

SUMMARY
Sustained angiogenesis, through either local sprouting (angiogenesis) or the recruitment of bone marrow-derived cells (BMDCs) (vasculogenesis), is essential to the development of a tumor. How BMDCs are recruited to the tumor and their contribution to the tumor vasculature is poorly understood. Here we demonstrate that both IL-8 and Angiogenin contribute to the complementary pathways of angiogenesis and BMDC mobilization to increase tumor growth. These two factors are regulated by PHD2 in a HIF-independent but NF-κB-dependent manner. PHD2 levels are decreased in human cancers compared to corresponding normal tissue and correlate with an increase in mature blood vessels. Thus, PHD2 plays a critical role in regulating tumor angiogenesis.
Keywords: Angiogenesis, Vasculogenesis, Tumor Vasculature, Bone Marrow-Derived Cells, Prolyl Hydroxylases, Hypoxia-inducible Factor, NF-κB
INTRODUCTION
Tumor growth is highly dependent on its vascular supply. However, tumor growth often exceeds the ability of the existing vasculature to supply it with nutrients and oxygen (Hanahan and Weinberg, 2000). Continued tumor growth therefore requires new blood vessels. This induction of angiogenesis by solid tumors encompasses two complementary processes: sprouting angiogenesis and neovascularization (Carmeliet, 2000). Sprouting angiogenesis is a local response, involving existing blood vessels. Neovascularization, or vasculogenesis, is the de novo formation of new blood vessels through the mobilization and recruitment of bone marrow-derived cells.
This angiogenic switch is mediated in part by the hypoxia-inducible factor (HIF) family of transcription factors, key regulators of oxygen homeostasis, composed of an oxygen-labile α subunit and a constitutive beta subunit. HIF positively contributes to the growth of solid tumors (Kondo et al., 2002; Ryan et al., 1998; Ryan et al., 2000) and its overexpression in a wide variety of tumor types correlates with poor prognosis, resistance to radiotherapy and chemotherapy, and increased patient mortality (Harris, 2002; Semenza, 2002; Unruh et al., 2003; Zhong et al., 1999). Given the importance of HIF in multiple facets of tumor progression, mutations that enhance HIF activity might be expected to be involved in a variety of human cancers. So far, no genetic mutations in any of the HIF subunits affecting the stability or activity of these proteins have been identified in human tumors. However, elevated HIF levels in cancers can result from mutations of key regulators of HIF stability. Mutation of the von Hippel-Lindau gene (VHL), an upstream negative regulator of HIF, accounts for a subset of tumors with elevated HIF (Ohh et al., 2000). VHL is a tumor suppressor gene and VHL disease is characterized by a distinct subset of highly vascular tumor (Kaelin, 2007).
PHD2 is one of three prolyl hydroxylases originally identified as negative regulators of HIF. Proline hydroxylation serves to regulate the stability of the α subunits of HIF, a critical step in the cellular response to low oxygen, but additional roles for these genes remain elusive (Ivan et al., 2001; Jaakkola et al., 2001; Yu et al., 2001). PHD2 (EGLN1, HPH-2) has been proposed to be the key oxygen sensor as transiently silencing PHD2 results in normoxic stabilization of HIF-1α (Berra et al., 2003). In contrast, silencing PHD1 or PHD3 has little effect on HIF levels. Furthermore, PHD2 is an essential gene as Phd2 knockout mice die between embryonic day 12.5 and 14.5, while Phd1 and Phd3 knockout mice appear normal (Takeda et al., 2006). Conditional inactivation of Phd2 increased serum erythropoietin levels and consequently, red blood cell production, leading to polycythemia and congestive heart failure (Minamishima et al., 2008). Specific knockout of PHD2 in endothelial cells affects cell proliferation, while heterozygosity of PHD2 in normal mouse stroma plays a role in vessel maturation (Mazzone et al., 2009; Takeda and Fong, 2007). Overall, these results suggest a key physiological role for PHD2. We hypothesized that deregulation of PHD2 might contribute to a number of other tumor types that have high levels of HIF. In this study, we examined the role of PHD2 in regulating tumorigenesis.
RESULTS
PHD2 Levels Decrease in Human Cancers
The important role of PHD2 in regulating HIF led us to investigate how normoxic PHD2, HIF-1, and HIF-2 protein levels relate to one another. The abundance of PHD proteins directly correlates with the amount of PHD activity (Appelhoff et al., 2004). Western blot analysis of the NCI60 panel, a set of 60 cell lines derived from nine distinct tissues, revealed that PHD2 levels are highly variable and do not necessarily inversely correlate with HIF levels as expected (Figure 1A). Nor did HIF levels always correlate with PHD1 or PHD3 levels. Tumors of the same tissue of origin also display disparate levels of PHD2. For example, the MCF7 breast cancer cell line has very little PHD2 compared to the BT-549 line. In some cases, a reduced level of PHD2 correlates with an increased in HIF-1α levels (e.g. HS 578T) or HIF-2α (e.g. SK-MEL-5). HIF levels are also sometimes elevated despite the presence of PHD2, as seen in 786-O cells, in which the loss of VHL results in HIF stabilization.
Figure 1.

PHD2 Levels Decrease in Human Cancers
(A) Variation of PHD2 levels in the NCI-60 panel of cell lines immunoblotted for PHD1, PHD2, PHD3, HIF-1α, HIF-2α, and α-tubulin.
(B) Immunohistochemical staining for PHD2 in colon adenocarcinoma (bottom panels) compared to corresponding normal adjacent colon tissue (top panels) from several different patients. Scale bar = 500 μm.
(C) Oncomine database analysis to examine PHD2 mRNA expression levels in adjacent, non-tumoral colon versus colon carcinoma (p<0.0005).
(D) HCT116 cells with shRNA designed to specifically target PHD2. Cells were treated with DFO (100 μM; 4 hours) or proteasome inhibitor (MG132: 10 μM; 4 hours) and harvested to examine protein levels of PHD2, HIF-1α, and α-tubulin.
The reduction of PHD2 in a number of cancer cell lines suggests the possibility that PHD2 loss contributes to tumorigenesis and represents a pathway for HIF activation in a subset of tumors. Our analysis of published human cancer gene expression datasets revealed that PHD2 expression is significantly decreased in tumors compared to normal tissue (Figure S2A) (Ramaswamy et al., 2001). In particular, PHD2 mRNA is specifically lower in colon carcinoma compared to normal colon tissue (Figure 1C) (Graudens et al., 2006). To further investigate the role of PHD2 in human tumors, we stained colorectal tumors and matched, non-involved adjacent colon tissue (Figure 1B). Four out of the ten patient samples had decreased PHD2 in colon tumors relative to normal tissue. Together these data suggest that disruption of PHD2 could contribute substantially to tumorigenesis in human cancers.
Loss of PHD2 Increases Tumor Growth
To directly compare the growth of tumors with and without PHD2, we designed short hairpin RNA constructs complementary to different regions of PHD2 to target PHD2 mRNA for degradation. Five constructs targeting five unique sequences of PHD2 were stably expressed in HCT116 cells, a colon carcinoma cell line that had moderate levels of PHD2 and an intact HIF response (Figures 1A and 1D). Constructs A and C, which were used in subsequent experiments, completely abrogated PHD2 protein and confirmed previous findings that PHD2 silencing is sufficient to stabilize HIF-1α protein in normoxic conditions (Berra et al., 2003). Knockdown of PHD2 was nearly as effectively as inhibiting the prolyl hydroxylases with desferrioxamine (DFO) but not as strong as the proteasome inhibitor MG132 (Figure 1D). Construct E, which did not alter PHD2 or HIF-1α protein levels, served as an shRNA control. In HIF reporter assays, PHD2 knockdown resulted in a seven-fold increase in HIF activity under normoxic conditions, which was further augmented by hypoxia, likely due to inhibition of PHD1, PHD3, or Factor Inhibiting HIF (FIH) (Figure S2B) (Lando et al., 2002a; Lando et al., 2002b; Mahon et al., 2001). Western blots demonstrated that the shRNA knockdown of PHD2 is specific and has no effect on the other HIF hydroxylases (Figure S2C). These data demonstrate an effective and specific disruption of PHD2 and subsequent stabilization of HIF-1α.
We next assessed the consequences of PHD2 disruption on tumor growth. Implanting PHD2 knockdown cells (construct A) into the flanks of SCID mice revealed that PHD2 loss confers a significant growth advantage over control tumors (Figure 2A). The effect on in vivo tumor growth is not due to intrinsic differences in proliferation rates as PHD2 knockdown and control cells had identical in vitro growth rates under normoxic and hypoxic conditions (Figure 2B). To further confirm the specificity of the PHD2 shRNA, cells expressing a control shRNA that does not affect PHD2 expression (construct E) or a second shRNA targeting a different region of PHD2 (construct C) were also implanted into SCID mice (Figure 2C). Tumors expressing the ineffective shRNA construct E exhibit wild-type tumor growth rates, whereas tumors expressing the effective construct C have a significant growth advantage. The efficiency of PHD2 knockdown is therefore directly correlated to the rate of tumor growth, demonstrating that the effects are specific to PHD2 silencing. The effect of shRNA on tumor growth is not unique to HCT116 cells as stable knockdown of PHD2 in a pancreatic carcinoma cell line (SU.86.86) and in two additional colorectal carcinoma cell lines (HT29 and RKO) also amplified tumor growth (Figures 2D, 2E, 2F, S2D, and S4A). Thus, loss of PHD2 increases tumor growth compared to control cells with wild-type PHD2, indicating a role of PHD2 in tumorigenesis.
Figure 2.
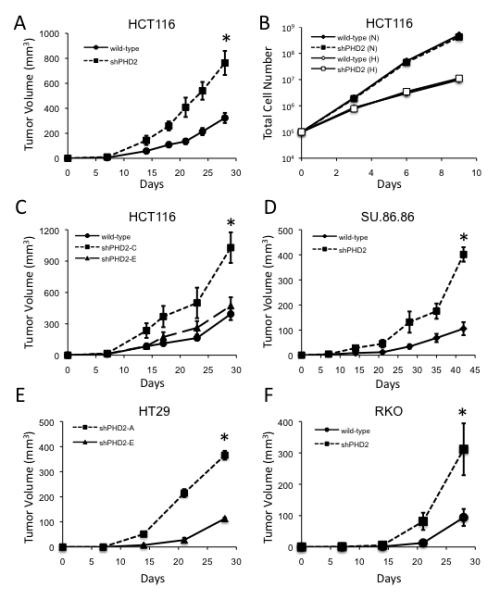
Loss of PHD2 Increases Tumor Growth
(A) In vivo growth of wild-type or PHD2 knockdown (Construct A) HCT116 xenograft tumors (*p<0.00001).
(B) In vitro growth curve of HCT116 cells with or without stable knockdown to PHD2 under either normoxic (N, 21% O2) or hypoxic (H, 2% O2) conditions.
(C) Growth of wild-type or PHD2 knockdown (construct C or E) HCT116 tumors in mice (*p<0.001).
(D) Growth of wild-type SU.86.86 or cells expressing PHD2 shRNA in SCID mice (*p<0.000005).
(E) In vivo growth of HT29 cells expressing shRNA to PHD2 (Construct A) or control (construct E)(*p<0.000005).
(F) Growth of wild-type RKO cells or cells expressing shRNA to PHD2 in SCID mice (*p<0.05). All error bars represent ±SEM.
HIF-Independent Tumor Suppressor Function of PHD2
As HIF-1α is the only functionally characterized target of PHD2 to date, we next determined whether the effects of PHD2 on tumorigenesis depended on HIF by employing HIF-deficient cells. Stable expression of an shRNA to HIF-1α in HCT116 cells prevented HIF-1α protein accumulation in response to either hypoxia or DFO (Figure S3A). Simultaneous knockdown of PHD2 and HIF-1α enabled us to examine HIF-dependent and HIF-independent effects by PHD2 disruption. Silencing HIF-1α in HCT116 cells is sufficient to eliminate all HIF activity in HIF-reporter assays (Figure S3B). Since the HIF-reporter used does not distinguish between HIF-1 and HIF-2, these data indicate that HIF-2α is nonfunctional in these cells. Western blot analysis further demonstrated a lack of HIF-2α protein induction under hypoxia in these cells (Figure S3C). In vitro growth under normal oxygenated conditions or hypoxia was unaffected by the different shRNA constructs (Figure S3D). These data demonstrate that HIF knockdown is effective in these cell lines, enabling us to examine whether the effects of PHD2 disruption on tumor growth are reliant on HIF.
In contrast to the expectation that PHD2 would affect tumor growth through its regulation of HIF, we found that the improved tumor growth did not depend on the presence of HIF. Cells with both PHD2 and HIF-1α disrupted exhibited increased tumor growth compared to cells that only had HIF-1α silenced (Figure 3A). These results indicate that PHD2 has HIF-independent functions that contribute to tumor growth. Western blot analysis demonstrated that PHD2 and HIF-1α knockdown was maintained in the corresponding tumors (Figure 3B). Moreover, PHD2 levels in vivo were uniformly high in control tumors but dramatically reduced in the tumors expressing shRNA to PHD2 as determined by immunostaining tumor sections (Figure S3E). Staining of PHD2-deficient tumor sections revealed high levels of CAIX, a specific HIF target, indicating that shRNA to PHD2 was sufficient to result in functional HIF in vivo, whereas wild-type tumors displayed a classic hypoxic staining pattern of CAIX (Figure 3C). Expression of HIF-1α shRNA in PHD2 knockdown tumors completely abrogated CAIX staining, demonstrating that HIF-1α silencing was maintained in vivo. It has been suggested that PHD2 itself is a HIF target (D’Angelo et al., 2003). However, PHD2 staining in wild-type tumors was homogeneous and did not exhibit a hypoxic pattern, revealing that PHD2 protein is unaffected by hypoxia in this cell type. Thus, while PHD2 knockdown results in upregulation of HIF activity, this activity is not necessary for the effects of PHD2 on tumorigenesis.
Figure 3.
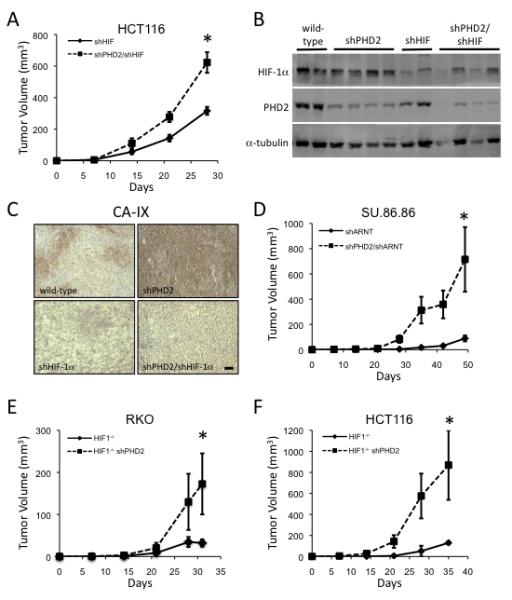
HIF-Independent Tumor Suppressor Function of PHD2
(A) Tumor growth of HCT116 cells expressing shRNA to HIF-1α alone or in combination with shRNA to PHD2 (*p<0.05).
(B) PHD2 and HIF-1α silencing is maintained in vivo. Protein extracts from tumors in Figure 3A were immunoblotted to examine levels of HIF-1α, PHD2, and α-tubulin.
(C) Tumor sections of the given genotype were stained for CA-IX expression. Scale bar = 200 μm.
(D) Tumor growth of SU.86.86 cells with shRNA to PHD2 or shRNA to PHD2 and HIF-1β (*p<0.05).
(E) In vivo growth of RKO cells deficient in HIF-1α or HIF-1α-deficient cells expressing PHD2 shRNA (*p<0.05).
(F) In vivo growth of HIF-1α-deficient HCT116 cells or those expressing PHD2 shRNA (p<0.05). All error bars represent ±SEM.
To further evaluate whether HIF is necessary for the enhanced tumor growth seen in PHD2-silenced cells, we next examined the dependence on HIF in three additional models, SU.86.86 cells, and RKO and HCT116 cells in which the HIF-1α gene has been deleted by homologous recombination (Figure S4A) (Dang et al., 2006). As SU.86.86 cells possess both HIF-1α and HIF-2α, we inhibited both HIF family members through expression of an shRNA to ARNT (HIF-1β), the constitutive binding partner of HIF-α (Figure S4B). Simultaneous knockdown of ARNT and PHD2 increases tumor growth over tumors expressing ARNT shRNA alone (Figure 3D). Silencing PHD2 in both HIF-1α-null HCT116 and RKO cell lines also increases tumor growth (Figures 3E, 3F, S2D, and S4A). Taken together, these data indicate that PHD2 functions as a tumor suppressor and that this activity does not require HIF in multiple settings.
PHD2 Regulates Angiogenesis and Recruitment of Bone Marrow-Derived Cells
In order to determine how PHD2 loss increases tumor growth, we performed immunohistochemical analyses on tumor sections. Examining sections from HCT116 and SU.86.86 tumors for the endothelial cell marker CD31 staining revealed that tumors expressing PHD2 shRNA have a significant increase in blood vessels (Figures 4A and S4C). Furthermore, in cells expressing shRNA to HIF-1α or ARNT, PHD2 silencing also increased blood vessels, indicating that PHD2 can affect angiogenesis, even in the absence of HIF-1α (Figures 4B and S4D). The increase in tumor volume could be attributed to an increase in proliferation of PHD2 knockdown tumors as determined by Ki-67 staining (Figures S5A and S5B). TUNEL staining of these tumors, however, is unaffected by PHD2 loss (Figure S5C). Together these data indicate that PHD2 normally functions to restrict the tumor vasculature, suggesting a mechanism by which the loss of PHD2 enhances tumor growth.
Figure 4.

PHD2 Regulates Angiogenesis and Recruitment of Bone Marrow-Derived Cells
(A) CD31 staining in sections from HCT116 tumors expressing PHD2 shRNA compared to wild-type tumors (*p<0.01). Scale bar = 500 μm.
(B) CD31 staining in sections from HCT116 tumors expressing shRNA to both PHD2 and HIF-1α compared to those expressing HIF-1α shRNA alone (*p<0.0005). Scale bar = 500 μm.
(C) In vitro tube formation of endothelial cells in conditioned media from wild-type HCT116 cells or cells expressing PHD2 (*p<0.000005). Scale bar = 1 mm.
(D) In vitro tube formation of endothelial cells in conditioned media from HIF-1α-deficient HCT116 or HIF-1α-deficient HCT116 cells expressing PHD2 shRNA (*p<0.00005). Scale bar = 1 mm.
(E) Staining of HCT116 tumors for bone marrow-derived myelomonocytic cells with CD11b (red) and nuclei with DAPI (blue) (*p<0.005). Scale bar = 100 μm.
(F) HCT116 tumor staining for CD45 (green) and nuclei with DAPI (blue) (*p<0.005). Scale bar = 100 μm. All error bars represent ±SEM.
There are two main contributors to the tumor vasculature, angiogenesis and vasculogenesis. To further delineate the effect of PHD2 loss on the tumor vasculature, we employed a short-term in vitro assay to measure angiogenesis. This assay assesses the propensity of primary endothelial cells to sprout, forming three-dimensional structures resembling capillaries, in response to appropriate cues. Conditioned media from HCT116 cells with silenced PHD2 under normoxic conditions significantly increases the number of cells in capillary branch points, a surrogate marker for elevated angiogenesis (Figure 4C). Similarly, conditioned medium from cells expressing PHD2 knockdown construct C also increases tube formation, whereas conditioned medium from cells expressing control construct E is unable to increase in vitro angiogenesis (Figures S5D and S5E). Conditioned medium from HIF-1α-deficient HCT116 cells with silenced PHD2 increases tube formation compared to that from cells with HIF-1α-deleted alone (Figure 4D). Silencing ARNT by siRNA in HT29 cells with PHD2 shRNA also demonstrated a HIF-independent effect of PHD2 on angiogenesis (Figures S5F and S5G). These results indicate that PHD2, regardless of the presence or absence of HIF, influences angiogenesis in multiple cancer cell types.
Vasculogenesis requires the de novo production of endothelial cells and is dependent on bone marrow-derived cells (BMDCs), in part to provide the appropriate cues to recruit progenitor cells and other vasculatory modulatory cells (De Palma et al., 2003; Shojaei et al., 2007; Yang et al., 2004). Thus, to determine whether recruitment of BMDCs also contributes to the augmented tumor growth, we stained tumors for the myeloid-monocyte lineage marker CD11b and the leukocyte lineage marker CD45. HCT116 tumors with reduced PHD2 display a 6-7 fold increase in infiltrating BMDCs, regardless of the presence or absence of HIF, compared to control tumors (Figures 4E and 4F). Both CD11b and CD45 are increased in tumors lacking PHD2 compared to control tumors, suggesting that PHD2 regulates the recruitment of BMDCs, which contribute to vasculogenesis.
Hydroxylase function of PHD2 is not necessary for regulating angiogenesis
We next investigated the role of hypoxia in the regulation of angiogenesis by PHD2. As expected, conditioned medium from hypoxia-treated HCT116 cells induces tube formation, demonstrating that hypoxia can increase angiogenesis through HIF-dependent mechanisms (Figure 5A). Hypoxic treatment of cells expressing PHD2 shRNA does not further increase tube formation (Figure 5A). Interestingly, although knockdown of PHD2 enhances tube formation induced by conditioned medium from HIF-1α-deficient HCT116 or RKO cells, this effect is not mimicked by hypoxia, a treatment that reduces PHD2 hydroxylase activity (Figures 5B and 5D). This result raises the possibility that unlike PHD2 regulation of HIF, the HIF-independent control of angiogenesis does not depend on the hydoxylase activity PHD2.
Figure 5.
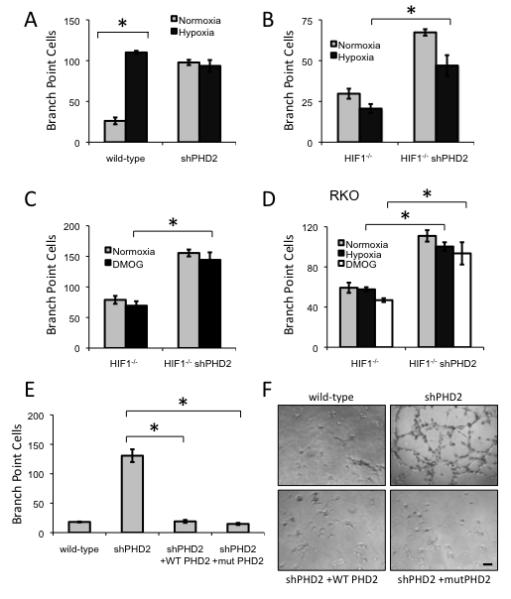
Hydroxylase function of PHD2 is not necessary for regulating angiogenesis
(A) The ability of conditioned media from wild-type or PHD2 shRNA expressing HCT116 cells with or without hypoxia condition (2% O2, 16 hours) in inducing in vitro tube formation (*p<0.001).
(B) The ability of conditioned media from HIF1-/- HCT116 cells or HIF1-/- HCT116 cells expressing PHD2 shRNA under normoxia or hypoxia (2% O2, 16 hours) in inducing in vitro tube formation (*p<0.01).
(C) Conditioned media from HCT116 HIF1-/- cells or cells expressing shRNA to PHD2 treated with DMOG (1 mM, 16 hours) was subjected to in vitro tube formation (*p<0.001).
(D) In vitro tube formation of conditioned media from RKO HIF1-/- cells or cells expressing shRNA to PHD2 treated with hypoxia (2% O2) or DMOG (1 mM) for 16 hours (*p<0.01).
(E) HCT116 cells expressing PHD2 shRNA were transfected with either wild-type PHD2 or hydroxylase mutant PHD2 (H313A H315D). Conditioned media from these cells were collected and subjected to a tube formation assay (*p<0.001). All error bars represent ±SEM.
(F) Representative photographs of primary endothelial cells seeded onto Matrigel in conditioned media from HCT116 cells stably expressing shRNA to PHD2 and transfected with either wild-type PHD2 or a hydroxylase mutant PHD2. Scale bar = 2 mm.
To assess this possibility pharmacologically, HIF-1α-deficient cells were treated with dimethyloxaloylglycine (DMOG), a pan-hydroxylase inhibitor. Conditioned media from DMOG-treated HCT116 or RKO cells fails to increase angiogenesis (Figures 5C and 5D). Furthermore, DMOG-treated HIF-1α-deficient cells with shRNA targeting PHD2 do not exhibit a further increase in tube formation (Figures 5C and 5D). These data also suggest that the hydroxylase activity is not mediating this function of PHD2.
Based on these findings, we next examined whether PHD2 lacking hydroxylase activity was able to rescue the tube formation capability of PHD2-silenced cells. We generated shRNA-resistant PHD2 constructs by introducing silent mutations in the region targeted by the shRNA. Transfection of this shRNA-resistant construct allowed overexpression of wild-type PHD2, which rescued the wild-type levels of angiogenesis, further confirming specificity that the shRNA is targeting PHD2. Importantly, an shRNA-resistant PHD2 construct with mutations at two critical residues, HD313 and D315, in the hydroxylase catalytic site (McDonough et al., 2006) also rescued wild-type levels of angiogenesis (Figures 5E and 5F). These data, along with the pharmacological and hypoxia findings, indicate that the hydroxylase function of PHD2 is not necessary for its angiogenic suppressor activity.
NF-κB regulates PHD2-mediated angiogenesis
Having identified a role of PHD2 in regulating tumor vasculature independent of its role in regulating HIF, we investigated whether another transcription factor also regulated by PHD2. Cummins et al. demonstrated that transiently silencing PHD1 or PHD2 increased NF-κB activity (Cummins et al., 2006). In agreement, we also find that shRNA to PHD2 is sufficient to increase NF-κB reporter expression (Figure 6A). Prolyl hydroxylase inhibition or hypoxia treatment of cells with an intact HIF pathway is able to increase NF-κB (Cummins et al., 2006) (Figure 6B). To more directly examine the requirement for the hydroxylase function of PHD2 in regulating NF-κB in a HIF-independent manner, we restored PHD2 levels in PHD2-silenced cells by transfecting either wild-type or hydroxylase mutant PHD2 into HIF-1α-deficient HCT116 cells. Both wild-type and mutant PHD2 decreased NF-κB activity and limited the induction of NF-κB (Figure 6C). Stimulation of NF-κB by TNF-α is similarly impaired when wild-type or hydroxylase mutant PHD2 is expressed (Figure 6C). Subjecting these HIF-deficient cells to hypoxia or pan-hydroxylase inhibition does not increase NF-κB activity, further confirming that in addition to the previously described hypoxia-mediated regulation of NF-κB, PHD2 can also regulate NF-κB in a hypoxia-, hydroxylase-, and HIF-independent manner.
Figure 6.

NF-κB regulates PHD2-mediated angiogenesis
(A) NF-κB reporter assay of wild-type or HCT116 cells expressing PHD2 shRNA (*p<0.05).
(B) NF-κB is hypoxia-responsive. HCT116 cells were transfected with a NF-κB luciferase reporter and treated with hypoxia (2% O2, 16 hours)(*p<0.005).
(C) Wild-type or hydroxylase mutant PHD2 were overexpressed along with a NF-κB luciferase reporter construct in HCT116 HIF-1-/- cells stably expressing shRNA to PHD2. Following transfection, these cells were not treated or treated with TNF-α (20 ng/ml; 6 hours), hypoxia (2% O2; 16 hours), or DMOG (1 mM; 16 hours) (*p<0.05).
(D) Quantitative real-time PCR to demonstrate knockdown of RELA in HCT116 cells with HIF-1α knocked out.
(E) Conditioned media from HCT116 HIF-1-/- cells expressing PHD2 shRNA transfected with pool of non-targeting siRNA or siRNA targeting RELA were subjected to a tube formation assay (*p<0.0005).
(F) Primary endothelial cells were seeded onto Matrigel in conditioned media from HIF-1α knockout cells or those expressing shRNA to PHD2 and assayed for tube formation. Scale bar = 1 mm.
(G) Angiogenesis antibody arrays were carried out in duplicate with conditioned media from either wild-type HCT116 or cells stably expressing PHD2 shRNA.
(H) Reporter assays with ANG or IL-8 promoters along with the promoters with mutant NF-κB sites. Two days after transfection, cells were treated with TNF-α (20 ng/ml; 6-8 hours) and luciferase activity was determined. (*p<0.005).
(I, J) RELA is recruited to the ANG promoter. HCT116 cells expressing shRNA to PHD2 were treated with TNF-α (20 ng/ml) for the given amount of time. ChIP with α-RELA antibodies was performed, and RELA occupancy is shown. MnSOD promoter is used as a positive control for recruitment of RELA to a NF-κB target promoter. IgG control ChIPs were also performed as a negative control. All error bars represent ±SEM.
To test whether NF-κB is necessary for enhanced angiogenesis, we transfected PHD2 silenced cells with siRNA to RELA, the p65 subunit of NF-κB, and examined the ability of conditioned media from these cells to stimulate tube formation in vitro (Figures S6A, S6B, and S6C). PHD2 silencing in HIF-1α-deficient cells is unable to enhance in vitro angiogenesis in the absence of RELA (Figures 6D, 6E, and 6F). In cells with HIF-1α, NF-κB is also necessary for this effect on angiogenesis (Figures S6B and S6C). Together these data support the conclusion that NF-κB mediates the HIF-independent tumor suppressor function of PHD2.
PHD2 regulates secreted pro-angiogenic factors
We sought to determine which secreted factors regulated by PHD2 could explain the effects on tumor vasculature. The secreted VEGF levels is unaffected by the lack of PHD2 or the lack of both PHD2 and HIF-1α and is induced by hypoxia normally (Figures S6D and S6E). This demonstrates that the effect of PHD2 on the tumor vasculature is distinct from both HIF and VEGF. We then probed antibody arrays with conditioned media from control HCT116 cells and cells expressing PHD2 shRNA (Figure S6F). In agreement with the ELISA, levels of VEGF were high in the conditioned media regardless of genotype (Figure 6F). However, we determined that PHD2 consistently regulates two angiogenic factors, namely interleukin-8 (IL-8) and angiogenin (ANG) (Figure 6E) (Fett et al., 1985; Koch et al., 1992). As we were also searching for a cytokine regulated by PHD2 in a HIF-independent manner, we probed arrays with conditioned media from HCT116 HIF-1 knockout cells and the corresponding cells with PHD2 knockdown (Figure S6G). Again, we identified that PHD2 knockdown increases secretion of IL-8 and ANG, regardless of HIF. Silencing of RELA in PHD2 shRNA cells reduced secreted ANG and IL-8 by about half in agreement with other reports that ANG and IL-8 can regulate angiogenesis and are regulated by NF-κB (Figure S6H) (Garkavtsev et al., 2004; Karashima et al., 2003; Wu et al., 1997).
To directly confirm the regulation of ANG and IL-8 by NF-κB, we performed reporter assays, using the respective promoters of ANG and IL-8 (Figure 6H). Stimulation of NF-κB with TNF-α increases activity of both ANG and IL-8 promoters, demonstrating that both are NF-κB responsive genes. Mutation of the NF-κB sites in their respective promoters eliminated NF-κB responsiveness. As IL-8 is already known NF-κB target (Karashima et al., 2003), we sought to further corroborate NF-κB regulation of ANG. Chromatin immunoprecipitation with RELA antibodies demonstrates that RELA is recruited to the promoter of ANG (Figure 6I). The MnSOD promoter, another NF-κB target, was used as a positive control (Figure 6J). These data further validate ANG and IL-8 as NF-κB target genes.
PHD2 Regulation of ANG and IL-8 Mediates Tumor Vasculature
We next examined whether ANG and IL-8 mediated the angiogenic phenotype conferred by PHD2 loss by silencing ANG or IL-8 in HCT116 with PHD2 knockdown (Figures S7A, S7B, S7E, and S7F). Knockdown of either ANG or IL-8 decreased the ability of the PHD2 knockdown cells to induce capillary branching, indicating that PHD2-mediated angiogenesis is directed in part by ANG and IL-8 (Figures 7A, 7B, S7C, and S7D). Staining HCT116 tumors for CD11b and CD45 revealed that mobilization of BMDCs by PHD2 silencing requires ANG and IL-8 (Figures 7C and 7D). CD11b-positive cells were associated with mature blood vessels as determined by staining pericytes with smooth muscle actin (Figure 7C, lower panel). The functional maturity was further verified by staining sections with CD31 and the hypoxia marker pimonidazole, which showed that CD31-positive blood vessels are in nonhypoxic regions (Figure 7E). We found a distinct, non-overlapping staining pattern between hypoxia and CD45, indicating that BMDCs are found in the better perfused areas of the tumor with access to the vasculature, further supporting the idea of a HIF/hypoxia-independent effect of PHD2 in recruiting BMDCs to a tumor (Figure 7D). To verify that this is a specific recruitment of BMDC and not a general infiltration of macrophages, we stained these tumors for F4/80, a pan-macrophage marker, finding equivalent numbers of macrophages in all tumors regardless of genotype and despite increased blood vessels in PHD2-silenced tumors (Figure S7G). Thus, in tumors that have reduced PHD2, mobilization of BMDCs may contribute to the vasculogenic tumor phenotype. To evaluate the effect of BMDC mobilization on tumor growth, we implanted ANG- and IL-8-deficient HCT116 cells in mice and found that disruption of ANG or IL-8 impaired the enhanced tumor growth induced by PHD2 loss. (Figure 7F). These data indicate that PHD2 loss increases tumor growth through ANG- and IL8-mediated angiogenesis and recruitment of BMDCs.
Figure 7.
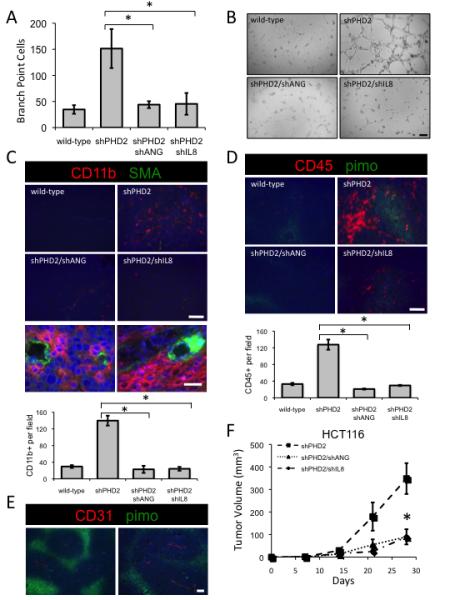
PHD2 Regulation of ANG and IL-8 Mediates Tumor Vasculature
(A) Conditioned media from HCT116 cells expressing PHD2 shRNA and shRNA targeting either ANG or IL-8 were subjected to a tube formation assay (*p<0.05).
(B) Tube formation with conditioned media from the given genotype. Scale bar = 2 mm.
(C) Tumor staining for CD11b myelomonocytic precursor cells (red) and pericytes with vascular smooth muscle actin (green). Scale bar = 100 microns. Bottom panels are higher magnification of PHD2-silenced tumors. Scale bar = 500 microns. Quantification of CD11b-positive cells (*p<0.001).
(D) Tumor staining for CD45 (red), hypoxia with pimonidazole (green) and nuclei with DAPI (blue). Scale bar = 100 microns. Quantification of CD45-positive cells (*p<0.001).
(E) Higher magnification images of wild-type HCT116 tumors stained for CD31 (red) and pimonidazole (green). Scale bar = 100 microns.
(F) Tumor growth of cells expressing the given shRNA in SCID mice (*p<0.005). All error bars represent ±SEM.
Decreased PHD2 Expression Correlates with Increases in NF-κB Activity and Tumor Angiogenesis in Human Cancers
To further evaluate the relationship between PHD2 and the tumor vasculature in human cancers, we extracted data on PHD2 mRNA expression level from two published studies of breast cancer composed of different patient populations. One study was on 286 patients with lymph node-negative breast cancers who did not receive adjuvant therapy whereas the other was on 159 population-derived breast cancer patients (Pawitan et al., 2005; Wang et al., 2005). Despite these differences in patients’ characteristics, we noted a statistically significant negative association between the expression level of PHD2 and that of the endothelial cell marker gene CD31 in both datasets, further supporting the in vivo relevance of the ability of PHD2 to inhibit tumor vasculature in human tumors (left panels of Figures 8A and 8B).
Figure 8.
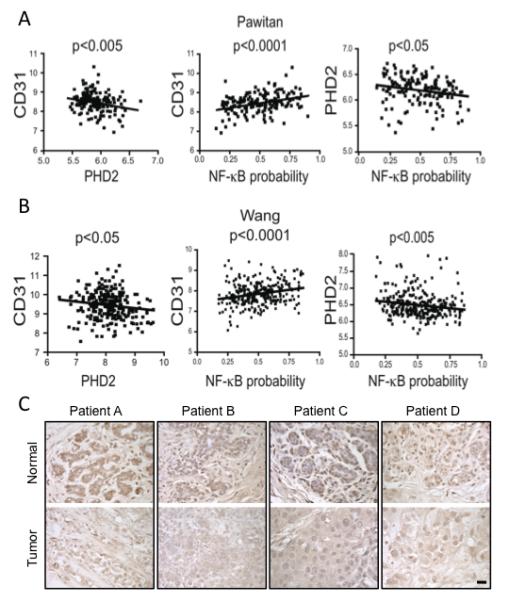
Decreased PHD2 Expression Correlates with Increases in NF-κB Activity and Tumor Angiogenesis in Human Cancers
(A, B) Left panels: Significant negative correlation between the expression level of PHD2 (x-axis) and CD31 (y-axis) in the breast cancer samples profiled in two expression studies of breast cancers. Middle panels: Significant positive correlation between NF-κB pathway activity (x-axis) and expression of CD31 (y-axis). Right panels: Significant negative correlation between the expression level of PHD2 (x-axis) and NF-κB activity (y-axis).
(C) Immunohistochemical staining for PHD2 in invasive breast carcinomas (bottom panels) compared to matched, uninvolved, adjacent breast tissue (top panels) from several different patients. Scale bar = 100 μm.
We next assessed the extent to which NF-κB activation is related to both PHD2 expression and markers of angiogenesis. We used binary logistic regression to estimate the probability of NF-κB activation as modeled on TNF-α stimulation training sets to generate NF-κB signatures (Bild et al., 2006; Mori et al., 2008). These signatures were used to approximate the likelihood of NF-κB activation in the two breast cancer datasets. We found a significant positive association between NF-κB activation and CD31 expression (Figures 8A and 8B, middle panels) as well as a significant negative correlation between NF-κB activation and PHD2 levels in both datasets (Figures 8A and 8B, right panels). These observations are consistent with the hypothesis that PHD2 represses the expression of CD31 through the inhibition of NF-κB.
We then examined PHD2 protein levels in clinical breast cancer samples by performing immunohistochemistry on breast tumors along with the corresponding normal tissue from the same patient (Figure 8C). Of the 11 patient samples, six had higher levels of PHD2 in the normal tissue compared to the matched tumor tissue, indicating that the loss of PHD2 occurs in clinically relevant situations and implicating PHD2 in tumorigenesis. PHD2 regulates tumor vasculature through both angiogenesis and neovascularization. Taken together our data suggests that PHD2 is at the intersection of multiple, complementary pathways. PHD2 controls both HIF-dependent and NF-κB-dependent pathways mediating both local sprouting and neovascularization, and consequently regulates tumor growth (Figure S1).
DISCUSSION
Induction of angiogenesis through local sprouting and neovascularization is essential to the rapid growth of solid tumors. The identity and regulation of the factors responsible for these processes are areas of active investigation as these signaling pathways could provide valuable avenues for therapeutic invention. HIF and VEGF have garnered considerable attention for their roles in mediating tumor angiogenesis. Recently, interest has grown in the regulators of HIF and in alternative mechanisms of pathological angiogenesis (Fischer et al., 2007; Shojaei et al., 2007). In this report, we demonstrate that PHD2 functions as a tumor suppressor by limiting ANG and IL-8, which play roles in both angiogenesis and BMDC recruitment contributing to vasculogenesis. Despite the role of PHD2 in regulating HIF stability, the tumor suppressor function of PHD2 observed here does not solely depend on the presence of HIF. Eliminating HIF did not reduce tumor growth in the PHD2-silenced background in multiple cancer types. PHD2 knockdown elevated normoxic NF-κB activity and consequently, increased IL-8 and ANG, both of which contribute to enhance tumor blood supply and thus tumor growth. Moreover, analysis of clinical data reveals a reduction of PHD2 in human cancers, which is also correlated with an increase in tumor vasculature. Together, these findings highlight the importance of HIF- and VEGF-independent pathways in regulating tumor angiogenesis.
Bone marrow-derived cells are increasingly appreciated to be important contributors to the expansion of tumor vasculature (Gao et al., 2008; Lyden et al., 2001; Shojaei et al., 2007; Yang et al., 2004). However, the mechanisms by which tumors recruit these cells are not yet fully understood. A few factors have been implicated, such as the Id proteins, MMP-9, and Bv8, but the entire repertoire of signals responsible for the recruitment is largely unknown. Our current data provide evidence that the release of IL-8 and ANG is enhanced in tumors with reduced PHD2, and that these factors are important for both recruitment of BMDCs and for local angiogenesis. The precise mechanisms by which PHD2 restricts release of IL-8 and ANG and how vasculogenesis is mediated by BMDC recruitment in response to IL-8 and ANG remains to be determined. We demonstrate that NF-κB is required for enhanced angiogenesis upon PHD2 silencing. NF-κB, in turn, regulates ANG and IL-8. Intriguingly, inhibition of either ANG or IL-8 appears to be sufficient to abolish the effect of losing PHD2, suggesting potential clinical applications. In particular, inhibiting either ANG or IL-8 may be potential avenues for treatment of tumors that have become resistant to anti-VEGF therapies. Our findings demonstrate that a biological consequence of NF-κB regulation by PHD2 is to impair tumor growth through disruption of the blood supply, suggesting that this pathway may be important in human cancers.
How does PHD2 come to be downregulated in some tumors? One possibility is that PHD2 itself might be mutated in a way that reduces its activity, expression, or stability. A recent report describes a patient with a PHD2 mutation who presents with both polycythemia and multiple tumors (Ladroue et al., 2008). In addition, PHD2 was found to be mutated at a high rate in endometrial cell lines (Kato et al., 2006). Another possibility is that methylation of the PHD2 promoter could result in epigenetic silencing of PHD2. Nevertheless, dysregulation of PHD2 has important consequences, contributing to tumorigenesis in multiple ways. Our analysis indicates that PHD2 levels are significantly reduced in certain clinical cases, such as colon and breast tumors. Although this reduction in PHD2 may contribute to tumor growth through enhanced stability of HIF, our work and the work of others indicate that PHD2 also has additional HIF-independent regulatory functions. For example, in agreement with the large variability of PHD2 protein levels in the NCI60 panel, simultaneous protein expression of HIF-1 and PHD2 has been found in a subset of head and neck tumors (Jokilehto et al., 2006). Our data suggests that PHD2 can regulate HIF and NF-κB in a complementary, additive fashion with both pathways likely contributing to tumor growth. Both the HIF-dependent hypoxia response and the HIF-independent function of PHD2 can influence NF-κB and IL-8. It therefore remains to be seen in which contexts one PHD2 function can prevail over the other and whether HIF-dependent or HIF-independent pathways come to dominate tumorigenesis. One possibility is that these pathways cooperate to provide redundant pathways for activating NF-κB and promoting angiogenesis. Alternatively, HIF-dependent mechanisms may be predominant in hypoxic regions, while disruption of PHD2 may allow for HIF-independent mechanisms that promote angiogenesis in nonhypoxic regions. Future studies will elucidate whether PHD2 regulation of HIF is important for tumor initiation while regulation of NF-κB is important for tumor progression or dissemination.
As the search for HIF inhibitors is an active area of cancer biology, this study suggests that PHD2 also plays a critical role in tumor growth. However, HIF inhibition may not be sufficient to impede tumor growth especially in tumors in which PHD2 activity is lost. PHD2 has at least two tumor suppressor activities: 1) negatively regulating HIF through hydroxylation and 2) negatively regulating NF-κB in a HIF-independent manner, both of which in turn modulates tumor vasculature through angiogenesis and vasculogenesis. Further studies on the mechanism of PHD2 loss will be essential to gain new insights into which tumors may benefit clinically from anti-angiogenic therapies. Identifying other PHD2 targets and binding partners, as well as delineating how they influence tumorigenesis, will be a burgeoning field of study and will be critical in furthering our understanding of tumor progression.
EXPERIMENTAL PROCEDURES
Cells
Cell pellets from the NCI60 panel of cell lines were provided by Dr. Giovanni Melillo (NCI-Frederick). HCT116 and RKO cells were cultured in DMEM, SU.86.86 in RPMI 1640, and HT29 in McCoy’s 5a media. HIF-1 knockout cells were a kind gift from Duyen and Long Dang (University of Michigan) and cultured in McCoy’s 5a media. All cell lines were maintained in 10% FCS. Primary human umbilical vein endothelial cells (HUVECs) were grown in complete Endothelial Growth Media (Clonetics). Transfection of plasmid DNA or RNA oligos were performed with Lipofectamine-Plus Reagent (Invitrogen) or DharmaFECT Reagent 1 (Dharmacon), according to manufacturer’s directions.
Reagents
MG132 was purchased from EMD, DFO from Sigma, DMOG from Biomol, ECMatrix from Millipore, and TNF-α from R&D, pimonidazole from NPI. ON-TARGETplus SMART pools against ANG, ARNT, IL-8, and RELA and non-targeting control (siCon2) were obtained from Dharmacon. Human angiogenesis antibody arrays were from Panomics. Immunoblotting was performed as described (Chan et al., 2005). Antibodies used are as follows: from Becton-Dickinson: HIF-1α (clone 54), HIF-1β (clone 29), CD31/PECAM (clone WM59), CD11b (clone M1/70); from Novus Biologicals: HIF-2α (clone ep190b), PHD1 (NB100-310), PHD2 (NB100-137), PHD3 (NB100-139), and FIH (NB100-428); from Santa Cruz, CAIX (Clone H); from Research Diagnostics (clone DM1A); and from AbD Serotec: CD45 (clone YW62.3). An Invivo2 hypoxia chamber (Ruskinn Inc.) was used for hypoxia treatment. Colon and breast tissue microarrays were purchased from US Biomax. All human tissues for the tissue microarrays were obtained with informed consent and under IRB- and HIPAA-approved protocols according to US federal laws and are exempt from consideration by the Stanford Administrative Panel on Human Subjects in Medical Research.
shRNA Constructs
Oligos for specific degradation of target RNA were designed using the Ambion siRNA Target Finder and cloned into RNAi-Ready pSiren RetroQ (BD Biosciences). All experiments refer to individual constructs and not a mixture of constructs.
Plasmids
Fusion PCR was used to make the catalytic hydroxylase mutations in PHD2 (H313A and D315A), which were confirmed by sequencing. A 1.3 kilobase sequence upstream of the transcriptional start site of ANG was cloned into pGL3-promoter. A NF-κB binding site was identified by TRES (http://bioportal.bic.nus.edu.sg/tres/).
Reporter Assays
Luciferase activity was determined by Dual-Glo Luciferase assay reagent (Promega) measured in a Monolight 2010 Luminometer (Analytical Luminescence Laboratory). Firefly luciferase was normalized to constitutive Renilla luciferase.
In Vivo Experiments and Immunohistochemistry
All animal experiments were approved by Stanford’s Administrative Panel on Laboratory Animal Care (APLAC) and in accordance with institutional and national guidelines. Two to five million cells were implanted subcutaneously into the flanks of CB17/SCID mice (four to six week old males supplied by the Stanford University Research Animal Facility or Charles River Laboratories). Tumors were measured with calipers. Volume was calculated by the following formula: width2 × length × 0.5. Five micron sections were cut for immunohistochemistry. Sections were counterstained with hematoxylin and eosin or nuclei were stained with DAPI. Five random fields were counted per section to determine the amount of a given stain.
In Vitro Growth Curve
One hundred thousand cells were plated on 60 mm dishes. Three days later, cells were trypsinized, counted on a hemacytometer and a hundred thousand cells were replated.
In Vitro Angiogenesis Assay
Fifty thousand cells of a given genotype were plated in 1 ml of Endothelial Basal Media with 2% FCS. Conditioned media was collected two days later. Five to ten thousand HUVECs were plated in 100 μl of conditioned media on Matrigel. Cells were incubated for four to six hours at 37°C and visualized by light microscopy. The amount of angiogenesis was quantified by counting the number of cells in branch point capillaries (≥3 cells per branch) in five random fields per replicate.
Real-Time Quantitative RT-PCR
Total RNA was extracted from cells with TRIzol (Invitrogen) as per manufacturer’s directions. Total RNA (1.5 μg) was reversed transcribed with random hexamer primers and MMLV-RT. Power SYBR Green PCR reactions were performed in triplicate for each sample and analyzed using the ABI Prism 7900HT sequence detection system. Data were normalized to TBP levels.
Chromatin Immunoprecipitation
Cells were treated with TNF-α (20 ng/ml) for the indicated time and crosslinked for 10 minutes with 1% formaldehyde. Crosslinking was stopped by the addition of 0.125 M glycine. Chromatin was sonicated to approximately 300 bp using a Bioruptor (Diagenode). Purified chromatin was incubated with an α-RELA antibody (Biomol International). Chromatin was then de-crosslinked and ChIP-associated sequences were detected by quantitative real-time PCR.
Gene Expression Analysis
PHD2 expression data were analyzed by Oncomine (www.oncomine.org). Data from two human clinical cancer studies were used (Graudens et al., 2006; Ramaswamy et al., 2001). The Ramaswamy study included 90 normal samples and 190 cancer samples. The Graudens study included 12 normal colon samples and 18 colorectal carcinomas. The expression levels of all probe sets for PHD2 and CD31 were extracted from the expression studies of indicated breast cancer samples after RMA normalization (Pawitan et al., 2005; Wang et al., 2005). The binary regression analysis was based on TNF-α-treated cells relative to control as training sets. This gene signature was then used to estimate the pathway activities of NF-κB for each breast cancer sample in both datasets relative to the expression values of PHD2 and CD31.
Statistical Analysis
Student’s t-test or one-way ANOVA with post-hoc Tukey-Kramer tests were used to determine significance. All error bars represent the standard error of the mean.
SIGNIFICANCE.
Virtually all solid tumors require a vascular supply to provide nutrients, oxygen and eliminate wastes. A tumor can exploit the existing vessels of the invaded tissue and can mobilize bone marrow-derived cells to induce neovascularization. Although PHD2 was originally identified as a negative regulator of the protein stability of the hypoxia-inducible factor-α transcription factors, we demonstrate that PHD2 has additional HIF-independent functions. PHD2 levels are altered in a subset of human tumors, suggesting a role in tumor progression. We identify a key role of PHD2 in mediating the angiogenic switch, regulating both angiogenesis and the recruitment of vascular modulatory cells. These studies provide evidence for an important role of PHD2 in controlling tumor growth.
Supplementary Material
ACKNOWLEDGEMENTS
We kindly thank Drs. Duyen T. Dang and Long H. Dang (University of Michigan Comprehensive Cancer Center) for the generous gift of HIF-1 knockout cells, Dr. Andrew Keates for IL-8 reporter constructs, Dr. Giovanni Melillo, Pauline Chu for help with immunohistochemical analyses, and Dr. Trent A. Watkins for helpful discussions. This investigation was supported a Silicon Valley Community Fellowship, NCI-CA-123823 (D.A.C.), NCI-CA-116685, and NCI-CA-67166 (A.J.G), and CA-125618 awarded by the National Institutes of Health (J.T.C.).
Footnotes
SUPPLEMENTAL DATA Supplemental Data includes Supplemental Experimental Procedures, including primer sequences and shRNA sequences, and 7 figures.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Appelhoff RJ, Tian YM, Raval RR, Turley H, Harris AL, Pugh CW, Ratcliffe PJ, Gleadle JM. Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. J Biol Chem. 2004;279:38458–38465. doi: 10.1074/jbc.M406026200. [DOI] [PubMed] [Google Scholar]
- Berra E, Benizri E, Ginouves A, Volmat V, Roux D, Pouyssegur J. HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1alpha in normoxia. Embo J. 2003;22:4082–4090. doi: 10.1093/emboj/cdg392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bild AH, Yao G, Chang JT, Wang Q, Potti A, Chasse D, Joshi MB, Harpole D, Lancaster JM, Berchuck A, et al. Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature. 2006;439:353–357. doi: 10.1038/nature04296. [DOI] [PubMed] [Google Scholar]
- Carmeliet P. Mechanisms of angiogenesis and arteriogenesis. Nat Med. 2000;6:389–395. doi: 10.1038/74651. [DOI] [PubMed] [Google Scholar]
- Chan DA, Sutphin PD, Yen SE, Giaccia AJ. Coordinate regulation of the oxygen-dependent degradation domains of hypoxia-inducible factor 1 alpha. Mol Cell Biol. 2005;25:6415–6426. doi: 10.1128/MCB.25.15.6415-6426.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummins EP, Berra E, Comerford KM, Ginouves A, Fitzgerald KT, Seeballuck F, Godson C, Nielsen JE, Moynagh P, Pouyssegur J, Taylor CT. Prolyl hydroxylase-1 negatively regulates IkappaB kinase-beta, giving insight into hypoxia-induced NFkappaB activity. Proc Natl Acad Sci U S A. 2006;103:18154–18159. doi: 10.1073/pnas.0602235103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Angelo G, Duplan E, Boyer N, Vigne P, Frelin C. Hypoxia up-regulates prolyl hydroxylase activity: a feedback mechanism that limits HIF-1 responses during reoxygenation. J Biol Chem. 2003;278:38183–38187. doi: 10.1074/jbc.M302244200. [DOI] [PubMed] [Google Scholar]
- Dang DT, Chen F, Gardner LB, Cummins JM, Rago C, Bunz F, Kantsevoy SV, Dang LH. Hypoxia-inducible factor-1alpha promotes nonhypoxia-mediated proliferation in colon cancer cells and xenografts. Cancer Res. 2006;66:1684–1936. doi: 10.1158/0008-5472.CAN-05-2887. [DOI] [PubMed] [Google Scholar]
- De Palma M, Venneri MA, Roca C, Naldini L. Targeting exogenous genes to tumor angiogenesis by transplantation of genetically modified hematopoietic stem cells. Nat Med. 2003;9:789–795. doi: 10.1038/nm871. [DOI] [PubMed] [Google Scholar]
- Fett JW, Strydom DJ, Lobb RR, Alderman EM, Bethune JL, Riordan JF, Vallee BL. Isolation and characterization of angiogenin, an angiogenic protein from human carcinoma cells. Biochemistry. 1985;24:5480–5486. doi: 10.1021/bi00341a030. [DOI] [PubMed] [Google Scholar]
- Fischer C, Jonckx B, Mazzone M, Zacchigna S, Loges S, Pattarini L, Chorianopoulos E, Liesenborghs L, Koch M, De Mol M, et al. Anti-PlGF inhibits growth of VEGF(R)-inhibitor-resistant tumors without affecting healthy vessels. Cell. 2007;131:463–475. doi: 10.1016/j.cell.2007.08.038. [DOI] [PubMed] [Google Scholar]
- Gao D, Nolan DJ, Mellick AS, Bambino K, McDonnell K, Mittal V. Endothelial progenitor cells control the angiogenic switch in mouse lung metastasis. Science. 2008;319:195–198. doi: 10.1126/science.1150224. [DOI] [PubMed] [Google Scholar]
- Garkavtsev I, Kozin SV, Chernova O, Xu L, Winkler F, Brown E, Barnett GH, Jain RK. The candidate tumour suppressor protein ING4 regulates brain tumour growth and angiogenesis. Nature. 2004;428:328–332. doi: 10.1038/nature02329. [DOI] [PubMed] [Google Scholar]
- Graudens E, Boulanger V, Mollard C, Mariage-Samson R, Barlet X, Gremy G, Couillault C, Lajemi M, Piatier-Tonneau D, Zaborski P, et al. Deciphering cellular states of innate tumor drug responses. Genome Biol. 2006;7:R19. doi: 10.1186/gb-2006-7-3-r19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Harris AL. Hypoxia--a key regulatory factor in tumour growth. Nat Rev Cancer. 2002;2:38–47. doi: 10.1038/nrc704. [DOI] [PubMed] [Google Scholar]
- Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG., Jr. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292:464–468. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- Jokilehto T, Rantanen K, Luukkaa M, Heikkinen P, Grenman R, Minn H, Kronqvist P, Jaakkola PM. Overexpression and nuclear translocation of hypoxia-inducible factor prolyl hydroxylase PHD2 in head and neck squamous cell carcinoma is associated with tumor aggressiveness. Clin Cancer Res. 2006;12:1080–1087. doi: 10.1158/1078-0432.CCR-05-2022. [DOI] [PubMed] [Google Scholar]
- Kaelin WG. Von hippel-lindau disease. Annu Rev Pathol. 2007;2:145–173. doi: 10.1146/annurev.pathol.2.010506.092049. [DOI] [PubMed] [Google Scholar]
- Karashima T, Sweeney P, Kamat A, Huang S, Kim SJ, Bar-Eli M, McConkey DJ, Dinney CP. Nuclear factor-kappaB mediates angiogenesis and metastasis of human bladder cancer through the regulation of interleukin-8. Clin Cancer Res. 2003;9:2786–2797. [PubMed] [Google Scholar]
- Kato H, Inoue T, Asanoma K, Nishimura C, Matsuda T, Wake N. Induction of human endometrial cancer cell senescence through modulation of HIF-1alpha activity by EGLN1. Int J Cancer. 2006;118:1144–1153. doi: 10.1002/ijc.21488. [DOI] [PubMed] [Google Scholar]
- Koch AE, Polverini PJ, Kunkel SL, Harlow LA, DiPietro LA, Elner VM, Elner SG, Strieter RM. Interleukin-8 as a macrophage-derived mediator of angiogenesis. Science. 1992;258:1798–1801. doi: 10.1126/science.1281554. [DOI] [PubMed] [Google Scholar]
- Kondo K, Klco J, Nakamura E, Lechpammer M, Kaelin WG., Jr. Inhibition of HIF is necessary for tumor suppression by the von Hippel-Lindau protein. Cancer Cell. 2002;1:237–246. doi: 10.1016/s1535-6108(02)00043-0. [DOI] [PubMed] [Google Scholar]
- Ladroue C, Carcenac R, Leporrier M, Gad S, Le Hello C, Galateau-Salle F, Feunteun J, Pouyssegur J, Richard S, Gardie B. PHD2 mutation and congenital erythrocytosis with paraganglioma. N Engl J Med. 2008;359:2685–2692. doi: 10.1056/NEJMoa0806277. [DOI] [PubMed] [Google Scholar]
- Lando D, Peet DJ, Gorman JJ, Whelan DA, Whitelaw ML, Bruick RK. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 2002a;16:1466–1471. doi: 10.1101/gad.991402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lando D, Peet DJ, Whelan DA, Gorman JJ, Whitelaw ML. Asparagine hydroxylation of the HIF transactivation domain a hypoxic switch. Science. 2002b;295:858–861. doi: 10.1126/science.1068592. [DOI] [PubMed] [Google Scholar]
- Lyden D, Hattori K, Dias S, Costa C, Blaikie P, Butros L, Chadburn A, Heissig B, Marks W, Witte L, et al. Impaired recruitment of bone-marrow-derived endothelial and hematopoietic precursor cells blocks tumor angiogenesis and growth. Nat Med. 2001;7:1194–1201. doi: 10.1038/nm1101-1194. [DOI] [PubMed] [Google Scholar]
- Mahon PC, Hirota K, Semenza GL. FIH-1: a novel protein that interacts with HIF-1alpha and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev. 2001;15:2675–2686. doi: 10.1101/gad.924501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzone M, Dettori D, de Oliveira R. Leite, Loges S, Schmidt T, Jonckx B, Tian YM, Lanahan AA, Pollard P, de Almodovar C. Ruiz, et al. Heterozygous Deficiency of PHD2 Restores Tumor Oxygenation and Inhibits Metastasis via Endothelial Normalization. Cell. 2009 doi: 10.1016/j.cell.2009.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonough MA, Li V, Flashman E, Chowdhury R, Mohr C, Lienard BM, Zondlo J, Oldham NJ, Clifton IJ, Lewis J, et al. Cellular oxygen sensing: Crystal structure of hypoxia-inducible factor prolyl hydroxylase (PHD2) Proc Natl Acad Sci U S A. 2006;103:9814–9819. doi: 10.1073/pnas.0601283103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minamishima YA, Moslehi J, Bardeesy N, Cullen D, Bronson RT, Kaelin WG., Jr. Somatic inactivation of the PHD2 prolyl hydroxylase causes polycythemia and congestive heart failure. Blood. 2008;111:3236–3244. doi: 10.1182/blood-2007-10-117812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori S, Rempel RE, Chang JT, Yao G, Lagoo AS, Potti A, Bild A, Nevins JR. Utilization of pathway signatures to reveal distinct types of B lymphoma in the Emicro-myc model and human diffuse large B-cell lymphoma. Cancer Res. 2008;68:8525–8534. doi: 10.1158/0008-5472.CAN-08-1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohh M, Park CW, Ivan M, Hoffman MA, Kim TY, Huang LE, Pavletich N, Chau V, Kaelin WG. Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von Hippel-Lindau protein. Nat Cell Biol. 2000;2:423–427. doi: 10.1038/35017054. [DOI] [PubMed] [Google Scholar]
- Pawitan Y, Bjohle J, Amler L, Borg AL, Egyhazi S, Hall P, Han X, Holmberg L, Huang F, Klaar S, et al. Gene expression profiling spares early breast cancer patients from adjuvant therapy: derived and validated in two population-based cohorts. Breast Cancer Res. 2005;7:R953–964. doi: 10.1186/bcr1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswamy S, Tamayo P, Rifkin R, Mukherjee S, Yeang CH, Angelo M, Ladd C, Reich M, Latulippe E, Mesirov JP, et al. Multiclass cancer diagnosis using tumor gene expression signatures. Proc Natl Acad Sci U S A. 2001;98:15149–15154. doi: 10.1073/pnas.211566398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan HE, Lo J, Johnson RS. HIF-1 alpha is required for solid tumor formation and embryonic vascularization. Embo J. 1998;17:3005–3015. doi: 10.1093/emboj/17.11.3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan HE, Poloni M, McNulty W, Elson D, Gassmann M, Arbeit JM, Johnson RS. Hypoxia-inducible factor-1alpha is a positive factor in solid tumor growth. Cancer Res. 2000;60:4010–4015. [PubMed] [Google Scholar]
- Semenza GL. Involvement of hypoxia-inducible factor 1 in human cancer. Intern Med. 2002;41:79–83. doi: 10.2169/internalmedicine.41.79. [DOI] [PubMed] [Google Scholar]
- Shojaei F, Wu X, Zhong C, Yu L, Liang XH, Yao J, Blanchard D, Bais C, Peale FV, van Bruggen N, et al. Bv8 regulates myeloid-cell-dependent tumour angiogenesis. Nature. 2007;450:825–831. doi: 10.1038/nature06348. [DOI] [PubMed] [Google Scholar]
- Takeda K, Fong GH. Prolyl hydroxylase domain 2 protein suppresses hypoxia-induced endothelial cell proliferation. Hypertension. 2007;49:178–184. doi: 10.1161/01.HYP.0000251360.40838.0f. [DOI] [PubMed] [Google Scholar]
- Takeda K, Ho VC, Takeda H, Duan LJ, Nagy A, Fong GH. Placental but not heart defects are associated with elevated hypoxia-inducible factor alpha levels in mice lacking prolyl hydroxylase domain protein 2. Mol Cell Biol. 2006;26:8336–8346. doi: 10.1128/MCB.00425-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unruh A, Ressel A, Mohamed HG, Johnson RS, Nadrowitz R, Richter E, Katschinski DM, Wenger RH. The hypoxia-inducible factor-1 alpha is a negative factor for tumor therapy. Oncogene. 2003;22:3213–3220. doi: 10.1038/sj.onc.1206385. [DOI] [PubMed] [Google Scholar]
- Wang Y, Klijn JG, Zhang Y, Sieuwerts AM, Look MP, Yang F, Talantov D, Timmermans M, Meijer-van Gelder ME, Yu J, et al. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. Lancet. 2005;365:671–679. doi: 10.1016/S0140-6736(05)17947-1. [DOI] [PubMed] [Google Scholar]
- Wu GD, Lai EJ, Huang N, Wen X. Oct-1 and CCAAT/enhancer-binding protein (C/EBP) bind to overlapping elements within the interleukin-8 promoter. The role of Oct-1 as a transcriptional repressor. J Biol Chem. 1997;272:2396–2403. [PubMed] [Google Scholar]
- Yang L, DeBusk LM, Fukuda K, Fingleton B, Green-Jarvis B, Shyr Y, Matrisian LM, Carbone DP, Lin PC. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell. 2004;6:409–421. doi: 10.1016/j.ccr.2004.08.031. [DOI] [PubMed] [Google Scholar]
- Yu F, White SB, Zhao Q, Lee FS. HIF-1alpha binding to VHL is regulated by stimulus-sensitive proline hydroxylation. Proc Natl Acad Sci U S A. 2001;98:9630–9635. doi: 10.1073/pnas.181341498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong H, De Marzo AM, Laughner E, Lim M, Hilton DA, Zagzag D, Buechler P, Isaacs WB, Semenza GL, Simons JW. Overexpression of hypoxia-inducible factor 1alpha in common human cancers and their metastases. Cancer Res. 1999;59:5830–5835. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.