

Abstract
Melatonin plays a neuroprotective role in models of neurodegenerative diseases. However, the molecular mechanisms underlying neuroprotection by melatonin are not well understood. Apoptotic cell death in the central nervous system is a feature of neurodegenerative diseases. The intrinsic and extrinsic apoptotic pathways and the antiapoptotic survival signal pathways play critical roles in neurodegeneration. This review summarizes the reports to date showing inhibition by melatonin of the intrinsic apoptotic pathways in neurodegenerative diseases including stroke, Alzheimer disease, Parkinson disease, Huntington disease, and amyotrophic lateral sclerosis. Furthermore, the activation of survival signal pathways by melatonin in neurodegenerative diseases is discussed.
Keywords: Alzheimer disease, Amyotrophic lateral sclerosis, Huntington disease, Melatonin, Mitochondrial cell death pathways, Parkinson disease, Stroke, Survival signal pathways
Introduction
Melatonin May Be Beneficial in Treatment of Neurodegenerative Diseases
Melatonin (N‐acetyl‐5‐methoxytryptamine) is a natural hormone secreted by the pineal gland. In clinical use for many years, melatonin is safe and well‐tolerated even at high doses [1] and easily crosses the blood–brain barrier. Besides being used to increase sleep efficiency, treat jet lag, improve the cardiovascular system [2], and as an antiaging drug [3, 4, 5] and a dietary supplement and cancer‐protective hormone [6], intensive research roughly in the past 10 years has indicated melatonin's beneficial effects in experimental models of neurodegenerative disorders. Brain oxidative damage has been implicated as a common link in the pathogenesis of such diseases. This small amphiphilic molecule acts as a free‐radical scavenger, and its broad spectrum of antioxidant activities in many central nervous system neurodegenerative diseases [7] has been well documented and reviewed [8]. There is growing evidence that its antiapoptotic effects play an important role in neurodegeneration as well. This review summarizes the antiapoptotic activities of melatonin via the inhibition of intrinsic apoptotic pathways and the activation of survival signal pathways in stroke, Alzheimer disease (AD), Parkinson disease (PD), Huntington disease (HD), and amyotrophic lateral sclerosis (ALS).
The Intrinsic and Extrinsic Apoptotic Pathways in Neurodegenerative Diseases
Two types of cell death occur in neurodegeneration: apoptosis and necrosis. Apoptosis (also called programmed cell death) occurs naturally under normal physiological conditions and in a variety of diseases, while necrosis is caused by external factors, such as infection, toxins, or trauma. Apoptosis is a feature of both acute and chronic central nervous system neurodegenerative diseases. There are two major apoptotic signaling pathways: extrinsic and intrinsic. The extrinsic apoptotic pathway (death receptor pathway) is initiated by death receptors (e.g., CD95/APO‐1/Fas; TNF receptor) on the surface of the cells, involving caspase‐8/Bid and caspase‐10 activation [9, 10]. Since there have been no obvious reports of the involvement of extrinsic pathways in the neuroprotection of melatonin, this review focuses only on the intrinsic pathway (the mitochondrial pathway) [11] (Fig. 1).
Figure 1.
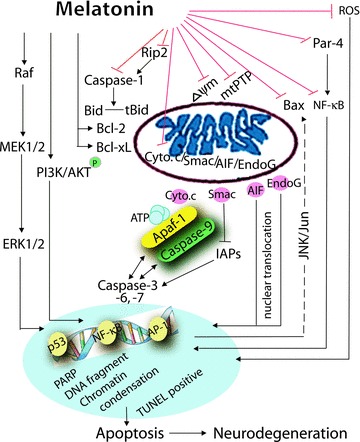
Scheme of neuroprotection of melatonin. The possible inhibition of the intrinsic cell death pathway and activation of the survival pathway by melatonin are schematized.
Proapoptotic mitochondria molecules, cytochrome c, Smac (second mitochondrion‐derived activator of caspase)/Diablo, AIF (apoptosis‐inducing factor), and Endo G (endonuclease G), when released into the cytoplasm from mitochondria, induce both caspase‐dependent and ‐independent mitochondrial death pathways in neurodegenerative diseases (Fig. 1) [12, 13, 14, 15, 16, 17, 18, 19]. The release of cytochrome c is pivotal in the activation of caspases [20]. During the progression of neurodegenerative diseases, once cytochrome c is released, it binds to Apaf‐1 and dATP, which stimulates the activation of caspase‐9, and then in turn cleaves the key effector caspase‐3 and two other effectors, caspase‐6 and ‐7 [12, 14, 15, 16, 17, 18, 21, 22, 23, 24]. In addition, DNA‐repairing enzyme poly(ADP‐ribose)polymerase (PARP) is cleaved [21], and transcription factors such as NF‐κB [5, 25, 26, 27], TNF‐α‐induced activator protein‐1 (AP‐1) [28, 29], and p53 [30, 31] are activated. Nuclear condensation and DNA fragmentation are induced, as shown by terminal deoxynucleotidyl transferase‐mediated DNA nick‐end labeling (TUNEL)‐positive cells, Hoechst 33342 stain, PI (propidium iodide), and 4′,6‐diamino‐2‐phenylindole dihydrochloride hydrate (DAPI) staining, as well as DNA ladder. These events ultimately cause neuronal cell death [13]. Other mitochondrial factors include mitochondrial permeability transition pores (mtPTP) and mitochondrial membrane potential (ΔΨm). mtPTP represents a multiprotein complex including inner and outer membrane components. The pores regulate transport of ions and peptides into and out of mitochondria. The activation of the permeability transition and in irreversible opening of mitochondria pores is a major step in the development of neurodegeneration [32, 33, 34]. ΔΨm reflects performance of the electron transport chain and can indicate a pathological disorder. The dissipation of ΔΨm and concomitant neuronal death have been reported in experimental models of neurodegeneration [14, 18, 34, 35, 36].
Caspase‐1 activation is an early event in neurodegenerative diseases [24, 37]. Caspase‐1 activator receptor interacting protein‐2 (Rip2) stimulates caspase‐1 to activate IL‐1β by truncating the proinflammatory cytokine. The release of mature IL‐1β indicates caspase‐1 activation [38]. The inhibition of pro‐IL‐1β cleavage and mature IL‐1β secretion are associated with inhibition of apoptosis in neurodegeneration [16, 18]. Rip2 upregulation has already been reported in AD [39], HD [40], and stroke [16].
Bcl‐2 family members include proapoptotic molecules (Bax, Bak, Bok, Bad, Bid, Bik, Blk, Hrk, BNIP3, and BimL) and antiapoptotic molecules (Bcl‐2, Bcl‐xL, Bcl‐w, Mcl‐1, and A1). Bcl‐2 family proteins participate in the modulation and execution of cell death [31] and can preserve or disrupt mitochondrial integrity by regulating the release of cytochrome c/Smac/AIF/endonuclease G [41, 42]. Cytosolic Bax translocates to mitochondria on death stimulus [23, 43], promoting cytochrome c release [43]. Besides the involvement of the Fas/caspase‐8/Bid cascade, Bid also mediates cytochrome c release while binding to both proapoptotic members (e.g., Bax) and antiapoptotic members Bcl‐2 and Bcl‐xL [44]; moreover, cleavage of Bid by caspase‐8 and caspase‐1 mediates the mitochondrial damage [45, 46]. Bax mediates cell death relates with mitochondrial permeability transition [11]. Bcl‐2 and Bcl‐xL bind to Apaf‐1, inhibiting the association of caspase‐9 with Apaf‐1 [47].
Prostate apoptosis response‐4 (Par‐4) induces mitochondrial membrane permeability changes and promotes mitochondrial dysfunction [48]. Par‐4 increases the secretion of β‐amyloid (Aβ) and neuronal degeneration [49]. Par‐4 levels are augmented in AD patients [50] and in models of stroke [51]. RNAi knockdown of Par‐4 inhibits neurosynaptic degeneration in ALS‐linked mice [52]. Par‐4 interacts with Bcl‐2, caspase‐8, and PKCζ, thus inhibiting NF‐κB‐dependent survival signaling [53].
The MAPK family includes three members: extracellular signal‐regulated kinase (ERK), p38 mitogen‐activated protein kinase (p38 MAPK), and c‐Jun NH(2)‐terminal kinase (JNK). Another kinase is MAP kinase kinase (MEK). JNK pathway has been observed in neurodegenerative diseases mostly by activating apoptosis [54, 55] and partly by inhibiting cell death [56]. DNA damage causes the JNK activation, which contributes to the mitochondrial transduction of Bax [57, 58]. The absence of JNK causes a defect in the mitochondrial death signaling pathway, including the failure to release cytochrome c[57]. Moreover, SP600125, a JNK inhibitor, enhances the activation of JNK pathway and attenuation of apoptosis through protection of mitochondrial dysfunction and reduction of caspase‐9 activity in PC12 cells [59].
The Survival Signaling Pathways in Neurodegenerative Diseases
During the progression of neurodegenerative diseases, the survival signaling cascades are activated by neuroprotective agents [60] including the phosphoinositol‐3 kinase (PI3K)/Akt pathway, the Bcl‐2 pathway, the NF‐κB pathway, as well as the MAPK pathway (Fig. 1). AKT (v‐Akt murine thymoma viral oncogene)/PKB (protein kinase‐B) has been identified as an important mediator of neuronal cell survival that helps counteract apoptotic stimuli. PI3K/Akt pathways play essential roles in neuronal cell survival. PI3K is activated and the membrane phospholipid phosphatidylinositol‐3,4,5‐trisphosphate is generated, which in turn recruits Akt to the membrane, where it becomes phosphorylated. Once Akt is activated, it phosphorylates survival‐mediated targets including Bcl‐2 family members, thereby promoting cell survival and inhibiting apoptosis [61]. The antiapoptotic Bcl‐2 family encodes Bcl‐2, Bcl‐xL, and BfI‐1 (A1) [62]. These antiapoptotic proteins repress mitochondrial death pathways through heterodimerization [62]. Depletion of the endogenous neuroprotective Bcl‐2 family signals directly contributes to neuronal loss in neurodegenerative diseases [62]. NF‐κB (nuclear factor kappa B) is an inducible transcription factor that exists in several dimeric forms, with the p50/p65 heterodimer predominant [63]. The NF‐κB pathway induces the expression of stress proteins, antioxidant enzymes, and calcium‐regulating proteins. The activation of NF‐κB not only induces apoptotic signaling [5, 25, 26, 64] but also has been known to activate survival signals in neurodegeneration [27]. Additionally, the phosphorylation of Raf‐1, MEK1/2, and ERK1/2 has been reported in neurodegeneration [65]. The JNK pathway is also involved in neurodegenerative diseases by inhibiting cell death [56].
Melatonin in Neurodegenerative Diseases
Melatonin in Experimental Stroke
Animal models of stroke include global, multifocal, and focal cerebral ischemia. Focal cerebral ischemia is divided into transient (with reperfusion) and permanent (without reperfusion). Middle cerebral artery occlusion (MCAO) is the most commonly used animal model in the study of melatonin. Primary cortical neurons (PCNs) are the cells most commonly used in cellular model of stroke. The ability of melatonin to reduce infarct volume and/or inhibit neuronal cell death in experimental models of stroke has been demonstrated in different mammalian species [18, 66, 67, 68], but the signaling mechanisms underlying melatonin's neuroprotective actions remain incompletely understood. We summarize reports of neuroprotection by melatonin gained through inhibiting mitochondrial cell death pathways (Table 1) and activating survival pathways (Table 2) in experimental models of stroke (Fig. 1).
Table 1.
Summary of inhibition of the antiapoptotic cell death pathway by melatonin
| Inhibits death pathway event | Diseases/ models | Effects of melatonin | Species/cell line | References |
|---|---|---|---|---|
| Cytochrome c | Neurodegeneration | Inhibits cytochrome c release from purified mitochondria | Mouse | [76] |
| Stroke/MCAO | Decreases cytochrome c release | Rat, mouse, PCN | [18, 71] | |
| PD | Prevents cytochrome c release | Astrocyte | [117] | |
| Smac/Diablo | HD | Neuroprotective in HD models | Mu‐htt ST14A | [unpublished data] |
| AIF | Stroke | Neuroprotective in PCN | PCN | [18] |
| ΔΨm | Stroke | Neuroprotective in PSN and PCN | PSN; PCN | [18, 71] |
| PD | Prevents ΔΨm depolarization | Astrocyte | [117] | |
| mtPTP | Stroke | Inhibits mtPTP in brain ischemia | PSN | [71] |
| PD | Prevents mtPTP opening | Astrocyte | [117] | |
| Bax | AD | Attenuates Aβ25‐35‐induced apoptosis | Microglial cells | [25] |
| Bad | Stroke/MCAO | Attenuates cerebral ischemic injury | Rat | [65, 78] |
| ROS | PD | Prevents ROS formation | Astrocyte | [117] |
| ALS | Reduces ROS in ALS model | NSC34 motoneuron | [1] | |
| PARP | Stroke/MCAO | Attenuates cerebral ischemic injury | Rat | [65] |
| Caspase‐3 | Stroke/MCAO | Prevents caspase‐3 activation | Rat, mouse, PCN | [18, 71, 77] |
| AD | Attenuates Aβ25‐35‐induced apoptosis | Microglial cells | [25] | |
| PD | Blocks caspase‐3 activation | Astrocyte, dopaminergic neuron; CGN | [116, 117, 118] | |
| Caspase‐9 | HD | Neuroprotective in HD models | Mu‐htt ST14A | [unpublished data] |
| Caspase‐1 | Stroke | Neuroprotective in PCN | PCN | [18] |
| IL‐1β | Stroke | Neuroprotective in PCN | PCN | [18] |
| Rip2 | HD | Neuroprotective in HD models | Mu‐htt ST14A | [unpublished data] |
| DNA | Stroke/MCAO | Displays decreased DNA | Rat, PCN | [18, 71] |
| Fragmentation | fragmentation, neuroprotective in PCN | |||
| AD | Attenuates Aβ25‐35‐ or Aβ1‐42‐induced apoptosis | Astroglioma C6 cell | [102] | |
| PD | Prevents DNA fragmentation | SK‐N‐SH cells, astrocyte, mesencephalic cells, striatal neuron; mouse; PC 12 cells | [54, 117, 121, 122] | |
| TUNEL‐positive | Neurodegeneration | Reduces number of DNA breaks | Rat | [80] |
| Stroke/MCAO | Decreases TUNEL‐positive cells | Rat | [65, 78, 79] | |
| Stroke/OGD | Neuroprotective in PCN | PCN | [18] | |
| AD/OVX | Improves spatial memory performance, reduces apoptosis | Rat | [89] | |
| AD | Protects the wortmannin‐induced tau hyperphosphorylation | N2a cells | [94] | |
| INK | PD | Inhibits cell death | SK‐N‐SH cells | [54, 55] |
| Par‐4 | AD | Reducts Par‐4 upregulation | Mouse | [92] |
| nf‐κb | AD | Blocks Aβ25‐35‐induced apoptosis | Microglial cells, mouse | [5, 25] |
| AD | Anti‐inflammatory effect on Aβ vaccination in mice | Mouse | [26] |
OVX, ovariectomized.
Table 2.
Summary of activation of antiapoptotic survival signal pathway by melatonin
| Activates element of survival pathway | Diseases/models | Effects of melatonin | Species/cell line | References |
|---|---|---|---|---|
| PI3‐K/Akt | Stroke/MCAO | Restores phosphorylated Akt | Mouse, rat | [56, 77, 78] |
| Protects against brain injury | Rat | [81] | ||
| AD | Impairs NADPH oxidase via PI3K/Akt signaling pathway | Microglia | [93] | |
| Bcl‐2 | Stroke/MCAO | Enhances Bcl‐2 upregulation | Rat | [79, 82] |
| AD/Ap25‐35 | Attenuates Ap25‐35‐induced apoptosis | Microglial cells | [25] | |
| Bcl‐xL | Stroke/MCAO | Elevates Bcl‐xL in brain injury | Mouse | [77] |
| JNK1/2 | Stroke/MCAO | Increases JNK1/2 phosphorylation | Mouse | [56] |
| ERK1/2 | Stroke/MCAO | Increases ERK1/2 phosphorylation | Mouse, rat | [56, 65] |
| Raf‐1 | Stroke/MCAO | Attenuates cerebral ischemic injury | Rat | [65] |
| MEK1/2 | Stroke/MCAO | Attenuates cerebral ischemic injury | Rat | [65] |
| nf‐κb | Stroke | Relates with NE‐κB‐me‐dialed protective signaling | Primary neurons | [27] |
The highest levels of melatonin are found in the mitochondria [69]. Mitochondria have been identified as a target for melatonin [70, 71]. Melatonin promotes mitochondrial homeostasis. Taken together, melatonin may be possible to treat neurodegenerative disorders by inhibiting mitochondrial cell death pathways [1, 72, 73, 74, 75]. We screened a library of 1040 FDA‐approved drugs assembled by the Neurodegeneration Drug Screening Consortium of the National Institute of Neurological Disorders and Stroke (NINDS) for their ability to inhibit release of cytochrome c from Ca2+‐stimulated mitochondria [76]. Melatonin occupied one of the top positions (14th) [76]. Furthermore, we and other laboratories demonstrated that melatonin has proved effective not only in the cell‐free purified mitochondrial system but also inhibits cytochrome c release in an MCAO mouse model [18, 71] and in PCN [18]. Melatonin prevents the release of cell death mediator AIF from mitochondria in PCNs on insult [71]. Thus, melatonin is likely to interfere with both caspase‐dependent (cytochrome c) and independent (AIF) mitochondrial cell death pathways. Proper ΔΨm is critical for appropriate cellular bioenergetic homeostasis, and dissipation of ΔΨm has been involved in stroke [18, 35]. Studies in both primary striatal neurons (PSNs) [71] and PCNs [18] showed that melatonin effectively inhibited oxygen/glucose deprivation (OGD)‐mediated dissipation of ΔΨm. These effects reflect the ability of melatonin to ameliorate the harmful reduction in the ΔΨm, which may trigger mitochondrial transition pore opening and the apoptosis cascade. mtPTP contributes to the pathology of ischemia. Further experiments indeed demonstrated that melatonin directly inhibits mtPTP in PSNs after OGD insult [71].
Caspase‐1 plays a critical role as an apical activator in models of stroke [16]. Interestingly, melatonin inhibits OGD‐induced caspase‐1 activation and mature IL‐1β release in PCNs [18]. In vitro and in vivo experiments have shown that melatonin prevents the activation of downstream caspase‐3 in OGD‐mediated PCN cell death [18], cerebral ischemia‐induced mouse injury, and the MCAO rat model [18, 72, 77]. Other experiments demonstrate significantly fewer TUNEL‐positive cells [65, 78], reduced levels of cleaved PARP [65], and less DNA fragments [71] are found with administration of melatonin in the rat MCAO model. In addition, melatonin prevents brain damage, with reduced TUNEL‐positive cells following transient cerebral artery occlusion (CerAO) [66] and transient MCAO model [79], as well as attenuating kainic acid‐induced neuronal death, and reduces the number of TUNEL‐labeled DNA breaks [80].
The neuroprotective role of melatonin is also mediated through the enhancement of the PI3‐K/Akt survival pathway [77, 81] and JNK pathway [56], and restores reduced phosphorylated Akt in a model of mouse intraluminal MCAO [77]. Melatonin protected neuronal cells from damage by enhancing the activation of Akt and its downstream target Bad, without affecting the expression of 14‐3‐3, which acts as an antiapoptotic factor through interaction with Bad, thus mediating antiapoptosis signals in a rat MCAO model [78]. Furthermore, in the same model, melatonin inhibits apoptotic signals by preventing the injury‐induced decrease of phosphorylation of Raf‐1, MEK1/2, and ERK1/2 and the downstream targets, including Bad and 90‐kDa ribosomal S6 kinase [65]. Melatonin effectively attenuated ischemic brain injury via the Bcl‐2‐related survival pathway by increasing the expression of Bcl‐2 [82] and Bcl‐xL [56] in the ischemic brain. Furthermore, related to melatonin, the constitutive activation of NF‐κB under physiological conditions protects neurons against physiological injury [27].
Intervention studies have identified a battery of approaches with potential benefits in reducing neuronal death in stroke patients, including antioxidant treatment. Clinical data report some alteration of the melatoninergic system in human stroke. On the basis of its lack of toxicity, melatonin may eventually be included in human stroke treatment.
Melatonin in Alzheimer Disease
AD, the most common neurodegenerative disease with progressive loss of memory and deterioration of comprehensive cognition, is characterized by extracellular senile plaques of aggregated β‐amyloid (Aβ) and intracellular neurofibrillary tangles that contain hyperphosphorylated tau protein. Aβ and tau therefore represent important therapeutic targets. The early phase of AD is treatable by inhibitors of β‐ and γ‐secretase, which degrade amyloid precursor protein (APP) to produce β‐amyloid peptide [83], and the late phase is amendable to treatment strategy by preventing or reversing tau phosphorylation [84, 85]. Mild cognitive impairment (MCI) is a transition stage between the cognitive decline of normal aging and the more serious problems caused by AD. Many people with MCI eventually develop AD. Studies show that melatonin levels are lower in AD patients compared with that in age‐matched control subjects [86, 87, 88]. The great advance has been currently conduced in studies of protection against AD by antioxidant melatonin inhibiting Aβ‐induced toxicity [25, 89, 90, 91, 92, 93] and attenuating tau hyperphosphorylation [85, 94, 95, 96, 97, 98, 99]. Besides the antioxidant properties, the antiamyloidogenic properties of melatonin for AD have been studied [100, 101]. Melatonin improved learning and memory deficits in an APP695 transgenic mouse model of AD in vivo[89]. In vitro experiments showed that Aβ‐treated cultures exhibited characteristic features of apoptosis, and melatonin attenuated Aβ‐induced apoptosis in a number of cellular models of AD including mouse microglial BV2 cells, rat astroglioma C6 cells, and PC12 cells [25, 89, 90, 91, 102].
It is known that melatonin scavenges oxygen and nitrogen‐based reactants generated in mitochondria, and mitochondria play a critical role in the neuroprotective function of melatonin in AD. As listed in table 1, studies in transgenic AD mice and cultured cells have suggested that administration of melatonin inhibited the Aβ‐induced increase in the levels of mitochondria‐related Bax [25, 92]. Furthermore, melatonin prevented upregulated expression of Par‐4 and suppressed Aβ‐induced caspase‐3 activity [92]. Another experiment in mouse microglial BV2 cells in vitro showed that melatonin also decreased caspase‐3 activity, inhibited NF‐κB activation, and reduced the generation of Aβ‐induced intracellular ROS (reactive oxygen species) [25]. In addition, in vivo observations showed that melatonin‐treated animals had diminished expression of NF‐κB compared to untreated animals [26]. Melatonin treatment significantly decreased the number of TUNEL‐positive neurons along with improving spatial memory performance in cognitively impaired, ovariectomized adult rats [89] and Alzheimer‐like tau hyperphosphorylation in wortmannin‐induced N2a cells [94].
On the other hand, melatonin may also activate the survival signal pathways. One such pathway is the Bcl‐2 pathway, which stabilizes mitochondrial function by antiapoptotic Bcl‐2 family modulators. It has been reported that Bcl‐2 expression was enhanced by melatonin concomitant with inhibition of Aβ‐induced cell death [25] (Table 2). Another experiment demonstrated that melatonin inhibited the phosphorylation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase via a PI3K/Akt‐dependent signaling pathway in microglia exposed to Aβ1‐42 [93] (Table 2). Taken together, the above‐mentioned evidence suggests that melatonin may provide an effective means of treatment for AD through its antiapoptotic activities.
It has been reported that administration of melatonin significantly delays the development of the signs of AD, prevents cognitive impairment, and ameliorates sundowning in AD patients [103, 104, 105, 106, 107]. In addition, light therapy or music therapy related with levels of melatonin may have effect on AD patients [108, 109]. On the contrary, some researchers report that the impact of melatonin would be relatively less in late stage of AD or fails to improve sleep or agitation; therefore, melatonin is not an effective soporific agent in patients with AD [110, 111].
Human trials in the relatively small scale suggest that melatonin can improve MCI [112, 113]. However, how melatonin affects disease initiation or progression of the neuropathology and if the antiapoptotic activity of melatonin is driving its function, remains to be answered. On the other hand, controversy reports suggest insufficient evidence to support the effectiveness of melatonin for managing cognitive impairment with low success rate [114]. Further, clinical phase II trial of the effect of melatonin on cognitive function in MCI patients is undergoing (ClinicalTrials.gov Identifier: NCT00544791).
Melatonin in Parkinson Disease
PD is the second most common neurodegenerative disease, affecting approximately 1.8% of people older than 65 years [115]. PD is characterized by a progressive loss of dopaminergic neurons and dopamine in the substantia nigra and striatum. Oxidative stress and free radicals from both mitochondrial impairment and dopamine metabolism are considered to play critical roles in the etiology of PD. In addition, neurodegeneration occurs in PD, at least in part, through the activation of the mitochondria‐dependent apoptotic molecular pathway [17]. 1‐Methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine (MPTP) has been reported to cause parkinsonism via its neurotoxic form, 1‐methyl‐4‐phenylpyridinium ion (MPP+), which inhibits mitochondrial complex I of the mitochondrial respiratory chain. MPP+ has been as a commonly used experimental model of PD [54, 116].
As shown in table 1 and figure 1, melatonin prevents H2O2‐induced mitochondrial calcium overload, ΔΨm depolarization, opening of mtPTP, avoidance of ROS formation, as well as blocked MPT‐dependent cytochrome c release in rat astrocytes, a model of PD [117]. Also in the same model, melatonin inhibited MPT‐dependent activation of caspase‐3 [117]. This conclusion is further supported by the finding that melatonin suppressed 3‐morpholinosydnonimine‐induced caspase‐3 activation in dopaminergic neurons [118] and diminished the activation of caspase‐3 enzyme activity in both MPP(+)‐treated SK‐N‐SH cultured cells [54] and cerebellar granule neurons (CGNs) [116]. In addition, melatonin exerted neuroprotective effects against MPP+‐induced apoptosis by inhibiting the calpain/cdk5 signaling cascade in CGNs [116]. Mounting evidence indicates that melatonin blocks the MPT‐dependent apoptotic fragmentation of nuclear DNA in rat astrocytes [117], rat mesencephalic cultures [119], and mouse striatal neurons [120]. Other experiments also indicate MPTP‐induced mouse brain cell DNA fragmentation in vivo[121], 6‐hydroxydopamine‐induced DNA fragmentation in neuronal PC12 cells [122], and MPP(+)‐mediated cleavage of DNA fragmentation factors in SK‐N‐SH cultured cells in vitro[54]. The JNK pathway is involved in PD by activating apoptosis [54, 55], and transcription factors play a role in PD, as shown in two experiments demonstrating the action of melatonin to inhibit JNK signaling cascade [54, 55] and diminish the induction of phosphorylation of c‐Jun in MPP(+)‐treated [54] and 6‐hydroxydopamine‐induced SK‐N‐SH‐cultured cells [54, 55]. To date, there are no reports of the activation of survival pathways by melatonin in PD.
Human trials for melatonin's effect on sleep disturbances in PD show significantly improved [123] or small improvement [124], but there are undetected differences in motor dysfunction [123].
Melatonin in Huntington Disease
HD, a hereditary disease affecting 30,000 Americans, is universally fatal with no effective treatment. HD is characterized by movement disorder (Huntington chorea), cognitive deterioration, emotional distress, and dementia [125]. HD is caused by expansion of cytosine—adenine–guanine (CAG) repeats in exon 1 of the huntingtin gene [126], initially affecting the striatum and then the cortex. Since oxidative stress plays an important role in the etiology of neuronal damage and degeneration in HD [127], therapeutic strategies against HD focus on antioxidant defense.
Mitochondrial complex II inhibitor 3‐nitropropionic acid can closely replicate the neurochemical, histological, and clinical features of HD and hence is used in an experimental model of HD [75, 128]. So far the antioxidant melatonin has been suggested to defer the signs of HD in a 3‐nitropropionic acid‐induced rat animal model of HD [75] and to reduce lipid peroxidation induced by quinolinic acid (a causative agent in HD) [74]. In addition, we report that melatonin is a remarkably potent neuroprotective agent in mutant‐hungtintin (mutant‐htt) ST14A cells, a cellular model of HD [76, 129, 130]. It protects 76.2% of mutant‐htt ST14A cell death from temperature shift‐induced cell death [76]. Furthermore, melatonin prevents cell death of PCNs that have been challenged with proapoptotic inducer [18]. One of our compelling findings underlying the mechanism of melatonin's action against HD is that it counters mitochondrial cell death pathways through the inhibition of the release of Smac and the activation of caspase‐9 in apoptotic mutant‐htt ST14A striatal cells (unpublished data). Furthermore, administration of melatonin also significantly inhibits the Rip2 upregulation in mutant‐htt ST14A cells under insult (unpublished data). Thus, our findings suggest that melatonin acts on initiated Rip2 (Fig. 1). On the other hand, there has been no report that melatonin activates survival pathways in HD yet.
In a human study with the addition of tryptophan, albeit melatonin levels rose significantly in both control and HD patients group, bigger increasing average mean occurs in HD patients group [131]. Moreover, the delayed onset of the diurnal melatonin rise in patients with HD in small scale has been currently reported [132]. Larger scale studies in detecting the level of melatonin in HD patients and further human trials on the impact of melatonin on HD are needed.
Melatonin in Amyotrophic Lateral Sclerosis
ALS is a fatal disease of varying etiology whose progression is characterized by a degeneration of motor neurons. Riluzole, an antagonist of the glutamate receptor, is the only approved treatment for ALS. However, it typically prolongs the patient's life by only 3 months. Since the common basis of cellular and extracellular alterations in this disease seems to be oxidative stress, the strategy for the treatment of ALS therefore emphasizes antioxidant molecules.
Rival et al. report that, exactly as administration of riluzole in dEAAT1 RNAi flies, administration of the antioxidant melatonin significantly enhanced performance in a Drosophila model, exhibiting remarkable similarity with some of the symptoms associated with ALS [133]. Furthermore, melatonin offers protection in human ALS. The first clinical trial of melatonin in three human ALS patients was reported in 2002 [73], and the second human trial in a group of 31 patients with sporadic ALS was reported in 2006 [1]. Importantly, circulating serum protein carbonyls, which provide a surrogate marker for oxidative stress, were elevated in ALS patients, but were reported to be normalized to control values by melatonin treatment in the second clinical trial [1]. In other words, reduced oxidative damage was reported in ALS trial using high‐dose enteral melatonin [1]. Chronic high‐dose (300 mg/day [1]) rectally administered melatonin was well tolerated in patients with sporadic ALS [1, 73]. In addition, the findings from both animal models in vivo and a cellular model in vitro support the results of human trials, in SOD1(G93A)‐transgenic mice, high doses of orally administered melatonin delayed disease progression, and extended survival in vivo[1]. However, Western blot analysis of spinal cord protein lysates in the same study found no differences in total amount or phosphorylation status of AKT or ERK1/2 in SOD1(G93A)‐transgenic mice with melatonin treatment compared with untreated controls [1] (Fig. 1). Another study showed that the administration of melatonin alters the expression of SOD1 in the lumbar spinal cord of neonatal rats [134]. Furthermore, melatonin attenuates superoxide‐induced cell death and modulates glutamate toxicity in cultured NSC‐34 motoneuron cells in vitro[1]. Although evidence indicates that mSOD1‐induced spinal cord motor neuron death and cultured motor neuronal cells involve apoptotic machinery [12, 135, 136, 137], to date, the neuroprotection afforded by melatonin through the inhibition of cell death pathways or activation of survival pathways remains essentially uninvestigated.
Because melatonin is neuroprotective in human, cellular, and animal models of ALS and is relatively nontoxic, it should be considered for further larger clinical trials as a novel pharmacotherapeutic agent to treat ALS.
Conclusion and Perspective
Given the fact that vigorous research efforts to date have achieved poor results in their efforts to identify effective treatments against neurodegenerative diseases, the combination of preclinical effectiveness and proven safety of melatonin in humans, animals, and cultured cells recommends it as a particularly interesting candidate of neuroprotectant in clinical trials seeking protection against neurodegeneration. Interestingly, melatonin is capable of interfering with mitochondrial cell death pathways and activating survival pathways, both of which would be useful in treating common events in stroke, AD, PD, ALS, and HD. In addition, blood concentrations of neurohormone melatonin are significantly decreased in patients with AD [87], while low levels of melatonin and a prolonged signal of melatonin are found in PD patients [138] and the delayed onset of the diurnal melatonin rise in HD patients [132]. Thus it is believed that reduced secretion of melatonin is associated with the development of neurodegenerative disease [87]. Knowing about the molecular mechanism of melatonin's declining potency should tell us about the pathogenesis of related neurodegenerative diseases and will guide the contemplated translation to the clinic. Pharmacological strategies to enhance melatonin levels may benefit those suffering from neurodegenerative diseases.
Cell death‐based therapies are becoming an active area of drug development. For a multidrug regimen to effectively protect neurons from inappropriate apoptosis, several pathways could be coactivated, including antiapoptotic pathways and survival pathways. This review gains deeper insights into the action mechanism of melatonin. Thus, it may provide a new perspective in our understanding of the regulation of apoptotic cell death in neurodegeneration by the pharmacotherapeutic indoleamine. Besides its traditional role as an antioxidant and free radical scavenger, melatonin proved to target a variety of pathways while its systemic effect correlates with the drug's disruption of the intrinsic mitochondrial cell death pathway, silencing of the Rip2/caspase‐1 pathway, and the activation of survival pathways. These actions may be synchronistic and complementary in models of HD. Effective treatment to prevent neurodegeneration could be achieved using a combination of melatonin and other pharmacological agents that act on different apoptosis targets.
Future therapeutic strategies could be directed at identifying and developing drugs from among the analogues of melatonin. Candidate drugs may have more powerful inhibitory effects on the mitochondrial cell death pathway and activate the survival pathway, thus slowing the progression of neurodegenerative diseases.
Conflict of Interest
The authors have no conflict of interest.
Acknowledgments
The author thanks Paul Guttry for editorial assistance. This work was supported by grants from the National Institutes of Health/National Institute of Neurological Disorders and Stroke (to X.W.) and the Hereditary Disease Foundation (to X.W.).
References
- 1. Weishaupt JH, Bartels C, Polking E, Dietrich J, Rohde G, Poeggeler B, Mertens N, Sperling S, Bohn M, Huther G, et al Reduced oxidative damage in ALS by high‐dose enteral melatonin treatment. J Pineal Res 2006;41:313–323. [DOI] [PubMed] [Google Scholar]
- 2. Sewerynek E. Melatonin and the cardiovascular system. Neuro Endocrinol Lett 2002;23(Suppl 1):79–83. [PubMed] [Google Scholar]
- 3. Rodriguez MI, Escames G, Lopez LC, Lopez A, Garcia JA, Ortiz F, Sanchez V, Romeu M, Acuna‐Castroviejo D. Improved mitochondrial function and increased life span after chronic melatonin treatment in senescent prone mice. Exp Gerontol 2008;43:749–756. [DOI] [PubMed] [Google Scholar]
- 4. Caballero B, Vega‐Naredo I, Sierra V, Huidobro‐Fernandez C, Soria‐Valles C, De Gonzalo‐Calvo D, Tolivia D, Pallas M, Camins A, Rodriguez‐Colunga MJ, et al Melatonin alters cell death processes in response to age‐related oxidative stress in the brain of senescence‐accelerated mice. J Pineal Res 2009;46:106–114. [DOI] [PubMed] [Google Scholar]
- 5. Gutierrez‐Cuesta J, Tajes M, Jimenez A, Coto‐Montes A, Camins A, Pallas M. Evaluation of potential pro‐survival pathways regulated by melatonin in a murine senescence model. J Pineal Res 2008;45:497–505. [DOI] [PubMed] [Google Scholar]
- 6. Ravindra T, Lakshmi NK, Ahuja YR. Melatonin in pathogenesis and therapy of cancer. Indian J Med Sci 2006;60:523–535. [PubMed] [Google Scholar]
- 7. Tan DX, Reiter RJ, Manchester LC, Yan MT, El‐Sawi M, Sainz RM, Mayo JC, Kohen R, Allegra M, Hardeland R. Chemical and physical properties and potential mechanisms: Melatonin as a broad spectrum antioxidant and free radical scavenger. Curr Top Med Chem 2002;2:181–197. [DOI] [PubMed] [Google Scholar]
- 8. Reiter RJ, Cabrera J, Sainz RM, Mayo JC, Manchester LC, Tan DX. Melatonin as a pharmacological agent against neuronal loss in experimental models of Huntington's disease, Alzheimer's disease and parkinsonism. Ann N Y Acad Sci 1999;890:471–485. [DOI] [PubMed] [Google Scholar]
- 9. Ashkenazi A, Dixit VM. Death receptors: Signaling and modulation. Science 1998;281:1305–1308. [DOI] [PubMed] [Google Scholar]
- 10. Viswanath V, Wu Y, Boonplueang R, Chen S, Stevenson FF, Yantiri F, Yang L, Beal MF, Andersen JK. Caspase‐9 activation results in downstream caspase‐8 activation and bid cleavage in 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine‐induced Parkinson's disease. J Neurosci 2001;21:9519–9528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jin Z, El‐Deiry WS. Overview of cell death signaling pathways. Cancer Biol Ther 2005;4:139–163. [DOI] [PubMed] [Google Scholar]
- 12. Friedlander RM. Apoptosis and caspases in neurodegenerative diseases. N Engl J Med 2003;348:1365–1375. [DOI] [PubMed] [Google Scholar]
- 13. Namura S, Nagata I, Takami S, Masayasu H, Kikuchi H. Ebselen reduces cytochrome c release from mitochondria and subsequent DNA fragmentation after transient focal cerebral ischemia in mice. Stroke 2001;32:1906–1911. [DOI] [PubMed] [Google Scholar]
- 14. Wang X, Zhu S, Drozda M, Zhang W, Stavrovskaya IG, Cattaneo E, Ferrante RJ, Kristal BS, Friedlander RM. Minocycline inhibits caspase‐independent and ‐dependent mitochondrial cell death pathways in models of Huntington's disease. Proc Natl Acad Sci USA 2003;100:10483–10487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhu S, Stavrovskaya IG, Drozda M, Kim BY, Ona V, Li M, Sarang S, Liu AS, Hartley DM, Wu du C, et al Minocycline inhibits cytochrome c release and delays progression of amyotrophic lateral sclerosis in mice. Nature 2002;417:74–78. [DOI] [PubMed] [Google Scholar]
- 16. Zhang WH, Wang X, Narayanan M, Zhang Y, Huo C, Reed JC, Friedlander RM. Fundamental role of the Rip2/caspase‐1 pathway in hypoxia and ischemia‐induced neuronal cell death. Proc Natl Acad Sci USA 2003;100:16012–16017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vila M, Przedborski S. Targeting programmed cell death in neurodegenerative diseases. Nat Rev Neurosci 2003;4:365–375. [DOI] [PubMed] [Google Scholar]
- 18. Wang X, Figueroa BE, Stavrovskaya IG, Zhang Y, Zhu S, Day AL, Kristal BS, Friedlander RM. Methazolamide and melatonin inhibit mitochondrial cytochrome c release and are neuroprotective in experimental models of ischemic injury. Stroke 2009;40:1877–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lee BI, Lee DJ, Cho KJ, Kim GW. Early nuclear translocation of endonuclease G and subsequent DNA fragmentation after transient focal cerebral ischemia in mice. Neurosci Lett 2005;386:23–27. [DOI] [PubMed] [Google Scholar]
- 20. Kroemer G, Reed JC. Mitochondrial control of cell death. Nat Med 2000;6:513–519. [DOI] [PubMed] [Google Scholar]
- 21. Ferrer I, Planas AM. Signaling of cell death and cell survival following focal cerebral ischemia: Life and death struggle in the penumbra. J Neuropathol Exp Neurol 2003;62:329–339. [DOI] [PubMed] [Google Scholar]
- 22. Graham RK, Deng Y, Slow EJ, Haigh B, Bissada N, Lu G, Pearson J, Shehadeh J, Bertram L, Murphy Z, et al Cleavage at the caspase‐6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin. Cell 2006;125:1179–1191. [DOI] [PubMed] [Google Scholar]
- 23. Guegan C, Vila M, Rosoklija G, Hays AP, Przedborski S. Recruitment of the mitochondrial‐dependent apoptotic pathway in amyotrophic lateral sclerosis. J Neurosci 2001;21:6569–6576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li M, Ona VO, Guegan C, Chen M, Jackson‐Lewis V, Andrews LJ, Olszewski AJ, Stieg PE, Lee JP, Przedborski S, et al Functional role of caspase‐1 and caspase‐3 in an ALS transgenic mouse model. Science 2000;288:335–339. [DOI] [PubMed] [Google Scholar]
- 25. Jang MH, Jung SB, Lee MH, Kim CJ, Oh YT, Kang I, Kim J, Kim EH. Melatonin attenuates amyloid beta25‐35‐induced apoptosis in mouse microglial BV2 cells. Neurosci Lett 2005;380:26–31. [DOI] [PubMed] [Google Scholar]
- 26. Jesudason EP, Baben B, Ashok BS, Masilamoni JG, Kirubagaran R, Jebaraj WC, Jayakumar R. Anti‐inflammatory effect of melatonin on a beta vaccination in mice. Mol Cell Biochem 2007;298:69–81. [DOI] [PubMed] [Google Scholar]
- 27. Kratsovnik E, Bromberg Y, Sperling O, Zoref‐Shani E. Oxidative stress activates transcription factor NF‐κB‐mediated protective signaling in primary rat neuronal cultures. J Mol Neurosci 2005;26:27–32. [DOI] [PubMed] [Google Scholar]
- 28. Jang JH, Surh YJ. AP‐1 mediates beta‐amyloid‐induced iNOS expression in PC12 cells via the ERK2 and p38 MAPK signaling pathways. Biochem Biophys Res Commun 2005;331:1421–1428. [DOI] [PubMed] [Google Scholar]
- 29. Akaji K, Suga S, Fujino T, Mayanagi K, Inamasu J, Horiguchi T, Sato S, Kawase T. Effect of intra‐ischemic hypothermia on the expression of c‐Fos and c‐Jun, and DNA binding activity of AP‐1 after focal cerebral ischemia in rat brain. Brain Res 2003;975:149–157. [DOI] [PubMed] [Google Scholar]
- 30. De La Monte SM, Sohn YK, Ganju N, Wands JR. P53‐ and CD95‐associated apoptosis in neurodegenerative diseases. Lab Invest 1998;78:401–411. [PubMed] [Google Scholar]
- 31. Deigner HP, Haberkorn U, Kinscherf R. Apoptosis modulators in the therapy of neurodegenerative diseases. Expert Opin Investig Drugs 2000;9:747–764. [DOI] [PubMed] [Google Scholar]
- 32. Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c‐dependent caspase activation by eliminating IAP inhibition. Cell 2000;102:33–42. [DOI] [PubMed] [Google Scholar]
- 33. Sas K, Robotka H, Toldi J, Vecsei L. Mitochondria, metabolic disturbances, oxidative stress and the kynurenine system, with focus on neurodegenerative disorders. J Neurol Sci 2007;257:221–239. [DOI] [PubMed] [Google Scholar]
- 34. Jordan J, Cena V, Prehn JH. Mitochondrial control of neuron death and its role in neurodegenerative disorders. J Physiol Biochem 2003;59:129–141. [DOI] [PubMed] [Google Scholar]
- 35. Iijima T. Mitochondrial membrane potential and ischemic neuronal death. Neurosci Res 2006;55:234–243. [DOI] [PubMed] [Google Scholar]
- 36. Wang H, Guan Y, Wang X, Smith K, Cormier K, Zhu S, Stavrovskaya IG, Huo C, Ferrante RJ, Kristal BS, et al Nortriptyline delays disease onset in models of chronic neurodegeneration. Eur J Neurosci 2007;26:633–641. [DOI] [PubMed] [Google Scholar]
- 37. Chen M, Ona VO, Li M, Ferrante RJ, Fink KB, Zhu S, Bian J, Guo L, Farrell LA, Hersch SM, et al Minocycline inhibits caspase‐1 and caspase‐3 expression and delays mortality in a transgenic mouse model of Huntington disease. Nat Med 2000;6:797–801. [DOI] [PubMed] [Google Scholar]
- 38. Li P, Allen H, Banerjee S, Franklin S, Herzog L, Johnston C, McDowell J, Paskind M, Rodman L, Salfeld J, et al Mice deficient in IL‐1 beta‐converting enzyme are defective in production of mature IL‐1 beta and resistant to endotoxic shock. Cell 1995;80:401–411. [DOI] [PubMed] [Google Scholar]
- 39. Engidawork E, Gulesserian T, Yoo BC, Cairns N, Lubec G. Alteration of caspases and apoptosis‐related proteins in brains of patients with Alzheimer's disease. Biochem Biophys Res Commun 2001;281:84–93. [DOI] [PubMed] [Google Scholar]
- 40. Wang X, Wang H, Figueroa BE, Zhang WH, Huo C, Guan Y, Zhang Y, Bruey JM, Reed JC, Friedlander RM. Dysregulation of receptor interacting protein‐2 and caspase recruitment domain only protein mediates aberrant caspase‐1 activation in Huntington's disease. J Neurosci 2005;25:11645–11654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Danial NN, Korsmeyer SJ. Cell death: Critical control points. Cell 2004;116:205–219. [DOI] [PubMed] [Google Scholar]
- 42. Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, Peng TI, Jones DP, Wang X. Prevention of apoptosis by Bcl‐2: Release of cytochrome c from mitochondria blocked. Science 1997;275:1129–1132. [DOI] [PubMed] [Google Scholar]
- 43. Gross A, Jockel J, Wei MC, Korsmeyer SJ. Enforced dimerization of BAX results in its translocation, mitochondrial dysfunction and apoptosis. Embo J 1998;17:3878–3885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Luo X, Budihardjo I, Zou H, Slaughter C, Wang X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 1998;94:481–490. [DOI] [PubMed] [Google Scholar]
- 45. Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 1998;94:491–501. [DOI] [PubMed] [Google Scholar]
- 46. Guegan C, Vila M, Teissman P, Chen C, Onteniente B, Li M, Friedlander RM, Przedborski S. Instrumental activation of bid by caspase‐1 in a transgenic mouse model of ALS. Mol Cell Neurosci 2002;20:553–562. [DOI] [PubMed] [Google Scholar]
- 47. Hu Y, Benedict MA, Wu D, Inohara N, Nunez G. Bcl‐XL interacts with Apaf‐1 and inhibits Apaf‐1‐dependent caspase‐9 activation. Proc Natl Acad Sci USA 1998;95:4386–4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mattson MP. Excitotoxic and excitoprotective mechanisms: Abundant targets for the prevention and treatment of neurodegenerative disorders. Neuromolecular Med 2003;3:65–94. [DOI] [PubMed] [Google Scholar]
- 49. Guo Q, Xie J, Chang X, Du H. Prostate apoptosis response‐4 enhances secretion of amyloid beta peptide 1‐42 in human neuroblastoma IMR‐32 cells by a caspase‐dependent pathway. J Biol Chem 2001;276:16040–16044. [DOI] [PubMed] [Google Scholar]
- 50. Xie J, Guo Q. PAR‐4 is involved in regulation of beta‐secretase cleavage of the Alzheimer amyloid precursor protein. J Biol Chem 2005;280:13824–13832. [DOI] [PubMed] [Google Scholar]
- 51. Culmsee C, Zhu Y, Krieglstein J, Mattson MP. Evidence for the involvement of Par‐4 in ischemic neuron cell death. J Cereb Blood Flow Metab 2001;21:334–343. [DOI] [PubMed] [Google Scholar]
- 52. Xie J, Awad KS, Guo Q. RNAi knockdown of Par‐4 inhibits neurosynaptic degeneration in ALS‐linked mice. J Neurochem 2005;92:59–71. [DOI] [PubMed] [Google Scholar]
- 53. Culmsee C, Landshamer S. Molecular insights into mechanisms of the cell death program: Role in the progression of neurodegenerative disorders. Curr Alzheimer Res 2006;3:269–283. [DOI] [PubMed] [Google Scholar]
- 54. Chetsawang J, Govitrapong P, Chetsawang B. Melatonin inhibits MPP+‐induced caspase‐mediated death pathway and DNA fragmentation factor‐45 cleavage in SK‐N‐SH cultured cells. J Pineal Res 2007;43:115–120. [DOI] [PubMed] [Google Scholar]
- 55. Chetsawang B, Govitrapong P, Ebadi M. The neuroprotective effect of melatonin against the induction of c‐Jun phosphorylation by 6‐hydroxydopamine on SK‐N‐SH cells. Neurosci Lett 2004;371:205–208. [DOI] [PubMed] [Google Scholar]
- 56. Kilic U, Kilic E, Reiter RJ, Bassetti CL, Hermann DM. Signal transduction pathways involved in melatonin‐induced neuroprotection after focal cerebral ischemia in mice. J Pineal Res 2005;38:67–71. [DOI] [PubMed] [Google Scholar]
- 57. Tournier C, Hess P, Yang DD, Xu J, Turner TK, Nimnual A, Bar‐Sagi D, Jones SN, Flavell RA, Davis RJ. Requirement of JNK for stress‐induced activation of the cytochrome c‐mediated death pathway. Science 2000;288:870–874. [DOI] [PubMed] [Google Scholar]
- 58. Lei K, Nimnual A, Zong WX, Kennedy NJ, Flavell RA, Thompson CB, Bar‐Sagi D, Davis RJ. The Bax subfamily of Bcl2‐related proteins is essential for apoptotic signal transduction by c‐Jun NH(2)‐terminal kinase. Mol Cell Biol 2002;22:4929–4942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Marques CA, Keil U, Bonert A, Steiner B, Haass C, Muller WE, Eckert A. Neurotoxic mechanisms caused by the Alzheimer's disease‐linked Swedish amyloid precursor protein mutation: Oxidative stress, caspases, and the JNK pathway. J Biol Chem 2003;278:28294–28302. [DOI] [PubMed] [Google Scholar]
- 60. Mattson MP, Culmsee C, Yu ZF. Apoptotic and antiapoptotic mechanisms in stroke. Cell Tissue Res 2000;301:173–187. [DOI] [PubMed] [Google Scholar]
- 61. Yang ZZ, Tschopp O, Baudry A, Dummler B, Hynx D, Hemmings BA. Physiological functions of protein kinase B/Akt. Biochem Soc Trans 2004;32:350–354. [DOI] [PubMed] [Google Scholar]
- 62. Lukiw WJ, Bazan NG. Survival signalling in Alzheimer's disease. Biochem Soc Trans 2006;34:1277–1282. [DOI] [PubMed] [Google Scholar]
- 63. Pande V, Ramos MJ. NF‐kappaB in human disease: Current inhibitors and prospects for de novo structure based design of inhibitors. Curr Med Chem 2005;12:357–374. [DOI] [PubMed] [Google Scholar]
- 64. Nakai M, Qin ZH, Chen JF, Wang Y, Chase TN. Kainic acid‐induced apoptosis in rat striatum is associated with nuclear factor‐kappaB activation. J Neurochem 2000;74:647–658. [DOI] [PubMed] [Google Scholar]
- 65. Koh PO. Melatonin attenuates the cerebral ischemic injury via the MEK/ERK/p90RSK/bad signaling cascade. J Vet Med Sci 2008;70:1219–1223. [DOI] [PubMed] [Google Scholar]
- 66. Joo JY, Uz T, Manev H. Opposite effects of pinealectomy and melatonin administration on brain damage following cerebral focal ischemia in rat. Restor Neurol Neurosci 1998;13:185–191. [PubMed] [Google Scholar]
- 67. Zhang J, Guo JD, Xing SH, Gu SL, Dai TJ. The protective effects of melatonin on global cerebral ischemia‐reperfusion injury in gerbils. Yao Xue Xue Bao 2002;37:329–333. [PubMed] [Google Scholar]
- 68. Letechipia‐Vallejo G, Gonzalez‐Burgos I, Cervantes M. Neuroprotective effect of melatonin on brain damage induced by acute global cerebral ischemia in cats. Arch Med Res 2001;32:186–192. [DOI] [PubMed] [Google Scholar]
- 69. Martin M, Macias M, Escames G, Leon J, Acuna‐Castroviejo D. Melatonin but not vitamins C and E maintains glutathione homeostasis in t‐butyl hydroperoxide‐induced mitochondrial oxidative stress. Faseb J 2000;14:1677–1679. [DOI] [PubMed] [Google Scholar]
- 70. Leon J, Acuna‐Castroviejo D, Escames G, Tan DX, Reiter RJ. Melatonin mitigates mitochondrial malfunction. J Pineal Res 2005;38:1–9. [DOI] [PubMed] [Google Scholar]
- 71. Andrabi SA, Sayeed I, Siemen D, Wolf G, Horn TF. Direct inhibition of the mitochondrial permeability transition pore: A possible mechanism responsible for anti‐apoptotic effects of melatonin. Faseb J 2004;18:869–871. [DOI] [PubMed] [Google Scholar]
- 72. Kilic E, Kilic U, Yulug B, Hermann DM, Reiter RJ. Melatonin reduces disseminate neuronal death after mild focal ischemia in mice via inhibition of caspase‐3 and is suitable as an add‐on treatment to tissue‐plasminogen activator. J Pineal Res 2004;36:171–176. [DOI] [PubMed] [Google Scholar]
- 73. Jacob S, Poeggeler B, Weishaupt JH, Siren AL, Hardeland R, Bahr M, Ehrenreich H. Melatonin as a candidate compound for neuroprotection in amyotrophic lateral sclerosis (ALS): High tolerability of daily oral melatonin administration in ALS patients. J Pineal Res 2002;33:186–187. [DOI] [PubMed] [Google Scholar]
- 74. Southgate G, Daya S. Melatonin reduces quinolinic acid‐induced lipid peroxidation in rat brain homogenate. Metab Brain Dis 1999;14:165–171. [DOI] [PubMed] [Google Scholar]
- 75. Tunez I, Montilla P, Del Carmen Munoz M, Feijoo M, Salcedo M. Protective effect of melatonin on 3‐nitropropionic acid‐induced oxidative stress in synaptosomes in an animal model of Huntington's disease. J Pineal Res 2004;37:252–256. [DOI] [PubMed] [Google Scholar]
- 76. Wang X, Zhu S, Pei Z, Drozda M, Stavrovskaya IG, Del Signore SJ, Cormier K, Shimony EM, Wang H, Ferrante RJ, et al Inhibitors of cytochrome c release with therapeutic potential for Huntington's disease. J Neurosci 2008;28:9473–9485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Kilic E, Kilic U, Reiter RJ, Bassetti CL, Hermann DM. Tissue‐plasminogen activator‐induced ischemic brain injury is reversed by melatonin: Role of iNOS and Akt. J Pineal Res 2005;39:151–155. [DOI] [PubMed] [Google Scholar]
- 78. Koh PO. Melatonin attenuates the focal cerebral ischemic injury by inhibiting the dissociation of pBad from 14‐3‐3. J Pineal Res 2008;44:101–106. [DOI] [PubMed] [Google Scholar]
- 79. Sun FY, Lin X, Mao LZ, Ge WH, Zhang LM, Huang YL, Gu J. Neuroprotection by melatonin against ischemic neuronal injury associated with modulation of DNA damage and repair in the rat following a transient cerebral ischemia. J Pineal Res 2002;33:48–56. [DOI] [PubMed] [Google Scholar]
- 80. Chung SY, Han SH. Melatonin attenuates kainic acid‐induced hippocampal neurodegeneration and oxidative stress through microglial inhibition. J Pineal Res 2003;34:95–102. [DOI] [PubMed] [Google Scholar]
- 81. Koh PO. Melatonin prevents the injury‐induced decline of Akt/forkhead transcription factors phosphorylation. J Pineal Res 2008;45:199–203. [DOI] [PubMed] [Google Scholar]
- 82. Ling X, Zhang LM, Lu SD, Li XJ, Sun FY. Protective effect of melatonin on injuried cerebral neurons is associated with bcl‐2 protein over‐expression. Zhongguo Yao Li Xue Bao 1999;20:409–414. [PubMed] [Google Scholar]
- 83. Rojas‐Fernandez CH, Chen M, Fernandez HL. Implications of amyloid precursor protein and subsequent beta‐amyloid production to the pharmacotherapy of Alzheimer's disease. Pharmacotherapy 2002;22:1547–1563. [DOI] [PubMed] [Google Scholar]
- 84. Iqbal K, Alonso Adel C, El‐Akkad E, Gong CX, Haque N, Khatoon S, Tsujio I, Grundke‐Iqbal I. Pharmacological targets to inhibit Alzheimer neurofibrillary degeneration. J Neural Transm Suppl 2002;62:309–319. [DOI] [PubMed] [Google Scholar]
- 85. Kostrzewa RM, Segura‐Aguilar J. Novel mechanisms and approaches in the study of neurodegeneration and neuroprotection. A review. Neurotox Res 2003;5:375–383. [DOI] [PubMed] [Google Scholar]
- 86. Liu RY, Zhou JN, Van Heerikhuize J, Hofman MA, Swaab DF. Decreased melatonin levels in postmortem cerebrospinal fluid in relation to aging, Alzheimer's disease, and apolipoprotein E‐epsilon4/4 genotype. J Clin Endocrinol Metab 1999;84:323–327. [DOI] [PubMed] [Google Scholar]
- 87. Ozcankaya R, Delibas N. Malondialdehyde, superoxide dismutase, melatonin, iron, copper, and zinc blood concentrations in patients with Alzheimer disease: Cross‐sectional study. Croat Med J 2002;43:28–32. [PubMed] [Google Scholar]
- 88. Mahlberg R, Walther S, Kalus P, Bohner G, Haedel S, Reischies FM, Kuhl KP, Hellweg R, Kunz D. Pineal calcification in Alzheimer's disease: An in vivo study using computed tomography. Neurobiol Aging 2008;29:203–209. [DOI] [PubMed] [Google Scholar]
- 89. Feng Z, Cheng Y, Zhang JT. Long‐term effects of melatonin or 17 beta‐estradiol on improving spatial memory performance in cognitively impaired, ovariectomized adult rats. J Pineal Res 2004;37:198–206. [DOI] [PubMed] [Google Scholar]
- 90. Pappolla MA, Sos M, Omar RA, Bick RJ, Hickson‐Bick DL, Reiter RJ, Efthimiopoulos S, Robakis NK. Melatonin prevents death of neuroblastoma cells exposed to the Alzheimer amyloid peptide. J Neurosci 1997;17:1683–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Feng Z, Chang Y, Cheng Y, Zhang BL, Qu ZW, Qin C, Zhang JT. Melatonin alleviates behavioral deficits associated with apoptosis and cholinergic system dysfunction in the APP 695 transgenic mouse model of Alzheimer's disease. J Pineal Res 2004;37:129–136. [DOI] [PubMed] [Google Scholar]
- 92. Feng Z, Qin C, Chang Y, Zhang JT. Early melatonin supplementation alleviates oxidative stress in a transgenic mouse model of Alzheimer's disease. Free Radic Biol Med 2006;40:101–109. [DOI] [PubMed] [Google Scholar]
- 93. Zhou J, Zhang S, Zhao X, Wei T. Melatonin impairs NADPH oxidase assembly and decreases superoxide anion production in microglia exposed to amyloid‐beta1‐42. J Pineal Res 2008;45:157–165. [DOI] [PubMed] [Google Scholar]
- 94. Deng YQ, Xu GG, Duan P, Zhang Q, Wang JZ. Effects of melatonin on wortmannin‐induced tau hyperphosphorylation. Acta Pharmacol Sin 2005;26:519–526. [DOI] [PubMed] [Google Scholar]
- 95. Wang JZ, Wang ZF. Role of melatonin in Alzheimer‐like neurodegeneration. Acta Pharmacol Sin 2006;27:41–49. [DOI] [PubMed] [Google Scholar]
- 96. Liu SJ, Wang JZ. Alzheimer‐like tau phosphorylation induced by wortmannin in vivo and its attenuation by melatonin. Acta Pharmacol Sin 2002;23:183–187. [PubMed] [Google Scholar]
- 97. Wang DL, Ling ZQ, Cao FY, Zhu LQ, Wang JZ. Melatonin attenuates isoproterenol‐induced protein kinase A overactivation and tau hyperphosphorylation in rat brain. J Pineal Res 2004;37:11–16. [DOI] [PubMed] [Google Scholar]
- 98. Wang XC, Zhang J, Yu X, Han L, Zhou ZT, Zhang Y, Wang JZ. Prevention of isoproterenol‐induced tau hyperphosphorylation by melatonin in the rat. Sheng Li Xue Bao 2005;57:7–12. [PubMed] [Google Scholar]
- 99. Li XC, Wang ZF, Zhang JX, Wang Q, Wang JZ. Effect of melatonin on calyculin A‐induced tau hyperphosphorylation. Eur J Pharmacol 2005;510:25–30. [DOI] [PubMed] [Google Scholar]
- 100. Pappolla MA, Chyan YJ, Poeggeler B, Frangione B, Wilson G, Ghiso J, Reiter RJ. An assessment of the antioxidant and the antiamyloidogenic properties of melatonin. implications for Alzheimer's disease. J Neural Transm 2000;107:203–231. [DOI] [PubMed] [Google Scholar]
- 101. Pappolla MA, Simovich MJ, Bryant‐Thomas T, Chyan YJ, Poeggeler B, Dubocovich M, Bick R, Perry G, Cruz‐Sanchez F, Smith MA. The neuroprotective activities of melatonin against the Alzheimer beta‐protein are not mediated by melatonin membrane receptors. J Pineal Res 2002;32:135–142. [DOI] [PubMed] [Google Scholar]
- 102. Feng Z, Zhang JT. Protective effect of melatonin on beta‐amyloid‐induced apoptosis in rat astroglioma C6 cells and its mechanism. Free Radic Biol Med 2004;37:1790–1801. [DOI] [PubMed] [Google Scholar]
- 103. Brusco LI, Marquez M, Cardinali DP. Monozygotic twins with Alzheimer's disease treated with melatonin: Case report. J Pineal Res 1998;25:260–263. [DOI] [PubMed] [Google Scholar]
- 104. Asayama K, Yamadera H, Ito T, Suzuki H, Kudo Y, Endo S. Double blind study of melatonin effects on the sleep‐wake rhythm, cognitive and non‐cognitive functions in Alzheimer type dementia. J Nippon Med Sch 2003;70:334–341. [DOI] [PubMed] [Google Scholar]
- 105. Cardinali DP, Brusco LI, Liberczuk C, Furio AM. The use of melatonin in Alzheimer's disease. Neuro Endocrinol Lett 2002;23(Suppl 1):20–23. [PubMed] [Google Scholar]
- 106. Olde Rikkert MG, Rigaud AS. Melatonin in elderly patients with insomnia. A systematic review. Z Gerontol Geriatr 2001;34:491–497. [DOI] [PubMed] [Google Scholar]
- 107. Brusco LI, Fainstein I, Marquez M, Cardinali DP. Effect of melatonin in selected populations of sleep‐disturbed patients. Biol Signals Recept 1999;8:126–131. [DOI] [PubMed] [Google Scholar]
- 108. Mishima K, Okawa M, Hishikawa Y, Hozumi S, Hori H, Takahashi K. Morning bright light therapy for sleep and behavior disorders in elderly patients with dementia. Acta Psychiatr Scand 1994;89:1–7. [DOI] [PubMed] [Google Scholar]
- 109. Kumar AM, Tims F, Cruess DG, Mintzer MJ, Ironson G, Loewenstein D, Cattan R, Fernandez JB, Eisdorfer C, Kumar M. Music therapy increases serum melatonin levels in patients with Alzheimer's disease. Altern Ther Health Med 1999;5:49–57. [PubMed] [Google Scholar]
- 110. Gehrman PR, Connor DJ, Martin JL, Shochat T, Corey‐Bloom J, Ancoli‐Israel S. Melatonin fails to improve sleep or agitation in double‐blind randomized placebo‐controlled trial of institutionalized patients with Alzheimer disease. Am J Geriatr Psychiatry 2009;17:166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Singer C, Tractenberg RE, Kaye J, Schafer K, Gamst A, Grundman M, Thomas R, Thal LJ. A multicenter, placebo‐controlled trial of melatonin for sleep disturbance in Alzheimer's disease. Sleep 2003;26:893–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Furio AM, Brusco LI, Cardinali DP. Possible therapeutic value of melatonin in mild cognitive impairment: A retrospective study. J Pineal Res 2007;43:404–409. [DOI] [PubMed] [Google Scholar]
- 113. Jean‐Louis G, Von Gizycki H, Zizi F. Melatonin effects on sleep, mood, and cognition in elderly with mild cognitive impairment. J Pineal Res 1998;25:177–183. [DOI] [PubMed] [Google Scholar]
- 114. Jansen SL, Forbes DA, Duncan V, Morgan DG. Melatonin for cognitive impairment. Cochrane Database Syst Rev 2006:CD003802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. De Rijk MC, Launer LJ, Berger K, Breteler MM, Dartigues JF, Baldereschi M, Fratiglioni L, Lobo A, Martinez‐Lage J, Trenkwalder C, et al Prevalence of Parkinson's disease in Europe: A collaborative study of population‐based cohorts. Neurologic Diseases in the Elderly Research Group. Neurology 2000;54:S21–S23. [PubMed] [Google Scholar]
- 116. Alvira D, Tajes M, Verdaguer E, Acuna‐Castroviejo D, Folch J, Camins A, Pallas M. Inhibition of the cdk5/p25 fragment formation may explain the antiapoptotic effects of melatonin in an experimental model of Parkinson's disease. J Pineal Res 2006;40:251–258. [DOI] [PubMed] [Google Scholar]
- 117. Jou MJ, Peng TI, Reiter RJ, Jou SB, Wu HY, Wen ST. Visualization of the antioxidative effects of melatonin at the mitochondrial level during oxidative stress‐induced apoptosis of rat brain astrocytes. J Pineal Res 2004;37:55–70. [DOI] [PubMed] [Google Scholar]
- 118. Ebadi M, Sharma SK, Ghafourifar P, Brown‐Borg H, El Refaey H. Peroxynitrite in the pathogenesis of Parkinson's disease and the neuroprotective role of metallothioneins. Methods Enzymol 2005;396:276–298. [DOI] [PubMed] [Google Scholar]
- 119. Iacovitti L, Stull ND, Johnston K. Melatonin rescues dopamine neurons from cell death in tissue culture models of oxidative stress. Brain Res 1997;768:317–326. [DOI] [PubMed] [Google Scholar]
- 120. Iacovitti L, Stull ND, Mishizen A. Neurotransmitters, KCl and antioxidants rescue striatal neurons from apoptotic cell death in culture. Brain Res 1999;816:276–285. [DOI] [PubMed] [Google Scholar]
- 121. Ortiz GG, Crespo‐Lopez ME, Moran‐Moguel C, Garcia JJ, Reiter RJ, Acuna‐Castroviejo D. Protective role of melatonin against MPTP‐induced mouse brain cell DNA fragmentation and apoptosis in vivo. Neuro Endocrinol Lett 2001;22:101–108. [PubMed] [Google Scholar]
- 122. Mayo JC, Sainz RM, Uria H, Antolin I, Esteban MM, Rodriguez C. Melatonin prevents apoptosis induced by 6‐hydroxydopamine in neuronal cells: Implications for Parkinson's disease. J Pineal Res 1998;24:179–192. [DOI] [PubMed] [Google Scholar]
- 123. Medeiros CA, Carvalhedo de Bruin PF, Lopes LA, Magalhaes MC, De Lourdes Seabra M, De Bruin VM. Effect of exogenous melatonin on sleep and motor dysfunction in Parkinson's disease. A randomized, double blind, placebo‐controlled study. J Neurol 2007;254:459–464. [DOI] [PubMed] [Google Scholar]
- 124. Dowling GA, Mastick J, Colling E, Carter JH, Singer CM, Aminoff MJ. Melatonin for sleep disturbances in Parkinson's disease. Sleep Med 2005;6:459–466. [DOI] [PubMed] [Google Scholar]
- 125. Kandel ER, Schwartz JH, Jessell TM. In: Principles of neural science, 4th edn New York : McGraw‐Hill, 2000. [Google Scholar]
- 126. Group. HsDCR . A novel gene containing a trinucleotide repeat that is expanded on Huntington's disease chromosomes. Cell 1993;72:971–983. [DOI] [PubMed] [Google Scholar]
- 127. Browne SE, Beal MF. Oxidative damage in Huntington's disease pathogenesis. Antioxid Redox Signal 2006;8:2061–2073. [DOI] [PubMed] [Google Scholar]
- 128. Schulz JB, Beal MF. Mitochondrial dysfunction in movement disorders. Curr Opin Neurol 1994;7:333–339. [DOI] [PubMed] [Google Scholar]
- 129. Rigamonti D, Sipione S, Goffredo D, Zuccato C, Fossale E, Cattaneo E. Huntingtin's neuroprotective activity occurs via inhibition of procaspase‐9 processing. J Biol Chem 2001;276:14545–14548. [DOI] [PubMed] [Google Scholar]
- 130. Rigamonti D, Bauer JH, De‐Fraja C, Conti L, Sipione S, Sciorati C, Clementi E, Hackam A, Hayden MR, Li Y, et al Wild‐type huntingtin protects from apoptosis upstream of caspase‐3. J Neurosci 2000;20:3705–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Christofides J, Bridel M, Egerton M, Mackay GM, Forrest CM, Stoy N, Darlington LG, Stone TW. Blood 5‐hydroxytryptamine, 5‐hydroxyindoleacetic acid and melatonin levels in patients with either Huntington's disease or chronic brain injury. J Neurochem 2006;97:1078–1088. [DOI] [PubMed] [Google Scholar]
- 132. Aziz NA, Pijl H, Frolich M, Schroder‐van der Elst JP, Van Der Bent C, Roelfsema F, Roos RA. Delayed onset of the diurnal melatonin rise in patients with Huntington's disease. J Neurol 2009: DOI DOI: 10.1007/s00415-009-5196-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Rival T, Soustelle L, Strambi C, Besson MT, Iche M, Birman S. Decreasing glutamate buffering capacity triggers oxidative stress and neuropil degeneration in the Drosophila brain. Curr Biol 2004;14:599–605. [DOI] [PubMed] [Google Scholar]
- 134. Rogerio F, Teixeira SA, De Rezende AC, De Sa RC, De Souza Queiroz L, De Nucci G, Muscara MN, Langone F. Superoxide dismutase isoforms 1 and 2 in lumbar spinal cord of neonatal rats after sciatic nerve transection and melatonin treatment. Brain Res Dev Brain Res 2005;154:217–225. [DOI] [PubMed] [Google Scholar]
- 135. Przedborski S. Programmed cell death in amyotrophic lateral sclerosis: A mechanism of pathogenic and therapeutic importance. Neurologist 2004;10:1–7. [DOI] [PubMed] [Google Scholar]
- 136. Sathasivam S, Grierson AJ, Shaw PJ. Characterization of the caspase cascade in a cell culture model of SOD1‐related familial amyotrophic lateral sclerosis: Expression, activation and therapeutic effects of inhibition. Neuropathol Appl Neurobiol 2005;31:467–485. [DOI] [PubMed] [Google Scholar]
- 137. Ryu H, Smith K, Camelo SI, Carreras I, Lee J, Iglesias AH, Dangond F, Cormier KA, Cudkowicz ME, Brown RH Jr., et al Sodium phenylbutyrate prolongs survival and regulates expression of anti‐apoptotic genes in transgenic amyotrophic lateral sclerosis mice. J Neurochem 2005;93:1087–1098. [DOI] [PubMed] [Google Scholar]
- 138. Blazejova K, Nevsimalova S, Illnerova H, Hajek I, Sonka K. Sleep disorders and the 24‐hour profile of melatonin and cortisol. Sb Lek 2000;101:347–351. [PubMed] [Google Scholar]