locations on the surface. The properties of these two points and the Euclidean distance d between them determine which PESD signature bin they will occupy. The graphical representation of the PESD signature of 1fbp is shown as a two dimensional grid of bins (Right). Darker circles indicate greater bin populations. Each row corresponds to specific endpoint color combinations while each column represents point-pair distances that increase from left to right.
The entire range of colors on the EP map was coarse-grained into 9 colors, and that of ALP map into 14 colors. These numbers come from down-sampling the 24 bit color scheme to a 6 bit color scheme. The final number of elements was thus 24 × 81 and 24 × 196 for EP and ALP surfaces, respectively. A representative EP mapped protein interaction surface of the PDB complex 1fbp and the corresponding PESD signature are shown in Figure 1. Each circle in the graphical representation of the PESD signature is a bin. Darker circles indicate greater bin populations. Each row is for a color combination, and each column represents a point-pair distance that increases from left to right. For each surface, a total of 100,000 pairs of points were selected. The population of each bin is thus proportional to the probability of a color (or property magnitude) being present at a certain distance from another color on the surface.
To eliminate bias in surface point selection, the procedure of Osada was utilized47. Within this scheme, the area of each triangle of the surface mesh was calculated and stored as an array of cumulative areas. A number between 0 and the total area was then randomly chosen, and the triangle corresponding to the cumulative area containing that value was selected. The use of a lookup table that segments the array of cumulative areas greatly increased the computational efficiency of the procedure. A co-planar point within this triangle was then selected from a random location within the part of the plane enclosed by the edges of the triangle as shown in eq 1, where r1 and r2 are random numbers and A, B and C are vertices of the selected triangle:
| (1) |
The color of the selected point was then set equal to the color of the nearest vertex of the triangle. Typical signature computation time of a Visual Basic program on a 2.66 GHz Intel Xeon running Windows XP with a look-up table is 8 to 33 seconds per surface out of which 5 to 20 seconds are for parsing a surface file. Running four jobs of signature computation in parallel, the maximum computation time for each complex is typically 33 seconds.
Datasets
Protein-ligand complexes from the publicly-available database PDBbind37,38 (version 2005) were used in this study. The ‘refined set’ of the PDBbind has 1296 good quality complexes. After the Protonate3D run, a total of 1255 complexes from the refined set were available for PESD signature generation. Experimental binding affinity for each complex was extracted from the PDBbind database. The binding affinities were either inhibition constants (Ki) or dissociation constants (Kd) which were used equivalently in this study, in keeping with what was done in earlier works11,39. The refined set of PDBbind complexes also has a subset called a ‘core set’ of 288 complexes. The core set is a non-redundant set of protein-ligand complexes separated from the refined set48 and includs three complexes per non-redundant protein. Out of 1255 complexes with adjusted protonation state, 278 are part of this core set (reduced from 288) and 977 are part of the core’ set (all remaining complexes of the refined set, reduced from 1008). The affinity values (pKd or pKi) ranged from 1.49 to 13.96 in the core set and 0.49 to 13 in the core’ set. The overlap between the two sets in terms of protein and/or ligand components is shown in Table 5. The core set was used as the training set, and the core’ set as the test set for Model I. To reduce the possibility of bias in choice of complexes for the training and test sets, three other training and test sets of the same size (training: 278, test: 977) were created from the 1255 complexes by random sampling without replacement. These formed training and test sets for models II, III and IV. Finally for Model V, the core’ set was used as the training and the core set as the test set.
Table 5.
Overlap between protonated core and core’ sets in terms of protein and ligand components of the complexes.
| Type | Number of complexes | RP |
|---|---|---|
| protein component in core’ not occurring in core | 343 | 0.492 |
| ligand component in core’ not occurring in core | 739a | 0.519 |
| Protein and ligand component in core’ not occurring in core | 263a | 0.496 |
| Protein and ligand component in core’ also occurring in core | 0 | |
| protein component in core not occurring in core’ | 37 | 0.710 |
| ligand component in core not occurring in core’ | 163 a | 0.632 |
| protein and ligand component in core not occurring in core’ | 17 a | 0.736 |
Similarity search was done by three letter ligand identifiers which did not include peptides. Actual numbers are therefore higher if peptides are taken into account.
Data for enthalpy and entropy analysis were obtained from the SCORPIO database20.
Modeling
Support Vector Machine (SVM) regression and classification models were built with the e1071 SVM package49 in R50 using PESD signatures of protein and ligand interaction surfaces as features (Figure 2). No subjective feature selection was employed for any of the models except for the removal of invariant columns prior to model building. Negative logarithms (base 10) of experimental Kd and Ki values were used as dependent variables (pKd and pKi respectively). For classification, individual pKd / pKi values were converted to class numbers (1 for weak binders (pKi/pKd < 5), 2 for medium binders (5 ≤ pKi/pKd ≤ 8) and 3 for strong binders (pKi/pKd > 8)). Only the “gamma” parameter of the default radial kernel and the “cost” (cost of constraints violation) parameter were tuned by a simple 5-fold cross-validation with replacement from within the training set, where 20% of the training set was randomly selected and held out for cross-validation. For each combination of parameter values in Table 1 a model was built from the remaining 80% and applied on the validation set. The sum of residuals and cross-validated correlation coefficients were then recorded for each iteration. For each parameter combination, ten such runs were made. For SVM classification, the parameter combination having the lowest sum of residuals was chosen to build the final tuned model. For SVM regression, the parameter combination with the highest average cross-validated correlation coefficient was chosen to build the final tuned model. In both cases, all other parameters were kept at their default values. In R, the default for SVM regression is “eps-regression” and the default for SVM classification is “C-classification”49.
Figure 2.
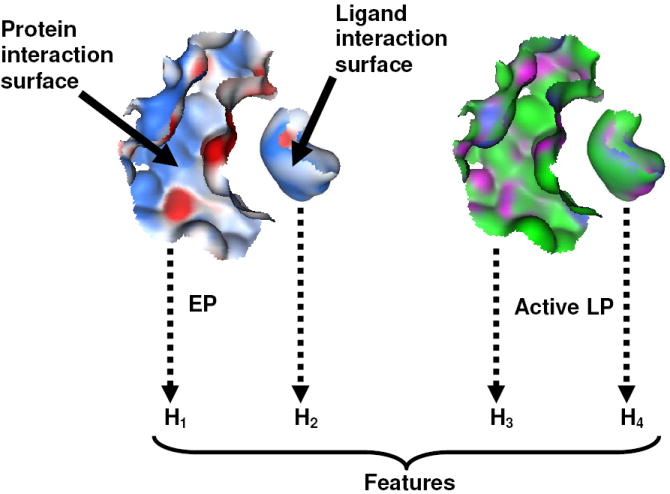
H1-4 depict protein and ligand interaction surfaces encoded with EP and Active LP maps. PESD signatures derived from these surfaces were used as features for building binding affinity SVM models.
Table 1.
Choice of parameter values for model parameter tuning
| Parameter | Values |
|---|---|
| Cost | 1, 10, 20, 30, 40, 50, 60, 70, 80, 90, 100 |
| Gamma | 1, 1/dimXa, 10/dimX a, 1/10dimXa. 1/100000 |
dimX = dimension of feature vector. Default values in R are cost=1 and gamma=1/dimX.
Chi-squared distance
We have shown earlier that the chi-squared distance between protein interaction surface PESD signatures is a good metric for assessing the similarity between pairs of protein active sites21, suggesting that this approach would provide a reasonable model applicability domain metric. Chi-squared distances were therefore computed for each pair of test and training protein interaction surfaces using the procedure shown in eq 2 where the dissimilarity distance d is assessed between two PESD signatures H and K. As shown below in eq 3, EP and ALP distances were combined using an ALP scaling factor of 0.7 since this weighting scheme gave the best set of clusters in a classification experiment of 40 active sites and was found to applicable to other active site comparisons as well21.
| (2) |
| (3) |
Quality metrics
While a number of metrics are available for comparing the performance of scoring functions, PESD-SVM predictions of pKd/pKi values were assessed against experimental data using Pearson’s correlation coefficient (RP), Spearman’s correlation coefficient (RS), standard deviation (SD) and mean error (ME). In eq 4 to 6, y represents the experimental value and x the predicted value. The predicted values were not scaled for SD and ME calculations (unlike in Wang et al.39) and instead the definitions in eq 5 and 6 were used. In addition to the statistical metrics above, the slope and the intercept, a and b, of the best-fit line for true and predicted affinities provide additional insight into model performance. In this case, a values close to 1.0 and b values close to 0.0 are considered favorable.
| (4) |
| (5) |
| (6) |
| (7) |
The ability of the models to correctly classify complexes into weak (pKi/pKd < 5), medium (5 ≤ pKi/pKd ≤ 8) and strong binders (pKi/pKd > 8) was judged by calculating the recovery rate and percentage true positive. The recovery rate is defined below.
| (8) |
Percentage true positive (% TP) defined below quantifies the reliability of a particular scoring function.
| (9) |
Scoring of docked poses
Given that the protein-ligand complex structures for affinity prediction were derived from crystal structures, it was of interest to determine how well the PESD-SVM approach would work to score poses obtained by docking simulation. In contrast to the usual PESD-SVM approach that separately encodes ligand and protein surface signatures, point pairs for PESD-DOCK signature generation were pooled in such a way that one endpoint was taken from the protein interaction surface and the other from the ligand interaction surface. Poses that arose during a docking experiment based on 1cbx (a complex of carboxypeptidase A and L-Benzyl succinate) were scored using a SVM regression model derived from PESD-DOCK signatures. This complex has previously been used in numerous benchmarking exercises in the literature,51, 52 providing us with a reasonable benchmark for evaluating the scoring efficacy of PESD-DOCK. To perform this evaluation, 50 docked poses were generated in MOE with docking site set to “Ligand” and rescoring set to “None”, with all other parameters being set to their default values. A SVM regression model was then trained on the PESD-DOCK signatures of 1253 complexes (1cbx and 1wht were excluded from the training data as these are complexes of L-benzyl succinate). The gamma and cost parameters were chosen from Table 1 to build a PESD-DOCK SVM model with the best cross-validated correlation coefficient (rTrain = 0.997, rCross-validated = 0.670; final model parameters: gamma = 1/Dimension of feature vector, cost=20).
Results
Regression statistics of training complexes
The tuned parameters chosen for the various PESD-SVM regression models I-V are shown in Table 2. The training set statistics for the models appear in Table 3. The correlation coefficients rTrain between actual and predicted affinities of the training complexes ranged between 0.879 and 0.997, indicating a significant level of overtraining. While usually not indicative of expected performance on test data, it is interesting to note that over-trained Models II and III with their 0.997 correlation coefficients were, in fact, found to perform better than Models I and IV on test data.
Table 2.
Tuned parameters for PESD-SVM regression models as determined by the cross-validation protocol
| Name | Cost | Gamma |
|---|---|---|
| Model I | 10 | 1/10dimX |
| Model II | 20 | 1/dimX |
| Model III | 10 | 1/dimX |
| Model IV | 10 | 1/100000 |
| Model V | 10 | 1/dimX |
Table 3.
Regression statistics of PESD-SVM regression models applied to respective training sets
| Name | NTrain | rTrain | rCross-validated |
|---|---|---|---|
| Model I | 278 | 0.879 | 0.588 |
| Model II | 278 | 0.997 | 0.482 |
| Model III | 278 | 0.997 | 0.565 |
| Model IV | 278 | 0.923 | 0.574 |
| Model V | 977 | 0.997 | 0.633 |
Regression statistics of test complexes
When applied to the respective test sets, PESD-SVM regression Models I-IV had Pearson’s correlation coefficients (RP) ranging from 0.517 to 0.574, and Spearman’s rank correlation coefficients (RS) from 0.535 to 0.597 (Table 4). The prediction accuracy for Model V was higher at RP = 0.638 and RS = 0.628. Plots of experimental binding affinities of test complexes versus predicted binding affinities from PESD-SVM regression models I, II and V are shown in Figure 3.
Table 4.
Regression statistics of performance of PESD-SVM regression models on respective test sets.
| Name | NTrain | NTest | Rp | Rs | ME | SD | a | b |
|---|---|---|---|---|---|---|---|---|
| Model I | 278 | 977 | 0.517 | 0.535 | 1.42 | 1.84 | 0.74 | 1.97 |
| Model II | 278 | 977 | 0.574 | 0.597 | 1.36 | 1.76 | 1.11 | -0.84 |
| Model III | 278 | 977 | 0.572 | 0.595 | 1.33 | 1.74 | 0.93 | 0.47 |
| Model IV | 278 | 977 | 0.531 | 0.550 | 1.39 | 1.84 | 0.75 | 1.76 |
| Model V | 977 | 278 | 0.638 | 0.628 | 1.45 | 1.86 | 1.23 | -1.52 |
| SFCscore(sfc_290m)a | 290 | 919 | 0.492 | 0.555 | ||||
| SFCscore(sfc_met)a | 341 | 919 | 0.540 | 0.582 |
SFCscore models with highest and lowest RP values11 are provided for comparison. Note that sfc_met was trained on an enriched training set.
Figure 3.
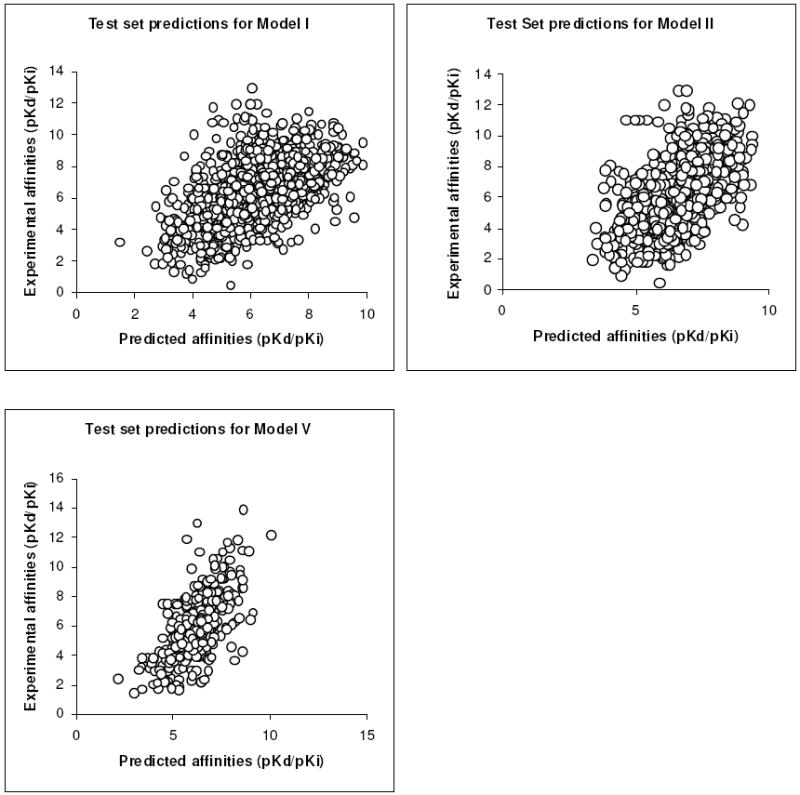
Plot of experimental affinities versus predicted affinities for PESD-SVM regression models I, II and V applied to their respective test sets.
For Model I, there were at least 263 complexes (Table 5) in the test set (core’) that had neither their protein component nor their ligand component common to complexes in the training set (core). Similarity search for ligands was done by three letter ligand identifiers which did not include peptides. Actual number of unique ligands is therefore slightly higher if peptides are taken into account. The unique 263 complexes gave an RP of 0.496 which is close to 0.517 observed when all complexes of the test set were considered. Interestingly, for Model V, an improvement in the value of RP was observed for complexes that had neither the ligand component nor the protein component common between test and training sets. This could be due to the relatively small number of complexes (only 17) that belonged to this category and/or due to a dominant enthalpic contribution (see Discussion). Overall, some transferability of the models is noted. Such a behavior is advantageous since the model need not be fitted with the same protein for which prediction is to be made.
Further analysis of the predictive performance of PESD-SVM regression models was done by breaking out prediction accuracies for several specific protein targets as shown in Table 6. For Model I, no significant correlations between predicted and true affinities were observed for carbonic anhydrase, HIV-1 protease and oligo-peptide binding protein. Moderate to good correlations were observed for trypsin, retinoic acid receptor, tyrosine phosphatase, and urokinase–type plasminogen activator. Overall prediction accuracy improved for these receptors in the case of Model II, with predictions for oligo-peptide binding protein and retinoic acid receptors showing large improvements in accuracy. Possible reasons for the improvements are provided in the Discussion section.
Table 6.
Prediction accuracy of PESD-SVM regression models I and II on different protein targets compiled from the respective test sets.
| Target Name | NTest | RP | RS |
|---|---|---|---|
| Model I | |||
| Trypsin | 91 | 0.737 | 0.687 |
| Carbonic anhydrase | 39 | 0.225 | 0.1 |
| HIV-1 protease | 71 | 0.02 | 0.022 |
| Oligo-peptide binding protein | 20 | 0.01 | -0.171 |
| Retinoic acid receptor (α, β and γ) | 6 | 0.470 | 0.657 |
| Retinoic acid receptor (α and γ) | 5 | 0.753 | 1.0 |
| Tyrosine phosphatase | 22 | 0.747 | 0.767 |
| Urokinase-type plasminogen activator | 23 | 0.714 | 0.738 |
| Model II | |||
| Trypsin | 74 | 0.746 | 0.636 |
| Carbonic anhydrase | 36 | 0.407 | 0.429 |
| HIV-1 protease | 60 | 0.298 | 0.132 |
| Oligo-peptide binding protein | 15 | 0.747 | 0.725 |
| Retinoic acid receptor (α, β and γ) | 7 | 0.874 | 0.929 |
| Retinoic acid receptor (α and γ) | 6 | 0.912 | 0.943 |
| Tyrosine phosphatase | 20 | 0.662 | 0.546 |
| Urokinase-type plasminogen activator | 20 | 0.844 | 0.767 |
Domain of applicability53-56
Consistently higher prediction accuracy for complexes within the domain of applicability of the model in question was observed. The domain of applicability was determined by a common similarity metric – chi squared distance between pairs of protein interaction surface signatures. Although both the protein and ligand interaction surface PESD signatures could be used for more accurately determining the difference between protein-ligand complexes, considering only the protein interaction surface PESD signature was found to be sufficient for demonstrating the trend. The domain of applicability was determined from the lowest value of the chi-squared distance of signatures between a test complex and the training complexes. All PESD-SVM regression models showed improvement in accuracy with decreasing chi-squared distance cutoffs (Table 7). A plot of RP and RS against cutoff distances for PESD-SVM regression Model II is shown in Figure 4.
Table 7.
Change in correlation coefficients with change in chi-squared cutoffs
| Name (Regression models) | Chi-squared cutoff | NTrain | NTest | RP | RS | Residual <1.5 pKd/pKi (% of NTest) |
|---|---|---|---|---|---|---|
| Model I | 6000 | 278 | 223 | 0.689 | 0.677 | 71.7 |
| Model II | 6000 | 278 | 266 | 0.762 | 0.727 | 75.9 |
| Model III | 6000 | 278 | 266 | 0.699 | 0.668 | 71.4 |
| Model IV | 6000 | 278 | 270 | 0.681 | 0.678 | 74.4 |
| Model I | 3000 | 278 | 33 | 0.792 | 0.853 | 78.8 |
| Model II | 3000 | 278 | 57 | 0.859 | 0.862 | 82.5 |
| Model III | 3000 | 278 | 51 | 0.715 | 0.630 | 76.5 |
| Model IV | 3000 | 278 | 41 | 0.778 | 0.697 | 85.3 |
Figure 4.
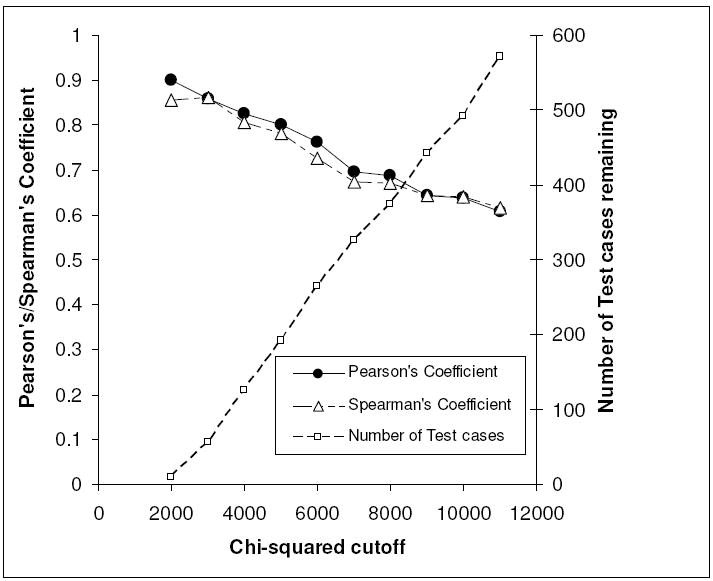
Plot of RP, RS and number of test cases against chi-squared cutoff distances for PESD-SVM regression Model II.
Classification
Differentiating weak and strong complexes from a set of weak, medium and strong complexes is a difficult task11, 39. The recovery rates (eq 8) of PESD-SVM classification models ranged from 30.0% to 62.1% for weak complexes and 18.9% to 47.6% for strong complexes (Table 8). We note that the recovery rate is not a complete indicator of true reliability of a classifier in a classification task. The reliability of a classifier is tied to its ability to report true positive values and this is shown in Table 9 for the PESD-SVM classification models. True positive percentages (eq 9) for PESD-SVM models ranged from 52.7% to 67% for weak binders and 40% to 78.8% for strong binders.
Table 8.
Recovery rates in classification with PESD-SVM classification models
| Name (Classification models) | Recovery rate |
||
|---|---|---|---|
| Weak (pKi/pKd < 5.0) | Medium (5 ≤ pKi/pKd ≤ 8) | Strong (pKi/pKd > 8) | |
| Model I | 139/224=62.1% | 319/527=60.5% | 78/226%=34.5% |
| Model II | 75/250=30.0% | 407/509=80.0% | 92/218=42.2% |
| Model III | 131/252=52.0% | 374/519=72.1% | 98/206=47.6% |
| Model IV | 128/248=51.6% | 421/512=82.2% | 41/217=18.9% |
Table 9.
True positive percentages in classification with PESD-SVM classification models
| Name (Classification models) | Percent True Positive |
||
|---|---|---|---|
| Weak (pKi/pKd < 5.0) | Medium (5 ≤ pKi/pKd ≤ 8) | Strong (pKi/pKd > 8) | |
| Model I | 139/264=52.7% | 319/518=61.2% | 78/195=40.0% |
| Model II | 75/112=67.0% | 407/693=58.7% | 92/172=53.5% |
| Model III | 131/213=61.5% | 374/578=64.7% | 98/186=52.7% |
| Model IV | 128/221=57.9% | 421/704=59.8% | 41/52=78.8% |
Comparison with SFCscore
SFCScore11 is a recently developed empirical scoring function that includes a number of descriptors including those accounting for polar and apolar surface areas and was previously shown to perform better than 14 other empirical , knowledge-based or force-field based scoring functions 39. Rigorous quantitative comparison between two empirical scoring functions requires that not only the test set but also the training set to be identical. This is because the choice of training sets can have a great impact on the performance of empirical scoring functions. Therefore the following comparison can only be considered semi-quantitative with comparably sized non-enriched training sets (models sfc_290m, sfc_229m, sfc_290p and sfc_229p and PESD-SVM models I-IV) unless otherwise noted. The reported correlation coefficients of predicted and experimental affinities with SFCscore are RP = 0.492 to 0.520, and RS = 0.547 to 0.565 on 919 test complexes in contrast to RP = 0.517 to 0.574, and RS = 0.535 to 0.595 on 977 test complexes with PESD-SVM models I-IV. There is considerable overlap between the two test sets. In version 2005 of PDBbind (used in this study), 209 new complexes were added and 4 older complexes were removed from the previous year’s version (used in SFCscore) resulting in an overlap of nearly 700 complexes. The PESD-SVM regression models I-IV gave RP = 0.491 to 0.551 and RS = 0.508 to 0.575 for the overlapping test complexes. The lower bound of RS was slightly lower and the upper bound of RP was slightly higher than SFCscore values. The ME and SD of PESD-SVM models I-IV (ME=1.33 to 1.42, SD=1.74 to 1.84) were comparable to slightly better than the four SFCscore models obtained on a different test set (ME=1.39 to 1.45, SD = 1.83 to 1.89). Although SFCscore showed an apparently higher accuracy for carbonic anhydrase (CA), the test and training sets of SFCscore had common complexes in this case: for example, 32 out of 37 test molecules in the CA data set and 14 out of 74 test HIV-1 protease molecules also belonged to the 290 complex SFCscore training set. In the present study no model had any overlapping complex between its test and training set. Two residual (|experimental affinity - predicted affinity|) cutoff values have been reported in the literature11, and for comparison, we utilize the same two criteria here: the percentage of complexes with residuals under 2.0 and 1.5 log units, respectively. For SFCscore, the best percentages under those residual cutoffs for the 919 test molecules were 72.4% and 60.8%. In contrast, for the model with the lowest RP in this study, PESD-SVM regression Model I, the percentages were higher at 75.5% and 62.2%. For PESD-SVM regression Model II, the percentages were 76.8% and 62.4%.
The highest recovery rate of weak complexes using PESD-SVM classifiers was 62.1% (classification model I) which is higher than the reported recovery rate for any other scoring functions 11, 39. However, given the recovery rate of strong binders for the Model I was not very high (only 34.5%), this could be due to a tendency to under-estimate affinities. The true positive percent of Model I for weak complexes was indeed the lowest among the four models at 52.7%. In contrast, Model III had good recovery rates for weak and strong complexes and its true positive rates were also relatively higher. The recovery rate of weak complexes by Model III at 52.0% was also higher than all SFCscore functions applied to the 919 unbiased test set. The highest recovery rate of strong complexes was 47.6% with PESD-SVM classifiers which is also significantly high compared to other reported values11,39. We note that this recovery rate is only exceeded by sfc_frag, which the authors had noted overestimated affinities11. The important point of difference between the SFCscore approach and the PESD-SVM approach is that SFCscore includes descriptors such as number of rotatable bonds, ring-metal interaction scores and ring-ring interaction scores (in addition to surface-based ones) that were absent in the PESD-SVM method although, in general, comparable results with were obtained with the PESD-SVM method.
Scoring of docked poses
Out of 50 poses generated for the complex 1cbx, the ligand pose with the highest PESD-DOCK SVM score had a root mean squared deviation (rmsd) of 1.46 Å with respect to the native crystal pose. It is significant to note that only one pose with rmsd > 2.0 Å (rmsd = 2.09 Å) had a PESD-DOCK SVM score higher than that obtained for the native crystal structure pose. A plot of the correlation between ligand pose (rmsd) and PESD-DOCK SVM score is shown in Figure 5. The Spearman’s correlation coefficient for this data was -0.524 (PESD-DOCK SVM model was trained on positive affinity values: pKd/pKi). Further study on several diverse protein targets needs to be made to assess the reliability of the PEST-DOCK SVM scoring method, and this is part of an ongoing effort which will be reported elsewhere.
Figure 5.
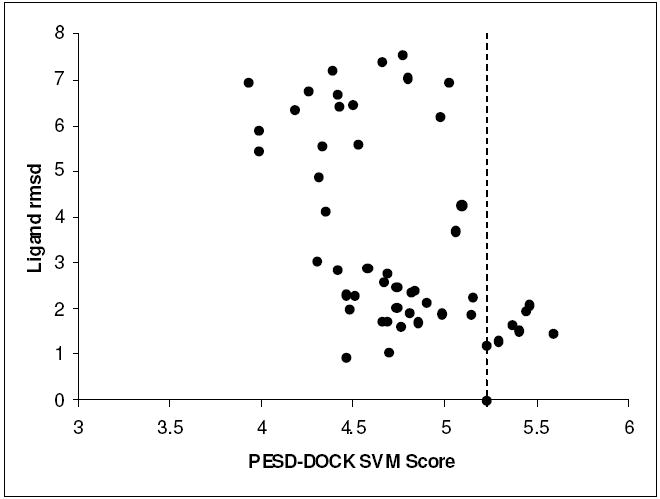
Correlation between ligand pose (rmsd from native pose) and PESD-DOCK SVM Score for docked conformations of L-Benzyl succinate in 1cbx. The score of the native pose (rmsd = 0) is shown as a dashed line. Since PESD-DOCK SVM models were trained on positive affinity values (pKd/pKi), higher scores indicate favorable interactions.
Discussion
Change in buried apolar surface area of protein and ligand together has been previously observed to have a good correlation with affinity20 and many approaches9,11,22,23,34 to binding affinity prediction have utilized surface area based descriptors and equations. PESD-SVM approach with only surface based signatures was able to achieve accuracy comparable to SFCscore that used a number of non-surface based descriptors in addition to surface-based ones. The population of different property combination bins in the PESD signatures are proportional not only to the surface area under different properties but also represent the relative locations of a surface under one property with respect to others under different properties. As noted by Golhke and Klebe57, “the burial of a part of a hydrophobic molecular surface at a binding site can induce a simultaneous cooperative enhancement of neighboring electrostatic interactions” 58, 59. Therefore the relative location of the areas under different magnitudes of property values is an important factor in binding that is captured by the PESD signatures and not by traditional sum of area descriptors.
In the present study however, a good correlation between ΔH/-TΔS and prediction accuracy was also noted. With Isothermal Titration Calorimetry (ITC) it is possible to determine the enthalpy value and hence determine the enthalpy/entropy contribution19,20. A database of energy values obtained from ITC experiments has also been set up20. Although limited in size, analysis of the data by Olsson and coworkers20 showed significant amount of the so-called “enthalpy-entropy compensation” 60 that resulted in a relatively small energy range for free-energy and large ranges for entropy and enthalpy. A plot of enthalpy versus free-energy of the data comprising of 322 entries (Figure 6a) showed no correlation between enthalpy and free-energy similar to earlier observations57. However, for about one-third of the entries (111 of 332) the ΔH/-TΔS was greater than 3, and this is where good correlation between free energy and enthalpy was observed (Figure 6b). Therefore, if the temperature at which the ITC data was obtained is assumed to be room temperature (298 K) under identical experimental conditions, a scoring function having poor treatment for entropy and trained on pKd/pKi obtained under those conditions, should achieve higher accuracy in predicting affinity constants for such entries at that temperature. The following analysis is not exhaustive due to the very small number of sample points, but some important trends are noted from the available data. Where energy values for a complex were available at multiple temperatures in SCORPIO, only the one closest to room temperature was used (such a procedure was also adopted in PDBbind38). Trypsin is a receptor where enthalpy on an average was found to be more than 4 times the magnitude of -TΔS based on entries in the SCORPIO database (1k1i, 1k1j, 1k1l, 1k1m, 1k1n, 1ce5 at ~298 K) and this is possibly a reason why most scoring functions including PESD-SVM perform well in this receptor. We also note a similar trend in prediction accuracy of complexes in the test set of Model I for which ITC data was available from SCORPIO and whose ΔH/-TΔS was greater than 3. The ten complexes (1a1c, 1k1j, 1k1l, 1k1m, 1kzn, 1swg, 1fdq, 1qy1, 1qy2, 1adl) had an RP of 0.685. Out of the ten complexes, 1qy1 and 1qy2 ITC values were obtained at 308 K and the rest were obtained within ± 5 K of room temperature. The range of chi-squared distances of these complexes with respect to training was 8643 to 15684, with an average of 12090, indicating their protein interaction surfaces were not very similar to those in the training. Complex 1adl was an outlier having a residual greater than three times the standard deviation of the residuals of the ten complexes (Figure 6d). On removal of this complex, the RP increased further to 0.865 while the average chi-squared distance decreased only slightly to 12074. The primary reference61 of 1adl indicated solvent participation in binding, and this could be a reason for the inaccuracy with the current PESD-SVM method. Similarly ITC data for HIV-1 protease (1hsg, 1ohr, 1a30, 1t7j, 1t7i; 1t7j and 1t7i at ~293 K, rest at ~298K) showed that entropy is a significant component with the average ΔH/-TΔS being 1.14 in the 5 complexes. Although polarization effects are important62,63, poor prediction accuracy with most scoring functions for HIV-1 protease could be because of inadequate treatment of entropy18. In fact taking into account entropy was shown to result in good prediction of affinity64. Entropic factors are also dominant in oligopeptide binding protein and solvent plays a significant role in binding57 which possibly explains the lack of correlation between predicted and experimental affinities for Model I. These trends support the hypothesis. Therefore, accounting for both entropy and solvent is necessary to improve the accuracy of the PESD-SVM method. Other possible sources of error are experimental conditions (such as temperature and pH) and techniques used for determining pKd/pKi65 and these can be reduced by ensuring consistency in the data.
Figure 6.
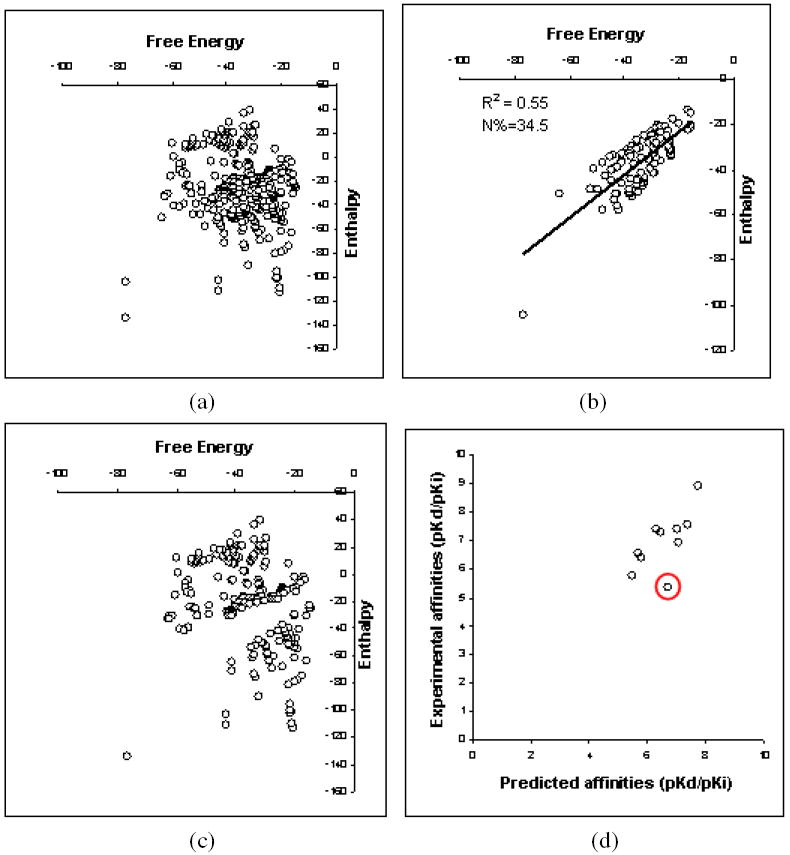
(a) Plot of free energy versus enthalpy for 322 entries from the SCORPIO database. (b) Plot of free energy versus enthalpy for 111 out of 322 entries from the SCORPIO database. The ΔH/-TΔS was greater than 3 for these complexes. (c) Difference plot of 6a and 6b (d) Plot of experimental versus predicted affinities of all entries in the core set for which ΔH/-TΔS was greater than 3 and could be obtained from the SCORPIO database. The complex 1adl is circled in red.
Interestingly, performance improved significantly for Model II in some receptors including oligo-peptide binding protein. The training set of Model II had 8 oligopepetide binding proteins in the training set as opposed to 3 in Model I. Recall that the protonated core set did not have more than 3 complexes per non-redundant protein. The improvement in accuracy could be due to the inclusion of more oligopeptide binding proteins in the training set of Model II resulting in enrichment. Similar trends with varying degrees of improvement are observed for other receptors. Enrichment and larger sized training sets were previously observed to improve accuracy of scoring functions10, 11, 66, 67. The domain of applicability filter applied to test set is complementary to training set enrichment, where we also see improved prediction accuracy with decreasing chi-squared distance cut-offs between test and train complexes.
Conclusions
The utility of the PESD signatures in affinity prediction has been demonstrated by its application to a large number of different proteins. A simple model building process was employed that generated models based on PESD signatures of two surface maps. The models had only modest accuracy but was comparable in general and slightly improved in some cases with respect to SFCscore. We have compared the results to those of SFCscore since the latter is a recently developed regression based scoring function, and included both surface and non-surface based descriptors.
However, the present results of the PESD-SVM approach show that only two surface maps are not adequate to achieve a higher degree of accuracy. Although enriching a training set or increasing its size had a positive effect of varying degrees on accuracy, factors such as entropy and solvent cannot be neglected. These terms need to be added in the future to improve the current models although this can be a difficult challenge68,69. Addition of specific interaction terms such as ring-ring and ring-metal descriptors11 can also be potentially beneficial. We also note that in certain receptors such as trypsin, tyrosine phosphatase and urokinase-type plasminogen activator, PESD-SVM performed consistently well and for most complexes with a dominant enthalpic contribution (ΔH/-TΔS > 3), a good correlation between true and predicted affinities was observed.
Supplementary Material
Acknowledgments
The authors would also like to acknowledge Dr. N. Sukumar and Dr. Dominic Ryan for valuable discussions. This work was supported by the National Institutes of Health, Grant number 1P20 HG003899 “Establishment of the Rensselaer Exploratory Center for Cheminformatics Research – RECCR” and was conducted in the RPI Center for Biotechnology and Interdisciplinary Studies (CBIS).
Footnotes
Supporting Information Available: PDB codes of training and test sets. This material is available free of charge via the Internet at http://pubs.acs.org.
Work was done at Department of Chemistry & Chemical Biology / RECCR Center, Center for Biotechnology and Interdisciplinary Studies, Rensselaer Polytechnic Institute, Troy, NY, USA
Availability: Source code for generating PESD signatures can be downloaded from http://breneman.chem.rpi.edu/PESDSVM
Contributor Information
Sourav Das, Department of Chemistry & Chemical Biology, Rensselaer Polytechnic Institute, 110-8th Street, Troy, NY 12180.
Michael P. Krein, Department of Chemistry & Chemical Biology, Rensselaer Polytechnic Institute, 110-8th Street, Troy, NY 12180
Curt M. Breneman, Department of Chemistry & Chemical Biology / RECCR Center Rensselaer Polytechnic Institute, 110-8th Street, Center for Biotechnology and Interdisciplinary Studies, Troy, NY 12180, Phone Number: 518-276-2678, Fax Number: 518-276-4887, brenec@rpi.edu
References
- 1.Gehlhaar DK, Verkhivker GM, Rejto PA, Sherman CJ, Fogel DR, Fogel LJ, Freer ST. Molecular Recognition of the Inhibitor AG-1343 by HIV-1 Protease: Conformationally Flexible Docking by Evolutionary Programming. Chem Biol. 1995;2:317–324. doi: 10.1016/1074-5521(95)90050-0. [DOI] [PubMed] [Google Scholar]
- 2.Muegge I, Martin YC. A General and Fast Scoring Function for Protein-Ligand Interactions: A Simplified Potential Approach. J Med Chem. 1999;42:791–804. doi: 10.1021/jm980536j. [DOI] [PubMed] [Google Scholar]
- 3.Böhm H-J. The development of a simple empirical scoring function to estimate the binding constant for a protein-ligand complex of known three-dimensional structure. J Comput-Aided Mol Des. 1994;8:243–256. doi: 10.1007/BF00126743. [DOI] [PubMed] [Google Scholar]
- 4.Rarey M, Kramer B, Lengauer T, Klebe G. A Fast Flexible Docking Method using an Incremental Construction Algorithm. J Mol Biol. 1996;261:470–489. doi: 10.1006/jmbi.1996.0477. [DOI] [PubMed] [Google Scholar]
- 5.Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. J Mol Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 6.Ewing TJA, Makino S, Skillman AG, Kuntz ID. DOCK 4.0: Search strategies for automated molecular docking of flexible molecule databases. J Comput-Aided Mol Des. 2001;15:411–428. doi: 10.1023/a:1011115820450. [DOI] [PubMed] [Google Scholar]
- 7.Eldridge MD, Murray CW, Auton TR, Paolini GV, Mee RP. Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J Comput-Aided Mol Des. 1997;11:425–445. doi: 10.1023/a:1007996124545. [DOI] [PubMed] [Google Scholar]
- 8.Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem. 1998;19:1639–1662. [Google Scholar]
- 9.Gohlke H, Hendlich M, Klebe G. Knowledge-based scoring function to predict protein-ligand interactions. J Mol Biol. 2000;295:337–356. doi: 10.1006/jmbi.1999.3371. [DOI] [PubMed] [Google Scholar]
- 10.Wang R, Lai L, Wang S. Further development and validation of empirical scoring functions for structure-based binding affinity prediction. J Comput-Aided Mol Des. 2002;16:11–26. doi: 10.1023/a:1016357811882. [DOI] [PubMed] [Google Scholar]
- 11.Sotriffer CA, Sanschagrin P, Matter H, Klebe G. SFCscore: Scoring functions for affinity prediction of protein-ligand complexes. Proteins: Struct Funct Bioinf. 2008;73:395–419. doi: 10.1002/prot.22058. [DOI] [PubMed] [Google Scholar]
- 12.Yin S, Biedermannova L, Vondrasek J, Dokholyan NV. MedusaScore: An Accurate Force Field-Based Scoring Function for Virtual Drug Screening. J Chem Inf Model. 2008;48:1656–1662. doi: 10.1021/ci8001167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang S, Golbraikh A, Tropsha A. Development of Quantitative Structure-Binding Affinity Relationship Models Based on Novel Geometrical Chemical Descriptors of the Protein-Ligand Interfaces. J Med Chem. 2006;49:2713–2724. doi: 10.1021/jm050260x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beveridge DL, Dicapua FM. Free Energy Via Molecular Simulation: Applications to Chemical and Biomolecular Systems. Annu Rev Biophys Biophys Chem. 1989;18:431–492. doi: 10.1146/annurev.bb.18.060189.002243. [DOI] [PubMed] [Google Scholar]
- 15.Kollman P. Free energy calculations: Applications to chemical and biochemical phenomena. Chem Rev. 1993;93:2395–2417. [Google Scholar]
- 16.Bash PA, Singh UC, Langridge R, Kollman PA. Free energy calculations by computer simulation. Science. 1987;236:564–568. doi: 10.1126/science.3576184. [DOI] [PubMed] [Google Scholar]
- 17.Dang LX, Merz KM, Kollman PA. Free energy calculations on protein stability: Thr-157 .fwdarw. Val-157 mutation of T4 lysozyme. J Am Chem Soc. 1989;111:8505–8508. [Google Scholar]
- 18.Cheng T, Li X, Li Y, Liu Z, Wang R. Comparative Assessment of Scoring Functions on a Diverse Test Set. J Chem Inf Model. 2009;49:1079–1093. doi: 10.1021/ci9000053. [DOI] [PubMed] [Google Scholar]
- 19.Ladbury JE, Williams MA. The extended interface: measuring non-local effects in biomolecular interactions. Curr Opin Struct Biol. 2004;14:562–569. doi: 10.1016/j.sbi.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 20.Olsson TSG, Williams MA, Pitt WR, Ladbury JE. The Thermodynamics of Protein-Ligand Interaction and Solvation: Insights for Ligand Design. J Mol Biol. 2008;384:1002–1017. doi: 10.1016/j.jmb.2008.09.073. [DOI] [PubMed] [Google Scholar]
- 21.Das S, Kokardekar A, Breneman CM. Rapid Comparison of Protein Binding Site Surfaces with Property Encoded Shape Distributions. J Chem Inf Model. doi: 10.1021/ci900317x. [Article ASAP], Published online: Nov 18 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stiefl N, Baumann K. Structure-Based Validation of the 3D-QSAR Technique MaP. J Chem Inf Model. 2005;45:739–749. doi: 10.1021/ci049683i. [DOI] [PubMed] [Google Scholar]
- 23.Wagener M, Sadowski J, Gasteiger J. Autocorrelation of Molecular Surface Properties for Modeling Corticosteroid Binding Globulin and Cytosolic Ah Receptor Activity by Neural Networks. J Am Chem Soc. 2002;117:7769–7775. [Google Scholar]
- 24.Renner S, Schneider G. Scaffold-Hopping Potential of Ligand-Based Similarity Concepts. ChemMedChem. 2006;1:181–185. doi: 10.1002/cmdc.200500005. [DOI] [PubMed] [Google Scholar]
- 25.Breneman CM, Sundling CM, Sukumar N, Shen L, Katt WP, Embrechts MJ. New developments in PEST shape/property hybrid descriptors. J Comput-Aided Mol Des. 2003;17:231–240. doi: 10.1023/a:1025334310107. [DOI] [PubMed] [Google Scholar]
- 26.Zauhar RJ, Moyna G, Tian L, Li Z, Welsh WJ. Shape Signatures: A New Approach to Computer-Aided Ligand- and Receptor-Based Drug Design. J Med Chem. 2003;46:5674–5690. doi: 10.1021/jm030242k. [DOI] [PubMed] [Google Scholar]
- 27.Noble WS. What is a support vector machine? Nat Biotechnol. 2006;24:1565–1567. doi: 10.1038/nbt1206-1565. [DOI] [PubMed] [Google Scholar]
- 28.Lapinsh M, Prusis P, Gutcaits A, Lundstedt T, Wikberg JES. Development of proteo-chemometrics: a novel technology for the analysis of drug-receptor interactions. Biochim Biophys Acta Gen Subj. 2001;1525:180–190. doi: 10.1016/s0304-4165(00)00187-2. [DOI] [PubMed] [Google Scholar]
- 29.Strömbergsson H, Daniluk P, Kryshtafovych A, Fidelis K, Wikberg JES, Kleywegt GJ, Hvidsten TR. Interaction Model Based on Local Protein Substructures Generalizes to the Entire Structural Enzyme-Ligand Space. J Chem Inf Model. 2008;48:2278–2288. doi: 10.1021/ci800200e. [DOI] [PubMed] [Google Scholar]
- 30.Ortiz AR, Pisabarro MT, Gago F, Wade RC. Prediction of Drug Binding Affinities by Comparative Binding Energy Analysis. J Med Chem. 1995;38:2681–2691. doi: 10.1021/jm00014a020. [DOI] [PubMed] [Google Scholar]
- 31.Datar PA, Khedkar SA, Malde AK, Coutinho EC. Comparative residue interaction analysis (CoRIA): a 3D-QSAR approach to explore the binding contributions of active site residues with ligands. J Comput-Aided Mol Des. 2006;20:343–360. doi: 10.1007/s10822-006-9051-5. [DOI] [PubMed] [Google Scholar]
- 32.Gohlke H, Klebe G. DrugScore meets CoMFA: adaptation of fields for molecular comparison (AFMoC) or how to tailor knowledge-based pair-potentials to a particular protein. J Med Chem. 2002;45:4153–4170. doi: 10.1021/jm020808p. [DOI] [PubMed] [Google Scholar]
- 33.Vijayan RSK, Bera I, Prabu M, Saha S, Ghoshal N. Combinatorial Library Enumeration and Lead Hopping using Comparative Interaction Fingerprint Analysis and Classical 2D QSAR Methods for Seeking Novel GABAA α3 Modulators. J Chem Inf Model. 2009;49:2498–2511. doi: 10.1021/ci900309s. [DOI] [PubMed] [Google Scholar]
- 34.Lindström A, Pettersson F, Almquist F, Berglund A, Kihlberg J, Linusson A. Hierarchical PLS modeling for predicting the binding of a comprehensive set of structurally diverse protein-ligand complexes. J Chem Inf Model. 2006;46:1154–1167. doi: 10.1021/ci050323k. [DOI] [PubMed] [Google Scholar]
- 35.Head R, Smythe ML, Oprea TI, Waller CL, Green SM, Marshall GR. VALIDATE: A New Method for the Receptor-Based Prediction of Binding Affinities of Novel Ligands. J Am Chem Soc. 1996;118:3959–3969. [Google Scholar]
- 36.Deng W, Breneman C, Embrechts MJ. Predicting Protein–Ligand Binding Affinities Using Novel Geometrical Descriptors and Machine-Learning Methods. J Chem Inf Comput Sci. 2004;44:699–703. doi: 10.1021/ci034246+. [DOI] [PubMed] [Google Scholar]
- 37.Wang R, Fang X, Lu Y, Yang CY, Wang S. The PDBbind Database: Methodologies and updates. J Med Chem. 2005;48:4111–4119. doi: 10.1021/jm048957q. [DOI] [PubMed] [Google Scholar]
- 38.Wang R, Fang X, Lu Y, Wang S. The PDBbind Database: Collection of Binding Affinities for Protein-Ligand Complexes with Known Three-Dimensional Structures. J Med Chem. 2004;47:2977–2980. doi: 10.1021/jm030580l. [DOI] [PubMed] [Google Scholar]
- 39.Wang R, Lu Y, Fang X, Wang S. An Extensive Test of 14 Scoring Functions Using the PDBbind Refined Set of 800 Protein–Ligand Complexes. J Chem Inf Comput Sci. 2004;44:2114–2125. doi: 10.1021/ci049733j. [DOI] [PubMed] [Google Scholar]
- 40.Labute P. Protonate3D: Assignment of ionization states and hydrogen coordinates to macromolecular structures. Proteins: Struct Funct Bioinf. 2009;75:187–205. doi: 10.1002/prot.22234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Molecular Operating Environment, Version 2007.09. Chemical Computing Group, Inc.; Montreal, QC: 2007. [Google Scholar]
- 42.Ryan MD, Hepburn T, Sukumar N, Das S, Breneman CM. TAE Augmented scoring functions: Two approaches, atom and surface based. Abstracts of Papers, 234th ACS National Meeting; Boston, MA, United States. August 19-23, 2007; 2007. COMP-42. [Google Scholar]
- 43.Das S, Breneman CM, Ryan MD. TAE Augmented Scoring Functions: Application to Enzymatic and Non-enzymatic proteins. Abstracts of Papers, 235th ACS National Meeting; New Orleans, LA. April 6-10, 2008; 2008. COMP-121. [Google Scholar]
- 44.Berman HM, Battistuz T, Bhat TN, Bluhm WF, Bourne PE, Burkhardt K, Feng Z, Gilliland GL, Iype L, Jain S, Fagan P, Marvin J, Padilla D, Ravichandran V, Schneider B, Thanki N, Weissig H, Westbrook JD, Zardecki C. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Labute P. An Integrated Application in MOE for the Visualization and Analysis of Protein Active Sites with Molecular Surfaces, Contact Statistics and Electrostatic Maps. [Dec 18, 2009];J Chem Comput Group. 2006 [Online] http://www.chemcomp.com/journal/f_surfmap.htm.
- 46.Santavy M, Labute P. Electrostatic Fields and Surfaces in MOE. [Dec 18, 2009];J Chem Comput Group. 1998 [Online] http://www.chemcomp.com/journal/grid.htm.
- 47.Osada R, Funkhouser T, Chazelle B, Dobkin D. Shape Distributions. ACM Trans Graph. 2002;21:807–832. [Google Scholar]
- 48.A Brief Introduction to the PDBbind bind Database v.2007. [Dec 18, 2009]; http://sw16.im.med.umich.edu/databases/PDBbind/pdfs/PDBbind_2007_intro.pdf.
- 49.Dimitriadou E, Hornik K, Leisch F, Meyer D, Weingessel A. Package ‘e1071’. [Dec 18, 2009]; http://cran.r-project.org/web/packages/e1071/e1071.pdf.
- 50.Ripley BD. The {R} project in statistical computing. MSOR Connections Newsletter of the LTSN Maths, Stats & OR Network. 2001;1:23–25. [Google Scholar]
- 51.Bursulaya B, Totrov M, Abagyan R, Brooks C. Comparative study of several algorithms for flexible ligand docking. J Comput-Aided Mol Des. 2003;17:755–763. doi: 10.1023/b:jcam.0000017496.76572.6f. [DOI] [PubMed] [Google Scholar]
- 52.Wang R, Lu Y, Wang S. Comparative Evaluation of 11 Scoring Functions for Molecular Docking. J Med Chem. 2003;46:2287–2303. doi: 10.1021/jm0203783. [DOI] [PubMed] [Google Scholar]
- 53.Dragos H, Gilles M, Alexandre V. Predicting the Predictability: A Unified Approach to the Applicability Domain Problem of QSAR Models. J Chem Inf Model. 2009;49:1762–1776. doi: 10.1021/ci9000579. [DOI] [PubMed] [Google Scholar]
- 54.Weaver S, Gleeson MP. The importance of the domain of applicability in QSAR modeling. J Mol Graph Model. 2008;26:1315–1326. doi: 10.1016/j.jmgm.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 55.Guha R, Schurer S. Utilizing high throughput screening data for predictive toxicology models: protocols and application to MLSCN assays. J Comput-Aided Mol Des. 2008;22:367–384. doi: 10.1007/s10822-008-9192-9. [DOI] [PubMed] [Google Scholar]
- 56.Guha R. On the interpretation and interpretability of quantitative structure–activity relationship models. J Comput-Aided Mol Des. 2008;22:857–871. doi: 10.1007/s10822-008-9240-5. [DOI] [PubMed] [Google Scholar]
- 57.Gohlke H, Klebe G. Approaches to the Description and Prediction of the Binding Affinity of Small-Molecule Ligands to Macromolecular Receptors. Angew Chem Int Ed. 2002;41:2644–2676. doi: 10.1002/1521-3773(20020802)41:15<2644::AID-ANIE2644>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 58.Sharman GJ, Searle MS, Benhamu B, Groves P, Williams DH. Burial of Hydrocarbon Causes Cooperative Enhancement of Electrostatic Binding. Angew Chem Int Ed. 1995;34:1483–1485. [Google Scholar]
- 59.Williams DH, Maguire AJ, Tsuzuki W, Westwell MS. An Analysis of the Origins of a Cooperative Binding Energy of Dimerization. Science. 1998;280:711–714. doi: 10.1126/science.280.5364.711. [DOI] [PubMed] [Google Scholar]
- 60.Gilli P, Ferretti V, Gilli G, Borea PA. Enthalpy-entropy compensation in drug-receptor binding. J Phys Chem. 1994;98:1515–1518. [Google Scholar]
- 61.LaLonde JM, Levenson MA, Roe JJ, Bernlohr DA, Banaszak LJ. Adipocyte lipid-binding protein complexed with arachidonic acid. Titration calorimetry and X-ray crystallographic studies. J Biol Chem. 1994;269:25339–25347. doi: 10.2210/pdb1adl/pdb. [DOI] [PubMed] [Google Scholar]
- 62.Hensen C, Hermann JC, Nam K, Ma S, Gao J, Höltje HD. A combined QM/MM Approach to Protein-Ligand Interactions: Polarization Effects of the HIV-1 Protease on Selected High Affinity Inhibitors. J Med Chem. 2004;47:6673–6680. doi: 10.1021/jm0497343. [DOI] [PubMed] [Google Scholar]
- 63.Das D, Koh Y, Tojo Y, Ghosh AK, Mitsuya H. Prediction of Potency of Protease Inhibitors Using Free Energy Simulations with Polarizable Quantum Mechanics-Based Ligand Charges and a Hybrid Water Model. J Chem Inf Model. doi: 10.1021/ci900320p. [Article ASAP], Published online: Nov 24, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Verkhivker G, Appelt K, Freer ST, Villafranca JE. Empirical free energy calculations of ligand-protein crystallographic complexes. I. Knowledge-based ligand-protein interaction potentials applied to the prediction of human immunodeficiency virus 1 protease binding affinity. Protein Eng Des Sel. 1995;8:677–691. doi: 10.1093/protein/8.7.677. [DOI] [PubMed] [Google Scholar]
- 65.Tame JR. Scoring functions - the first 100 years. J Comput-Aided Mol Des. 2005;19:445–451. doi: 10.1007/s10822-005-8483-7. [DOI] [PubMed] [Google Scholar]
- 66.Ajay, Murcko MA. Computational Methods to Predict Binding Free Energy in Ligand-Receptor Complexes. J Med Chem. 1995;38:4953–4967. doi: 10.1021/jm00026a001. [DOI] [PubMed] [Google Scholar]
- 67.Sales AP, Tomaras G, Kepler T. Improving peptide-MHC class I binding prediction for unbalanced datasets. BMC Bioinformatics. 2008;9:385. doi: 10.1186/1471-2105-9-385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Leach AR, Shoichet BK, Peishoff CE. Prediction of Protein-Ligand interactions. Docking and Scoring: Successes and Gaps. J Med Chem. 2006;49:5851–5855. doi: 10.1021/jm060999m. [DOI] [PubMed] [Google Scholar]
- 69.Tirado-Rives J, Jorgensen WL. Contribution of Conformer Focusing to the Uncertainty in Predicting Free Energies for Protein-Ligand Binding. J Med Chem. 2006;49:5880–5884. doi: 10.1021/jm060763i. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.