

Summary
Neurofibromatosis type 1 (Nf1) mutation predisposes to benign peripheral nerve (glial) tumors called neurofibromas. The point(s) in development when Nf1 loss promotes neurofibroma formation are unknown. We show that inactivation of Nf1 in the glial lineage in vitro at embryonic day 12.5 + 1, but not earlier (neural crest) or later (mature Schwann cell) results in colony-forming cells capable of multi-lineage differentiation. In vivo, inactivation of Nf1 using a DhhCre driver beginning at E12.5 elicits plexiform neurofibromas, dermal neurofibromas, and pigmentation. Tumor Schwann cells uniquely show biallelic Nf1 inactivation. Peripheral nerve and tumors contain transiently proliferating Schwann cells that lose axonal contact, providing insight into early neurofibroma formation. We suggest that timing of Nf1 mutation is critical for neurofibroma formation.
Introduction
Neurofibromatosis type 1 (NF1) is an autosomal dominant inherited disease, affecting 1:3500 individuals worldwide. Nearly all (90%) of NF1 patients develop plexiform and/or dermal neurofibromas (Friedman, 1999), composed of a mixture of axons, Schwann cells, fibroblasts, perineurial cells, and mast cells. Diffuse plexiform neurofibromas develop in childhood and often extend deeply along nerves and can involve all levels of skin, fascia, muscle, bone, and even viscera, whereas dermal (variously designated dermal/cutaneous and subcutaneous in the literature and which we group as dermal) neurofibromas associated with small nerve branches mainly develop with puberty and into adulthood (Huson, 1998). Schwann cells are believed to be the primary pathogenic cells in neurofibromas because they show biallelic mutation at NF1, and cell autonomous angiogenic and invasive properties (Serra et al., 1997; Sheela et al., 1990). However, other cell types present in tumors, in the NF1+/− state, also show cell autonomous defects (Cichowski et al., 2003; Kim et al., 1995; 1997; Yang et al., 2003). The role of these cells in human neurofibromas is not known.
The NF1 tumor suppressor gene, located on human chromosome 17q11.2, encodes a Ras GTPase activating protein (GAP). Ras is hyperactivated in NF1-deficient human neurofibromas and Schwann cells lacking NF1 (Cichowski and Jacks, 2001; DeClue et al., 1992; Guha et al., 1996; Sherman et al., 2000). Mutations affecting NF1 and Ras also induce a global negative feedback response that potently suppresses Ras and/or its effectors (Courtois-Cox et al., 2006). Nomenclature for mouse nerve tumors was defined at a consensus conference; mouse tumors are preceded by a genetically engineered mouse (GEM) designation (Stemmer-Rachamimov et al., 2004). GEM-neurofibromas and their aggressive derivates, GEM-peripheral nerve sheath tumors (GEM-PNST) developed by 3 months in visible locations (ear, tail) when the human T-lymphotropic virus type 1 trans-regulatory gene, tax, was expressed under the control of a viral long terminal repeat. Tax decreased Nf1 transcription though an element upstream of the Nf1 start site (Feigenbaum et al., 1996). In transgenic mouse using a CAMKinaseII promoter to drive expression of an activated N-Ras allele, mice developed GEM- dermal neurofibromas (Saito et al., 2007). More accurate, Nf1-driven models mimicking as closely as possible human dermal and plexiform neurofibroma formation are needed to clarify mechanisms underlying tumorigenesis and to provide a platform for therapeutic testing.
Nf1−/− mouse embryos die at E13.5, and Nf1+/− mice do not develop neurofibromas (Brannan et al., 1994; Jacks et al., 1994). Nf1+/− mice serve as an important therapeutic model for GEM-PNST when crossed to p53 mutant mice (Cichowski et al., 1999; Vogel et al., 1999). Chimeric mice partially composed of Nf1 homozygous (Nf1−/−) embryonic stem cells developed numerous neurofibromas resembling human plexiform neurofibromas, but not dermal neurofibromas (Cichowski et al., 1999). A breakthrough came from conditionally ablating Nf1 in the Schwann cell lineage, using Krox20-cre (Zhu et al., 2002), This promoter is expressed transiently in developing glial and neuronal cells in the boundary cap at E10.5 and developing peripheral nerve cells at E15.5. Boundary cap cells but not E15.5 nerve cells have stem cell properties (Aquino et al., 2006; Maro et al., 2004; Stemple and Anderson, 1993; Zorick et al., 1999). In the Nf1flox/flox; Krox20-cre model, while neurofibromas did not form, microscopic regions of nerve hyperplasia were noted. On the Nf1+/− background, all mice developed GEM-neurofibroma of the nerve roots and cranial nerves by 15 months of age (Zhu et al., 2002). Based on this data and because most NF1 patients have somatic cells heterozygous for NF1 mutation, it was proposed that Nf1+/− endothelial cells, fibroblasts, Schwann cells and/or mast cells are required for neurofibroma formation.
The existence of patients who are somatic mosaic for NF1 mutations, and the occurrence of neurofibromas in otherwise normal individuals suggested that at least in some cases a NF1+/− environment might not be required for neurofibroma formation. Here we evaluated another factor, developmental time, in neurofibroma formation. We report that inactivation of Nf1 at embryonic day 12.5 (E12.5) in developing glial cells in vitro elicits formation of colonies containing bi-potent precursors and in vivo recapitulates human neurofibroma formation in a wild type background.
Results
Loss of Nf1 in E12.5 glial cells promotes colony formation in vitro
To define the point(s) in peripheral glial cell lineage(s) at which loss of Nf1 might amplify a cell population we infected cells from mice with a targeted insertion of loxP sites flanking exon 31 of the mouse Nf1 gene with adenovirus-mediated Cre recombinase (adeno-Cre). All trunk peripheral glial cells are derived from the neural crest, a multi-potent cell population. Trunk neural crest cells develop into boundary cap cells in the region of the dorsal and ventral roots, satellite cells associated with neuronal cell bodies in the peripheral ganglia, and Schwann cell precursors and then Schwann cells in the nerve proper (Jessen and Mirsky, 2005; Maro et al., 2004). We effected Cre-recombinase mediated recombination of the floxed Nf1 gene in cultured cell preparations from E12.5 dorsal root ganglia (DRG) 16 hours after plating cells. We chose the dorsal root ganglia from mice, together with associated nerve roots and developing nerves, for these studies as they contain all classes of trunk peripheral glia. In these cultures (E12.5 + 1D.I.V.), we observed colonies in preparations from single embryos after 1 – 2 weeks in vitro (Figure 1A, B). In three independent experiments, an average of 58 colonies per 5×105 cells was obtained when cells were infected with adeno-Cre, while 8 colonies were observed when cells were infected with adeno-GFP; only cells infected with adeno-Cre could be replated and expanded for up to 5 passages in DMEM: F-12 (1:1) with N2 supplements medium (data not shown). In contrast, induced loss of Nf1 in neural tube derived neural crest cells (E8.5 + 1D.I.V.), did not elicit colonies (Figure 1A). Likewise, no colonies were identified when Nf1 flox/flox cells from E12.5 DRG were allowed to differentiate into Schwann cells (using a seven-day exposure to β heregulin) and then exposed to adenoviral-Cre recombinase (Figure 1A). At each developmental stage, control cultures infected with adeno-GFP showed 80–90% of cells infected by virus; in cultured post-migratory neural crest cells at the point of adeno-Cre infection cells were P75+, Lineage (Gfap, S100β, β3Tub) negative cells with the exception of rare SMA+ cells (fibroblasts); mixed E12.5 DRG cells are neurons, a few fibroblasts, and progenitors; Schwann cell cultures are 98–99% S100β+ Schwann cells (data not shown). Cre-mediated Nf1 recombination efficiency was confirmed by PCR (Figure 1C).
Figure 1. Acute loss of Nf1 in dorsal root ganglion cells at E12.5+1 results in colony formation in vitro.

(A). Quantification of colonies resulting from acute loss of Nf1 in E8.5 neural tube-derived crest cells, E12.5 dorsal root ganglion, (DRG) cells or differentiated Schwann cells (denoted E18.5) derived from Nf1 flox/flox mice cells. A few colonies were observed in the E12.5 DRG cells infected by adeno-GFP, but could not be expanded. (Mean ± SD). (B). Phase micrograph of expandable adherent colonies resulting from E12.5 DRG cells infected by adeno-Cre. (C). Adeno-Cre mediated recombination confirmed by PCR. N: Nf1 flox allele, R-Cre: recombined Nf1 allele. MOI = multiplicity of infection. Cells replated from colonies after E12.5 DRG-derived recombination (D) or adeno-GFP (E) immunostained with anti-Blbp and staining detected with a fluorescently tagged anti-rabbit antibody. (F, G). Differentiation of E12.5 DRG-derived colony-forming cells in vitro. E12.5 DRG-derived colonies were replated and cells cultured under gliogenic and smooth muscle/myofibroblast generating culture conditions for 12 days, immunostained with anti-MBP (F) or anti-SMA (G) antibodies and staining detected with appropriate second antibodies. Bars, B=50μm, D, E=26μm, F, G =15μm.
Staining of cells from E12.5-derived colonies demonstrated that they expressed Blbp (Figure 1D), a marker of the Schwann cell precursor and immature Schwann cell stages (Jessen and Mirsky, 2005). Cells present in nerve at the Schwann cell precursor stage can show multilineage differentiation in vitro (Joseph et al., 2004; Morrison et al., 1999; White et al., 2001). To test if the cells in colonies are multipotent, we cultured E12.5 colony forming cells under conditions that drive cells toward neuron, Schwann cell or fibroblast differentiation (Fernandes et al., 2004; Joseph et al., 2004). While no cells expressed lineage markers before treatment, twelve days later marker analysis by immunostaining indicated that >50% differentiated into either elongated glial cells (Myelin basic protein, Mbp+) in response to β-heregulin and forskolin, or large flat fibroblasts (SMA+) in response to TGFβ1 (Figure 1F, G). No β-tubulin+ neurons formed even after exposure to FGF2 and then neurotrophins. Thus, loss of Nf1 in dorsal root ganglion cells E12.5 + 1D.I.V. but not earlier or later caused colony formation, and cells within colonies could form fibroblasts and Schwann cells. Further analysis will be necessary to assess clonal relationships among derivatives.
DhhCre is expressed in boundary cap cells and Schwann cell precursors beginning at E12.5 in vivo
To test if loss of Nf1 post neural crest and before terminal glial cell differentiation might elicit a tumorigenic phenotype in vivo we chose the DhhCre mouse in which Cre recombinase is expressed from Desert Hedgehog (Dhh) regulatory sequences to effect recombination of the Nf1 flox/flox allele. This promoter has been reported to drive expression beginning at E12.5 in developing glial cells, but not neural crest cells, neurons, or CNS cells (Jaegle et al., 2003; Joseph et al., 2004). The DhhCre allele was also bred to a Nf1 flox/flox background EGFP reporter mouse in which the CMV-β actin promoter and loxP flanked CAT gene are upstream of the enhanced green fluorescent protein cassette (Nakamura et al., 2006). Hereafter, this allele is designated EGFP. To precisely define the onset of DhhCre expression in populations of peripheral glia, we bred Nf1 flox/flox background EGFP mice (Nf1 flox/flox; EGFP) to Nf1 flox/+; DhhCre transgenic mice. We isolated embryos from these crosses and viewed the spinal cord with nerve roots, DRGs and peripheral nerve directly under fluorescence optics or after staining cryosections with anti-GFP. No green cells (EGFP+) were detected in embryos at embryonic days 11.5 in early-developing boundary cap cells (Figure 2A). At day E12.5, EGFP+ cells were present in the dorsal and ventral root boundary cap zones, and proximal peripheral nerve, but not within the DRG (Figure 2B, K). In adult mice, EGFP+ cells were present in the DRG, peripheral nerve and nerve roots (Figure 2C, M). These studies were done in the Nf1 flox/flox background, so EGFP expression might have been altered due to Nf1 recombination. However, our data are completely consistent with a previous report that Dhh is not expressed in the neural crest, and is expressed beginning at E12.5 in the peripheral nerve (Jaegle et al., 2003).
Figure 2. The DhhCre driver elicits recombination beginning at E12.5 in boundary cap cells and embryonic Schwann cells.

Cryostat sections from Nf1 flox/flox; DhhCre; EGFP embryos dissected at E11.5 (A), E12.5 (B), and a section through spinal cord, nerve roots and peripheral nerve at postnatal day 165 (C). A – C, sections were stained with anti-EGFP antibody. B, arrows show position of EGFP+ dorsal root (BC-Dr) and ventral root (BC-Vr) boundary cap cells adjacent to the neural tube (Nt). The position of the largely unstained dorsal root ganglion (Drg) is marked, and some green cells are visible lateral to the DRG in the proximal nerve (PN). In D–J, Confocal analysis of adult DRG showing EGFP+ cells are S100β+ (D), Gfap-negative (G) and β(III)-Tubulin-negative (H). YZ view (E) and XZ view (F) of cell indicated by arrow in D. YZ view (I) and XZ view (J) of cell indicated by arrow in H. Note Gfap-positive (red) astrocytes in the spinal cord (G) and β(III)-Tubulin-positive (red) DRG neurons and spinal cord fibers (H). K – M are gross images. K, a developing spinal cord (E 12.5) with EGFP+ root cells is shown. L, a postnatal day 1 sciatic nerve is shown. M, strong expression is retained in P90 sciatic nerves. L and M, endoneurium containing Schwann cells is EGFP+ (double headed black arrow) and perineurium (adjacent to white arrows) is negative. N, DNA (50ng) isolated from designated tissues from a representative adult Nf1 flox/flox; DhhCre mouse was PCR amplified. An ethidium bromide stained gel demonstrates Nf1 recombination in sciatic nerve using the DhhCre driver with little recombination in other organs. FL/FL = Nfl flox allele. Bars: A–C, K–M=26μm, D–J=20μm.
A few cells in the dorsal root ganglion were also EGFP+. These cells were S100β+;Gfap- Schwann cells (Figure 2D–F). β-tubulin + neurons were EGFP-negative (Figure 2H–J). The perineurium surrounding the nerve was EGFP negative at postnatal day 1 (Figure 2L) and postnatal day 90 (Figure 2M). We also analyzed the DhhCre induced recombination efficiency in dissociated P1 sciatic nerve cells by FACS analysis. We found 19.9% of the total live cells were EGFP positive (data not shown). About 80% of cells in nerve are endoneurial Schwann cells, and about 10% are endoneurial fibroblasts (Asbury, 1967). Thus about 20–30% of the endoneurial cells (Dhh expressing) showed Cre-mediated recombination of the EGFP allele. Taken together the data demonstrate the specificity of the transgene for post neural crest developing glial cells and predict that DhhCre will effect recombination of Nf1 in glial cells at the embryonic Schwann cell stage in peripheral nerve and in boundary cap glia but not in satellite cells. We monitored Cre-induced Nf1 flox recombination by PCR. PCR DNA analysis of brain, spinal cord, lung, heart, liver, and spleen and sciatic nerve indicated that the peripheral nerve showed robust levels of recombination. The variable and low level of recombination in the other tissues is consistent with published data (Jaegle et al., 2003) and may indicate the presence of peripheral nerves in these tissues (Figure 2N).
Loss of Nf1 in Schwann cells at E12.5 is sufficient to develop both plexiform and dermal neurofibromas
To test whether DhhCre mediated loss of Nf1 could elicit neurofibroma formation we bred Nf1 flox/+; DhhCre mice to Nf1 flox/flox mice, and obtained Nf1 flox/flox; DhhCre mice in the expected Mendelian ratios. The life span of Nf1 flox/flox; DhhCre mice was greatly reduced. When Nf1 flox/flox; DhhCre mice (n=28) were aged using littermates as controls, we found that Nf1 flox/flox; DhhCre mice began to die at five and one-half months. All Nf1 flox/flox; DhhCre mice in the cohort required sacrifice by 13 months of age, while littermate controls remained healthy except that four mice were sacrificed with severe dermatitis; these mice were excluded from the control group. Kaplan-Meier analysis of survival data is shown in Figure 3A. Four – 8 weeks preceding death, Nf1 flox/flox; DhhCre mice became paralyzed in one or both hindlimbs. Nf1 flox/flox; DhhCre mice. Eight of 28 (28.6%) Nf1 flox/flox; DhhCre mice were paralyzed at the age of 6 months. Mice required sacrifice due to lethargy, weight loss, and/or dehydration secondary to paralysis that correlated with tumor formation (Table 1).
Figure 3. Nf1 flox/flox; DhhCre mice show early lethality and develop tumors.

(A) Kaplan-Meyer survival curve. Red, Nf1 flox/flox; DhhCre (n=28); blue, Nf1 flox/+; DhhCre (n=20); green, Nf1 flox/flox (n=9); brown, Nf1 flox/+ (n=8). To facilitate view of control mouse survival, blue green and brown lines are shown offset. (B–F) Gross dissections of tumors and areas of pigmentation in Nf1 flox/flox; DhhCre mice. In gross images B–E, a ruler showing 1mm markings is included. (B) Nf1 flox/flox (wild type) spinal cord from 9 month old mouse, with pairs of spinal roots attached. (C) A similar region from an Nf1 flox/flox; DhhCre mouse, showing three paraspinal GEM-neurofibromas (white arrows) and spinal cord compression. (D, E) Dermal GEM-neurofibromas (white arrows) in Nf1 flox/flox; DhhCre mice in anterior (D) and lateral views (E). (F) Pigmentation was often detected on the surface tumors or adjacent to tumor; a dramatic example is shown. White arrows point to black pigmentation. Bars=1mm.
Table 1. Tumor Phenotype of Nf1 flox/flox; DhhCre mice.
The table summarizes the sex; strain background (note that the FVB/N deriving from the DhhCre allele was re-derived onto CD1 [see text for details]); age at sacrifice; tumor grade; age at time of partial paralysis; months from partial paralysis to sacrifice; reason for sacrifice; at autopsy, gross presence of plexiform GEM-neurofibroma; at autopsy, gross presence of dermal GEM-neurofibroma; at autopsy, gross presence of pigmentation; at autopsy, location of plexiform GEM-neurofibroma.
| A | B | C | D | E | F | G | H | I | J | K | L | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Age at | Tumor | Paralysis first | Months to | Plexiform | Dermal | Plexiform neurofibroma | |||||
| 2 | ID# | Sex | Background | sacrifice | Grade | appears, months | sacrifice | Reason for Sacrifice | Neurofibromas | Neurofibromas | Pigmentation | location |
| 3 | NFC407 | F | 129/BL 6/FVBN | 9.2 | I* | 7.1 | 2.1 | paralysis | Y | N | N | Cervical |
| 4 | NFC412 | F | 129/BL 6/FVBN | 7.6 | I* | 4.8 | 2.8 | paralysis | Y | Y | N | Cervical |
| 5 | NFC421 | M | 129/BL 6/FVBN | 11.0 | I | 8.1 | 2.9 | paralysis+ dermatitis | Y | N | Y | Cervical |
| 6 | NFC422 | F | 129/BL 6/FVBN | 10.8 | I* | 8.2 | 2.6 | paralysis | Y | Y | Y | Cervical, thoracic |
| 7 | NFC423 | M | 129/BL 6/FVBN | 7.6 | I | 5.5 | 2.1 | found dead | Y | N | N | Cervical, thoracic |
| 8 | NFC425 | F | 129/BL 6/FVBN | 5.6 | I | 5.1 | 0.5 | paralysis | Y | Y | Y | Cervical, thoracic |
| 9 | NFC426 | M | 129/BL 6/FVBN | 11.0 | I | 7.1 | 3.9 | paralysis+ dermatitis | Y | Y | N | Cervical, thoracic |
| 10 | NFC430 | M | 129/BL 6/FVBN | 7.4 | I/II* | 4.7 | 2.7 | paralysis | Y | Y | Y | Cervical |
| 11 | NFC434 | F | 129/BL 6/FVBN | 8.2 | I* | 6.1 | 2.1 | paralysis | Y | N | N | Cervical, thoracic |
| 12 | NFC436 | F | 129/BL 6/FVBN | 9.2 | I* | 8.1 | 1.1 | paralysis | Y | N | N | Cervical, thoracic |
| 13 | NFC443 | F | 129/BL 6/FVBN | 9.2 | I | 7.7 | 1.5 | paralysis | Y | N | N | Thoracic |
| 14 | NFC446 | F | 129/BL 6/FVBN | 7.8 | I | 7.0 | 0.8 | paralysis | Y | N | N | Cervical, thoracic |
| 15 | NFC450 | M | 129/BL 6/FVBN | 9.4 | I | 7.0 | 2.4 | paralysis | Y | N | N | Cervical, thoracic, lumbar |
| 16 | NFC454 | F | 129/BL 6/FVBN | 7.8 | I | 6.2 | 1.6 | paralysis | Y | N | Y | Cervical, cauda equina |
| 17 | NFC459 | M | 129/BL 6/FVBN | 8.4 | I | 7.2 | 1.2 | paralysis | Y | N | N | Cervical |
| 18 | NFC470 | F | 129/BL 6/FVBN | 12.5 | I* | 10.6 | 1.9 | paralysis + abdominal tumor | Y | Y | N | Cervical, thoracic |
| 19 | NFC473 | M | 129/BL 6/FVBN | 5.4 | I* | 5.3 | 0.1 | paralysis | Y | Y | Y | Cervical, thoracic, cauda equina |
| 20 | NFC477 | M | 129/BL 6/FVBN | 9.8 | I* | 8.1 | 1.7 | paralysis | Y | N | N | Cervical, thoracic |
| 21 | NFC478 | F | 129/BL 6/FVBN | 8.2 | I | 6.7 | 1.5 | paralysis | Y | N | Y | Cervical, lumbar |
| 22 | NFC493 | M | 129/BL 6/FVBN | 12.5 | I | 11.0 | 1.5 | paralysis | Y | Y | Y | Cervical |
| 23 | NFC495 | F | 129/BL 6/FVBN | 7.1 | I* | 5.3 | 1.8 | paralysis | Y | N | N | Cervical, thoracic |
| 24 | NFC502 | M | 129/BL 6/FVBN | 12.6 | I* | 10.0 | 2.6 | paralysis | Y | Y | Y | Cervical, thoracic (also liver lymphoma) |
| 25 | NFC512 | M | 129/BL 6/FVBN | 11.3 | I* | 8.0 | 3.3 | paralysis+ dermatitis | Y | Y | N | Cervical |
| 26 | NFC516 | M | 129/BL 6/FVBN | 8.9 | I | 8.7 | 0.2 | paralysis+ dermatitis | Y | Y | N | Cervical |
| 27 | NFC519 | M | 129/BL 6/FVBN | 9.7 | I | 5.5 | 4.2 | lethargy + paralysis | Y | N | N | Cervical, thoracic |
| 28 | NFC528 | M | 129/BL 6/FVBN | 6.9 | I* | 5.5 | 1.4 | paralysis | Y | Y | Y | Cervical, thoracic |
| 29 | NFC534 | M | 129/BL 6/FVBN | 11.8 | I | 11.0 | 0.8 | skin ulcer + dermatitis | Y | Y | N | Cervical, thoracic |
| 30 | NFC536 | M | 129/BL 6/FVBN | 7.6 | I | 6.1 | 1.5 | paralysis | Y | N | Y | Cervical, thoracic |
Y = yes; N= no.
denotes S100β stain. All cases analyzed for S100β stain were positive for S100β staining.
We performed gross dissections of each Nf1 flox/flox; DhhCre mouse to determine whether tumors were present. The peripheral and cranial nerve trunks and nerve roots were all significantly enlarged in every animal. An exception was the optic nerve, in which DhhCre is not expressed because Schwann cells are not present (not shown). An example of nerve enlargement in spinal nerve roots is shown in Figure 3C.
We detected visible plexiform tumors at gross dissection of each Nf1 flox/flox; DhhCre mouse (n=28; Table 1). Although DhhCre is expressed in sertoli cells, we did not observe any tumors in testis (not shown). The plexiform tumor number ranged from three to 24 per mouse. Most large plexiform tumors were close to dorsal root ganglia, predominately at lower cervical or upper thoracic levels, accounting for paralysis. We found dermal neurofibromas in 13 of 28 (46.4%) of Nf1 flox/flox; DhhCre mice (Figure 3D, E), with older mice having increased numbers of dermal neurofibromas. Most dermal tumors were located under the dermal muscle layer in the back in the thoracic and lumbar areas. Specifically, in one five month old mouse one dermal tumor was found. At six months, one mouse had 3 dermal tumors. At 8 months, one mouse had >5 dermal tumors. At ten months, 3 mice each had numerous dermal tumors, and by >11 months, Seven of 8 showed numerous dermal tumors. Two tumors protruded through the epidermis. These tumors, as in NF1 patients, do not contribute to mortality (Table 1). We also found pigmentation overlying the dorsal aspect of the spinal cord or tumors in 11 of 28 (39.3%) Nf1 flox/flox; DhhCre mice (Figure 3F). Melanocytes and Schwann cells are both derived from the neural crest. The presence of pigmentation in these mice is consistent with Dhh-mediated loss of Nf1 in a common progenitor
We performed histological analysis on plexiform and dermal tumors and used published criteria to define grades of mouse peripheral nerve tumors (Stemmer-Rachamimov et al., 2004). Paraffin sections from >40 tumors were evaluated by H & E and S100β staining. Plexiform neurofibromas involved large nerve trunks, and showed classic neurofibroma histology (Grade 3) with a heterogeneous cell population containing S100β+ cells and S100β− cells (Figure 4A, B). The dermal tumors were also histologically classic neurofibroma grade 3, with numerous spindle shaped S100β+ cells (Figure 4C, D). Nf1 flox/flox; DhhCre mouse tumors contain abundant collagen (Figure 4E) and many mast cells (Figure 4F). Only one of the 40 tumors analyzed showed areas of hypercellularity that may correspond to early malignant transformation (grade 1/2) (Table 1).
Figure 4. Nf1 flox/flox; DhhCre mice: histological and electron microscopic analysis.

Paraffin embedded tissue sections from plexiform and dermal GEM-neurofibromas stained with hematoxylin and eosin (H&E) (A and C). Adjacent sections were stained with anti-S100β antibody visualized with DAB [brown] (B and D). (E) Plexiform neurofibroma sections stained with Masson’s trichrome showing abundant collagen (blue) in the tumor. (F) Toluidine blue staining showing metachromatic mast cell infiltration (black arrows). Electron micrographs of plexiform (G) and dermal GEM-neurofibromas (H); small black arrowheads point to non-myelinating Schwann cells that are apparently not wrapping multiple axons. White arrows in G and H indicate intact myelinated fibers. In I, a higher magnification electron micrograph shows abundant collagen deposition (C) and prominent Schwann cell cytoplasmic processes identified by continuous basal lamina (black arrows). Fibroblasts are identified by prominent intracellular vesicles, prominent endoplasmic reticulum and patchy basal lamina. (Bar: A–F, 20μm; G, H=10μm; I=1μm).
To better define the cells within these tumors, we performed ultrastructural analysis on dermal and plexiform lesions. As shown in Figure 4G – 4I, the tumors contained abundant collagen and prominent Schwann cell cytoplasmic processes as defined by continuous basal lamina. Remak bundles, groups of small axon diameter within membrane delineated cytoplasm of a non-myelinating Schwann cell, were disrupted, but myelinated fibers appeared normal. Mast cells were confirmed by presence of intracellular granules. Fibroblasts, identified by numerous intracellular vesicles and patchy basal lamina, were also present. The data demonstrate a close similarity between the lesions in Nf1 flox/flox; DhhCre mouse tumors and previously described human neurofibromas (Dickersin, 1987). These results indicated that loss of Nf1 in Dhh-expressing cell(s) causes neurofibroma formation.
To determine whether any cell types in addition to glial cells expressed Dhh at some time in their history, we first indirectly assessed the status of the Nf1 gene in multiple cell types in the Nf1 flox/flox; DhhCre; EGFP mouse sciatic nerves, by determining which cell types express EGFP. As shown in Figure 5, all of the EGFP positive cells are negative for the endothelial marker von Willebrand Factor (vWF) and for the mast cell marker FcE R3 in P80 Nf1 flox/flox; DhhCre; EGFP sciatic nerves. As shown in Figure 2, perineurial fibroblasts are EGFP-negative in P1 and P90 sciatic nerve, indicating that perineurial fibroblast cells are unlikely to show recombination of Nf1.
Figure 5. Only p75+ cells recombine Nf1 in Nf1 flox/flox; DhhCre mouse nerves and tumors.

EGFP expression (B, D) in sections from adult Nf1 flox/flox; DhhCre; EGFP sciatic nerves, cryo-sectioned and stained with anti-von Willebrand factor to mark endothelial cells (red, A), or anti IgE Fc receptor to mark mast cells (red, C). In B and D show merged images of EGFP fluorescence and immunostains. FACS sorted EGFP+ (E, F) and EGFP- (G, H) cytospun cells were immunostained with anti-p75 antibodies (E, G). All EGFP+ cells are p75+, but some EGFP-are p75+ while other are p75-negative cells (H, white arrows). FACS sorted EGFP+ cells from P1/P2 sciatic nerves (I–L) or tumor were stained with Blbp ( I, M, red) and S100β (J, N, green). Bars, A–P: 13 μm. FACS sorted cells from P1 sciatic nerve or tumor were analyzed for Cre mediated recombination of Nf1 by PCR (Q, R). Q, Endothelial cells (CD31+/CD45−; EC) or mast cells (c-kit+/IgE-FcR+; MC) from dissociated tumor cells did not recombine Nf1. DNA from cells from two Nf1 flox/flox; DhhCre; EGFP tumors (T1, T2) show recombination. Nf1fl allele (FL/FL), recombined Nf1 allele (R-Cre). R, postnatal day 1 EGFP+ cells (EGFP+) showed Nf1 recombination, as did tumor DNA (T1). Water is shown as a negative control.
To identify EGFP-expressing cells, we FACS sorted EGFP positive and negative cells from Nf1 flox/flox; DhhCre; EGFP mouse P1/P2 sciatic nerves, and from Nf1 flox/flox; DhhCre; EGFP neurofibromas. In the sorted tumor cells, all EGFP positive cells expressed p75 (79/79). Sixty- four percent of the EGFP negative cells (272/425) were also p75 positive. P75 marks neural crest derived Schwann cells and their progenitors in nerve, but also stains nerve axons and mast cells (Jippo et al., 1997; Obata et al., 2006). Endoneurial fibroblasts derive from the neural crest, and a shared glial-endoneurial fibroblast progenitor expresses Dhh (Joseph et al., 2004). A specific endoneurial fibroblast marker has not been described but endoneurial fibroblasts lose p75 after differentiation (Joseph et al., 2004). To more specifically identify the p75+;EGFP+ cells, FACS sorted EGFP+ cells were cytospun and stained for the Schwann cell marker S100β which marks immature and mature Schwann cells. In EGFP+ tumor cells, 94.5% (433/458) of the cells were S100β+. EGFP+ FACS sorted P1/P2 sciatic nerve cells were 98.1% (370/377) S100β. We also tested whether significant numbers of precursor/immature Schwann cells are present in the nerves or tumors of Nf1 flox/flox; DhhCre mice, by FACS sorting P1/P2 sciatic nerve cells or tumor cells and staining with anti-Blbp antibody, an early glial cell lineage marker. Interestingly, 47.5% and 53.3% of these sorted EGFP+ cells are Blbp+;S100β+ in P1/P2 (Figure 5I–L) and tumor cells (Figure 5M–P) respectively. Thus p75+; S100β cells, express EGFP. The expression of Blbp in many of the cells supports the idea that they are similar to immature Schwann cells as identified by Jessen and Mirksy (2005). Figure 7 shows the relative expression of these markers in normal cells.
Figure 7. Proposed model of neurofibroma initiation in the neural crest-derived glial lineage.

In the paraspinal region of the trunk, migrating neural crest (NC) cells (E8.5–9) give rise to boundary cap (BC) cells (E10.5–11) in the spinal roots and Schwann cell precursors (SCP, E12–13) in the peripheral nerve. The diagram shows a crest cell and general appearance of the nerve at subsequent developmental stages. Markers used in this report and expression over time are designated by horizontal boxes (P75, Dhh, Blpb, S100β, Gfap). We place the neurofibroma-initiating cell at the Schwann cell precursor/immature Schwann cell boundary.
Because EGFP expression is indirectly measures inactivation of the Nf1 gene, we also dissociated cells from neurofibromas from Nf1 flox/flox; DhhCre mice and performed FACS sorting to sort the endothelial cells (CD31+/CD45−) or mast cells (c-kit+/FcE R3 +) from the dissociated tumor cells. As a positive control we sorted EGFP positive cells (putative immature Schwann cells (Jessen and Mirsky, 2005)) from EGFP; Nf1 flox/flox; DhhCre mouse P1/P2 sciatic nerve cells. We carried out PCR reactions using primers for the non-recombined or recombined Nf1 alleles on these sorted cells. Only the P1/P2 nerve-derived EGFP+ cells showed Nf1 recombination (Figure 5R). Both endothelial cells and mast cells maintained Nf1 flox/flox (wild type) status (Figure 5Q). Because Schwann cells with Nf1 mutations have been reported to express c-kit (Ryan et al., 1994), we confirmed the results by PCR genotyping of bone marrow-derived mast cells from these mice. After 5 weeks of culture, >98% of the cells showed metachromatic granules characteristic of mast cells. These mast cells derived from bone marrow progenitors also maintained Nf1 flox/flox (wild type) status (data not shown).
Nf1 flox/flox; DhhCre mice exhibit increased Schwann cell proliferation in sciatic nerves and disruption of axon-glial interactions in peripheral nerves
Hyperproliferative non-neoplastic lesions have been described in various tumors and can provide insight into the biology of early tumor formation. We cut paraffin sections of nerves from Nf1 flox/flox; DhhCre mice. In histological section, these changes do not satisfy criteria for diagnosis of GEM-neurofibroma when these mice were six and one-half months old (Figure 6A, B). However, the enlarged nerves in Nf1 flox/flox; DhhCre are not normal, but show disorganization on histological section confirmed by electron microscopy, with loss of association between axons and non-myelin forming Schwann cells (Figure 6C, D). Specifically, the Remak bundles (bundles of small axon diameter within membrane delineated cytoplasm of a non-myelinating Schwann cell; arrows in Figure 6C, D) are well defined in normal nerves but not Nf1 flox/flox; DhhCre nerves. The disruption of axon-Schwann interaction in these nerves is similar to that observed in tumors (see Figure 4G–I).
Figure 6. Nf1 flox/flox; DhhCre mouse nerves show disrupted morphology and transiently increased Schwann cell proliferation.

Paraffin sections from 6.5 month old wild type (A) and Nf1 flox/flox; DhhCre (B) mouse sciatic nerves. Bars in A, B = 50μm. Electron micrograph from wild type (C) and Nf1 flox/flox; DhhCre (D) saphenous nerve. Black arrows in C indicate well organized wild type non-myelinated axons; those in D show disorganized non-myelinated axons. Bars in C, D = 10 microns. (E) Quantification of BrdU positive cells in Nf1 flox/flox; DhhCre P1, P15, and P60 sciatic nerves (white bars) and wild type littermate controls (black bars). Proliferation in the Nf1 flox/flox; DhhCre mouse nerve compared to wild type controls differs significantly in P1 sciatic nerves (p<0.01, n=3) and P15 sciatic nerves (p<0.001, n=3), but not in P60 sciatic nerves (p=0.17, n=3). Data analyzed by Student’s t-test are presented as mean values ± standard deviation. (F) Quantification of apoptotic cells in Nf1 flox/flox; DhhCre P15 sciatic nerves (white bars) and wild type littermate controls (black bars). There is no significant difference in TUNEL staining between Nf1 flox/flox; DhhCre mice and their littermates (p= 0.42, n=4, data analyzed by Student’s t-test are presented as mean values ± standard deviation. A representative image of double staining is shown in I (BrdU: red; P75: green). (G) FACS analysis of BrdU labeled P1/P2 Nf1 flox/flox; DhhCre sciatic nerve cells indicating BrdU+/EGFP+ and BrdU+/EGFP− cells (G, H). (J) Sorted BrdU+/EGFP+ cells are S100β positive (red).
To determine whether tumor formation is preceded by cell division in the Nf1 flox/flox; DhhCre model we injected mice prior to sacrifice with BrdU and analyzed tissue sections by staining with anti-BrdU and p75. Proliferation of cells was measured in P1, P15, and P60 sciatic nerves and GEM-plexiform neurofibromas. Proliferation of BrdU+;p75+ cells in tissue sections in the P1 and P15 Nf1 flox/flox; DhhCre mouse sciatic nerves was increased 3.2 fold and 4.5 fold compared to nerves from wild type littermates. Proliferation of BrdU+;p75+ cells in tissue sections in the P60 Nf1 flox/flox; DhhCre mouse sciatic nerves did not differ from normal (Figure 6E). In tissue sections from P15 nerve we did not detect Blbp expression (data not shown), indicating that most cells have matured into differentiated Schwann cells at this later stage. We also measured apoptosis in P15 mice sciatic nerves. There was no significant difference in TUNEL+ sciatic nerve cells from Nf1 flox/flox; DhhCre mice as compared to littermate controls (p= 0.42, n=4, Figure 6F).
To definitively determine if the BrdU+ cells are Schwann cells, we FACS sorted BrdU+;EGFP+ or BrdU+;EGFP− cells from Nf1 flox/flox; DhhCre; EGFP P1/P2 mouse sciatic nerves. We cytocentrifuged the cells and stained for S100β. All BrdU+/EGFP+ cells were S100β+, indicating these proliferating cells are Schwann cells (Figure 6G, I, J). BrdU+/EGFP− cells in P1/P2 sciatic nerve were also S100β+, suggesting affects of the abnormal nerve environment on surrounding cells. Alternatively, DhhCre may have elicited Nf1 but not EGFP recombination in these cells.
We also monitored BrdU incorporation in GEM-neurofibroma. Cell division varied greatly from region to region within neurofibromas, ranging from 1.4% to 29.9% of cells, even within the same tumor (Supplemental Figure 1). Thus tumor formation in this model is preceded by disruption of Remak bundles, and early but transient nerve Schwann cell proliferation. Proliferation resumes in the GEM-neurofibromas.
Discussion
Our results support the relevance of Nf1 inactivation in a Dhh expressing precursor cell to development in mice of manifestations characteristic of NF1 patients. Inactivation of the Nf1 gene in dorsal root ganglion-derived cells in vitro at embryonic day 12.5 + 1 results in colony formation, while inactivation of Nf1 in neural crest cells or in mature Schwann cells does not. In vivo, inactivation of the Nf1 gene by the DhhCre driver beginning at E12.5 elicits plexiform neurofibromas, dermal neurofibromas, and pigmentation.
Nf1 inactivation in the neural crest is apparently not required for neurofibroma formation. In vitro, colony formation of neural crest cells does not occur following acute loss of Nf1, and the DhhCre driver is not expressed in neural crest cells in vivo. Neural crest drivers Mpz-Cre (E9.5 – E10.5 in the region of the presumptive DRG), Pax3-Cre (E10.5 in dorsal neural tube and the region of the presumptive DRG), and Wnt1-Cre (E9.5 in migrating neural crest), also did not elicit neurofibroma formation in Nf1 flox/flox mice (Gitler et al., 2003).
What is the post-neural crest target cell in which Nf1 inactivation causes plexiform neurofibroma formation in vivo? The paraspinal region contains nerve roots, the dorsal root ganglion, and the proximal part of the peripheral nerve, and is the site of most plexiform GEM-neurofibromas in the Nf1 flox/flox; DhhCre model (Figure 7). The neural crest derived glial cells in this region are boundary cap cells and embryonic Schwann cells. The DRG also contains satellite cells derived from neural crest and boundary cap cells, which we tentatively exclude. Although cells within the DRG expressed DhhCre as assessed by EGFP expression, cells with the morphology of satellite cells (apposed to neuronal somata) did not. Rather, the few DRG cells expressing EGFP expressed the Schwann cell marker S100β, not expressed by satellite cells, and had the morphology of Schwann cells with linear processes apposed to nerve axons. However, until a specific satellite cell marker is available for mouse we cannot formally exclude this cell type as a target for DhhCre mediated Nf1 recombination.
A possible target cell population is boundary cap cells in the spinal roots of the mouse trunk, present by day E10.75. Boundary cap cells develop into dorsal root glial (Schwann-like) cells, some DRG neurons and satellite cells and proximal Schwann cells of peripheral nerve based on expression of Egr2/Krox20 (Maro et al., 2004), consistent with our finding of tumors in these locations. It is not yet known whether some distal nerve Schwann cells derive from boundary cap cells. Like Schwann cell progenitors, with neuregulin-induced differentiation in vitro boundary cap cells become Schwann cells (S100β +; Mbp+) capable of myelination (Aquino et al., 2006). The hypothesis that boundary cap cells are a target for tumorigenesis is consistent with the observation that plexiform neurofibromas form, albeit in an Nf1+/− genetic background, when the Krox20Cre driver is used to ablate Nf1 (Zhu et al., 2002). A detailed comparison of the Nf1 flox/flox; Krox20Cre and Nf1 flox/flox; DhhCre models is shown in Supplemental Table 1.
Embryonic and immature Schwann cells also express Dhh (Bitgood and McMahon, 1995; Jessen and Mirsky, 2005; Parmantier et al., 1999). While defined in sciatic nerve, similar cells are likely to exist in all peripheral and cranial nerves. The relevance of these cells to neurofibroma formation is consistent with the placement of some neurofibromas distal to the DRG in the Nf1 flox/flox; DhhCre model. Schwann cell precursors are present in mouse sciatic nerve beginning at E12.5 and have been referred to as neural crest stem cells because in vitro and when transplanted into avian host they can develop into neurons, Schwann cells and SMA+ fibroblasts (Morrison et al., 1999; White and Anderson, 1999; White et al., 2001). However, in vivo they are restricted to Schwann cell or endoneurial fibroblast lineages as shown by fate-mapping in vivo (Joseph et al., 2004). We use the term Schwann cell precursor here to distinguish them from BC cells and migrating neural crest cells. Supporting Schwann cell precursors as a target for tumorigenesis, in the Nf1 flox/flox; DhhCre model nerves containing Schwann cells derived from Schwann cell precursors are hypertrophied throughout their lengths, show elevated proliferation, and disrupted organization including mast cell recruitment and loss of axon-glial interaction. However, tumors do not form in nerves. The environment surrounding the DRG may facilitate tumor formation, or boundary cap cells may be the only cells with tumorigenic potential.
We defined the neurofibroma-initiating cell using markers of Schwann cell precursors and their progeny (Jessen and Mirsky, 2005). Markers for stages in boundary cap cell development have not been delineated. Our analysis places the neurofibroma initiating cell at the Schwann cell precursor/immature Schwann cell boundary based on a) tumor formation subsequent to DhhCre expression; b) Blpb, S100β, and Gfap expression concomitant with absence of β III-tubulin expression in EGFP-expressing cells in peripheral nerves; and c) the ability of E12.5+1 D.I.V cells in vitro to form colonies and the evidence that they are biopotent (Figure 7). While we cannot completely exclude the relevance of a progenitor with wide potential in the Nf1 flox/flox; DhhCre model, our inability to detect Nf1 recombination in vivo in p75-negative cells, which should include endoneurial fibroblasts (Joseph et al., 2004) is consistent with a more restricted progenitor (see below).
Neurofibromas formed under the skin in association with small nerves in 48% of Nf1 flox/flox; DhhCre mice. In humans cutaneous and subcutaneous neurofibromas, some of which are called dermal neurofibromas, form mainly beginning at puberty. In the Nf1 flox/flox; DhhCre mice visible dermal tumors developed in only adults. Another difference from human NF1 patients is that the mouse dermal tumors rarely raised the skin. This likely reflects the presence of the panniculus carnosus muscle under the epidermis in mouse but not human. We designate these Nf1 flox/flox; DhhCre tumors GEM-dermal neurofibromas to distinguish from GEM-plexiform neurofibromas, but recognize their position is not precisely identical to any one category of human neurofibroma.
The progenitor cell for dermal GEM-neurofibroma may be the hair follicle neural crest-derived cells with stem cell features, recently shown to express Dhh (Fernandes et al., 2004; Sieber-Blum et al., 2004; Wong et al., 2006). The normal role of these cells is postulated to be replenishing neural crest derived melanocytes in skin. We often observed melanocytes near tumors in the Nf1 flox/flox; DhhCre mice, and grafting E12.5 Nf1−/− DRG-derived cells into nerve generated pigmented melanocytes (Rizvi et al., 2002). Some data support the existence of a post neural crest bi-potential glial-melanocyte progenitor cell (Le Douarin and Dupin, 2003). A study of a patient who was somatic mosaic for NF1 mutation in whom café-au-lait macule melanocytes and neurofibroma Schwann cells shared identical biallelic NF1 mutations supports a role for such a progenitor in human NF1(Maertens et al., 2007).
Inactivation of Nf1 using DhhCre enables tumor formation in the absence of an Nf1+/− background. In a previous study plexiform neurofibromas were detected in an Nf1-driven mouse model, but only in the presence of an Nf1+/− genetic background (Zhu et al., 2002). In some but not all instances an Nf1+/− environment may contribute to neurofibroma formation. Another alternative, which we favor, is that because the Krox20-Cre driver is expressed in developing boundary cap cells beginning at E10.5 and is not expressed in embryonic Schwann cells (Maro et al., 2004) this driver does not cause Nf1 loss in a sufficiently large pool of glial cell progenitors, so that an Nf1+/− genetic background enhances the target cell pool. In either case Nf1+/− perineurial cells, Nf1+/− mast cells, and Nf1+/− endothelial cells are not always required for neurofibroma formation.
A model in which mice routinely develop GEM-neurofibroma allowed us to study the histology of nerves prior to tumor formation. We found that interactions between Schwann cells and axons in Remak bundles are dramatically perturbed. The finding that neurofibromas in NF1 patients more frequently form on sensory roots with many Remak bundles as compared to motor nerve roots is consistent with a pronounced effect on non-myelin forming Schwann cells. Furthermore, Schwann cell proliferation, normally very low in postnatal nerves is transiently increased in the first postnatal weeks in the Nf1 flox/flox; DhhCre mice. Transient proliferation was also reported in a mouse in which the EGFR was expressed in Schwann cells (Ling et al., 2005; Wu et al., 2006). Nerves from NF1 patients have not been analyzed, precluding direct comparison between the species. The relationship between proliferating cells in GEM-neurofibromas and proliferating cells within nerves also remains to be determined.
In conclusion, our results indicate that biallelic Nf1 mutations in post neural crest cells expressing Dhh can cause neurofibroma formation, and provide a model to study neurofibroma formation and test therapies.
Experimental Procedures
Animals
We housed mice in a temperature- and humidity-controlled vivarium on a 12-hour dark-light cycle with free access to food and water. The animal care and use committee of Cincinnati Children’s Hospital Medical Center approved all animal use. The Nf1 flox/flox mouse has been described previously (Zhu et al., 2002) and was on a mixed background 129/BL/6 background. The DhhCre transgenic mouse line was generated on the FVB/N background, re-derived and maintained on the CD1 (I129/BL/6) background for several generations (Jaegle et al., 2003), then maintained by breeding with C57BL/6. We bred DhhCre mice onto Nf1 flox/flox background to obtain the F1 generation (Nf1 flox/+; DhhCre/+); we bred F1 mice with Nf1 flox/flox mice to obtain Nf1 flox/flox; DhhCre mice. We bred a β-actin flox/stop/flox-EGFP mouse reporter line maintained on C57BL/6 (Nakamura et al., 2006) onto the Nf1 flox/flox background to obtain the EGFP− Nf1 flox/flox mice. We genotyped mice by PCR as described (Jaegle et al., 2003; Nakamura et al., 2006; Zhu et al., 2002). Littermates were used for controls. For analysis of embryos, we established timed pregnancies with the morning of a vaginal plug defined as E0.5.
Cell culture
We dissected E8.5 neural tubes from Nf1flox/flox embryos (Zhu et al., 2002), allowed neural crest cells to migrate from the neural tubes plated on poly-(L)-lysine (PLL) and fibronectin-coated or PLL and laminin coated dishes tissue culture dishes, in DMEM + 10% FBS and 1% pen/strep. After 24 hours, neural tubes were carefully removed (Williams et al., 2004). Cells remaining on the substrate were infected with adenovirus (see below) and medium changed to DMEM: F-12 (1:1) with N2 supplements (Invitrogen; Carlsbad, CA) and 50 μg/ml gentamycin. We established cultures from Nf1flox/flox embryos by dissociating cells from E12.5 DRG as described (Ratner et al., 2005) and plated cell suspensions on PLL and laminin coated dishes for 16 hours prior to adenoviral infection in DMEM: F-12 (1:1) with N2 supplements (Invitrogen) and 50 μg/ml gentamycin. To study Schwann cells, we exposed cells from E12.5 DRG to β-heregulin (10ng/ml) and forskolin (1μM) for 7 days in DMEM: F-12 (1:1) with N2 supplements (Invitrogen) and 50 μg/ml gentamycin, on PLL and laminin coated dishes, before infection with adenovirus (Kim et al., 1997). After infection, we monitored cultures from each time point for colony formation. We trypsinized and replated colonies that arose in E12.5 cultures.
Differentiation Assays
We passaged Adeno-Cre infected E12.5 DRG Nf1flox/flox cells which grew as colonies, plated on PLL and laminin, and exposed to cells in basal medium of DMEM + 10% FBS + 1% Pen/Strep, with lineage-driving growth factors (Fernandes et al., 2004; Stemple and Anderson, 1993; Wong et al., 2006). For neurogenesis, we cultured cells for 5 days with bFGF (20 ng/ml) followed by 7 days with BDNF, NT3, NGF (10 ng/ml each). Gliogenic conditions were β-heregulin and forskolin (5ng/ml and 2 μM, respectively) for 12 days. Smooth muscle/myofibroblast generating culture assays included TGF-β1 (1 ng/ml) for 12 days. Cells were then fixed and stained with rat anti-MBP (1:100 Serotec, Raleigh, NC), mouse anti-β3tubulin (1:200, Chemicon, Temecula, CA) or mouse anti-smooth muscle actin (SMA) (1:500, Sigma-Aldrich, St. Louis, MO) and appropriate secondary antibodies.
Adenovirus Infection
Cells were exposed to CsCl2-purified Cre adenovirus for 2 hours in DMEM + 1% FBS, then returned to normal growth medium. We infected cells with a range of concentrations of virus to generate 80 – 90% infected cells as assessed by expression of EGFP. For Schwann cells, a multiplicity of infection of 100 MOI generated EGFP+ cells and recombination. This concentration of virus is toxic to E12.5 cells and neural crest cells, which required MOI = 10. Cells in colonies were stained with anti-Blbp (1:1000).
Fluorescence-activated cell sorting (FACS)
We generated cell suspensions of GEM-neurofibroma by incubation in 0.5μg/ml collagenase (Worthington, Lakewood, NJ) and 2.5μg/ml dispase II (Roche) for 4 hours. After 2 washes in ice-cold PBS, we strained cells through a 70μm cell strainer (Becton-Dickinson; San Jose, CA). We incubated cells with anti-mouse CD45 (Becton-Dickinson) bound to allophycocyanin (APC), CD31 (Becton-Dickinson ) bound to phycoerythrin (PE), CD-117 (c-kit, Becton-Dickinson) bound to APC, and/or rat anti-mouse FcE RI (eBioscience, San Diego, CA) bound to PE on ice in a solution containing PBS/0.2%BSA for 30 minutes. After washing, we resuspended cells in PBS/0.2%BSA/2μg/mL 7-aminoactinomycin D (7-AAD, Invitrogen). We carried out isotipic controls containing irrelevant mouse IgG1-APC and mouse-IgG1-PE in parallel. Cells were acquired in a FACSVantage DiVa equipped with a 488 nm-Argon laser, a 633 nm-HeNe laser and a tunable Argon-UV laser (Becton-Dickinson). For EGFP tagged GEM-neurofibroma cells, we sorted the cells directly to identify EGFP high and negative cells. A total number of 5×106 cells were sorted in each experiment.
For BrdU FACS, P1/P2 Nf1 flox/flox; DhhCre; EGFP mice were injected (intraperitoneally) with bromodeoxyuridine (BrdU, Sigma-Aldrich) in water (50 mg BrdU per kg of body weight) three times at 2-hour intervals (total 6 hours) before sacrifice. Sciatic nerves were dissected and cells dissociated. We fixed, permeabilized, and stained cells with APC-conjugated anti-BrdU (1:50, BD Biosciences, San Jose, CA) according to the manual and FACS sorted BrdU+/EGFP+ or BrdU+/EGFP− cells as above.
Histology
Were perfused mice intracardially with 4% paraformaldehyde (w/v) in PBS. We dissected sciatic nerves, cryoprotected, and cut in blocks 12-μm sections for immunofluorescence staining (Ling et al., 2005; Wu et al., 2006). Sections were stained with anti-von Willebrand factor (vWF) (Chemicon), anti-IgE FcR 3 (CD23, Santa Cruz Biotechnology, Santa Cruz, CA), anti-EGFP antibodies (Invitrogen) overnight at 4°C followed by incubation with FITC or TRITC labeled appropriate secondary antibodies. We viewed sections with a fluorescence microscope (Carl Zeiss, Thornwood, NY) equipped with a digital imaging system. We embedded some tumor specimens in paraffin, cut. 6-μm paraffin sections and stained with hematoxylin and eosin, toluidine blue, or Masson’s trichrome, or with anti-S100β (Dako, Carpinteria, CA) and HRP-conjugated goat anti-rabbit followed by diaminobenzidine reaction to highlight Schwann cells.
Cell proliferation
We injected mice intraperitoneally with BrdU (Sigma-Aldrich) in water (50 mg BrdU per kg of body weight) three times at 2-hour intervals (total 6 hours) before intracardiac perfusion. Sciatic nerves or tumors were dissected, paraffin-embedded, and cut into 6-μm sections. Deparaffinized sections were treated with 2 mol/L HCl for 30 minutes at 37°C, blocked in PBS containing 0.15% Triton X-100 and 5% donkey serum and incubated with a rat anti-BrdU antibody (1:200; Zymed Laboratories Inc., South San Francisco, CA) and rabbit anti-p75 (1:500, Chemicon) overnight at 4°C followed by TRITC and FITC -conjugated secondary antibodies. We stained nuclei with 4, 6-diamidino-2-phenylindole (DAPI) (Sigma) for 5 minutes. We counted BrdU-labeled cells and blue nuclei in at least three cross sections made through each sciatic nerve. Data are presented as average numbers of BrdU-labeled cells per section.
Tunel assay
Tunel assay was performed according to Promega manual instruction by DeadEnd™ Fluorometric TUNEL System (Promega, San Luis, CA), on deparaffinized sections. Nuclei were stained with DAPI as described above. We counted TUNEL-positive cells and the DAPI nuclei in at least three cross sections made through each sciatic nerve. Data are presented as average numbers of TUNEL-positive cells per section.
Electron Microscopy
We perfused mice intracardially with Karnovsky’s fixation solution (3% paraformaldehyde and 3% glutaraldehyde in 0.1 mol/L phosphate buffer, pH 7.4 to 7.6). We dissected saphenous nerves, embedded, and evaluated them as described (Ling et al., 2005; Wu et al., 2006).
Significance
Neurofibromatosis type 1 (NF1) is an autosomal dominant inherited disease, affecting 1:3500 individuals worldwide. Nearly all (90%) of NF1 patients develop plexiform and/or dermal neurofibromas composed of axons, Schwann cells, fibroblasts, perineurial cells, endothelial cells and mast cells. Schwann cells are believed to be the primary pathogenic cells in neurofibromas because they show biallelic mutation at NF1. However, other cell types present in tumors, in the NF1+/− state, also show cell autonomous defects and can contribute to neurofibroma formation. Our data support an additional mechanism for tumor development, in which loss of Nf1 at the correct time in development facilitates tumor formation in a wild type environment.
Supplementary Material
Nf1flox/flox:DhhCre and Nf1flox/flox:Krox20-Cre mouse models were compared in strain background, promoter expression, mouse survival, environment, tumor type, location and size, and pigmentation.
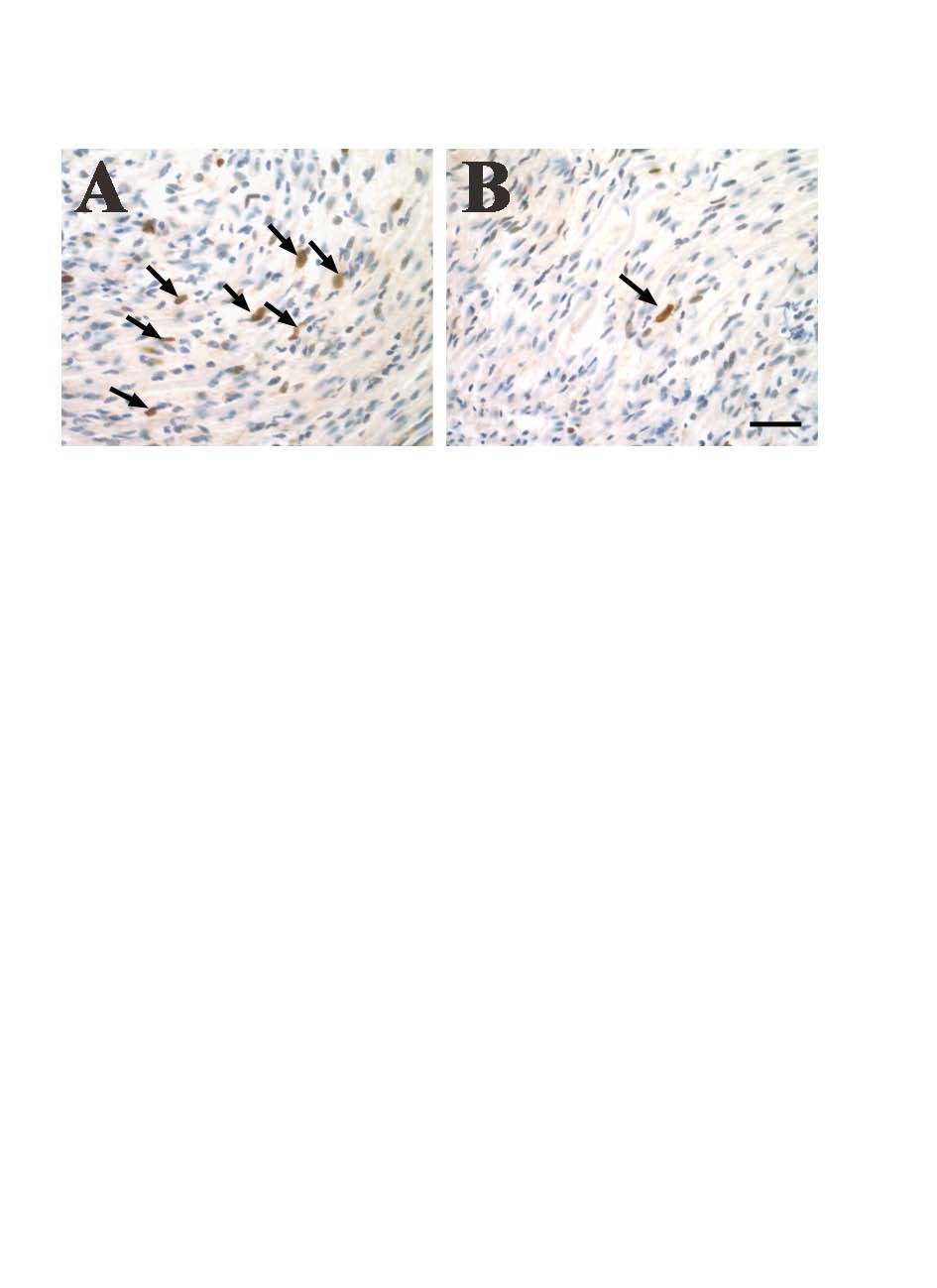
Cell division varied greatly from region to region within GEM-neurofibromas, ranging from 1.4% to 29.9% of BrdU+ cells (black arrow), even within the same tumor. Sections were count staining with hematoxylin (blue). Representative images of high (A) and low (B) proliferation are shown. Bars=50μm.
{kind=link}
Acknowledgments
We are very grateful to Dr. Luis F. Parada (University of Texas Southwestern) for providing Nf1 flox/flox mice. We also thank Dr. Melissa Colbert (Cincinnati Children’s Hospital Medical Center) for EGFP flox/stop/flox mice, Dr. Ting Xie (Stowers Institute) for re-deriving the DhhCre mice, Dr. Nathaniel Heintz (Rockefeller University) for the generous donations of anti-BLBP antibodies, and Ms. Karen Niehaus for assistance with mouse work. A grant from the National Institutes of Health supported this work (1R01 NS28840 to N.R.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aquino J, Hjerling-Leffler J, Koltzenburg M, Villar M, Ernfors P. In vitro and in vivo differentiation of boundary cap neural crest stem cells into mature Schwann cells. Exp Neurol. 2006;198:438–449. doi: 10.1016/j.expneurol.2005.12.015. [DOI] [PubMed] [Google Scholar]
- Asbury A. Schwann cell proliferation in developing mouse sciatic nerve. A radioautographic study. J Cell Biol. 1967;34:735–743. doi: 10.1083/jcb.34.3.735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitgood M, McMahon A. Hedgehog and Bmp genes are coexpressed at many diverse sites of cell-cell interaction in the mouse embryo. Dev Biol. 1995;172:126–138. doi: 10.1006/dbio.1995.0010. [DOI] [PubMed] [Google Scholar]
- Brannan C, Perkins A, Vogel K, Ratner N, Nordlund M, Reid S, Buchberg A, Jenkins N, Parada L, Copeland N. Targeted disruption of the neurofibromatosis type-1 gene leads to developmental abnormalities in heart and various neural crest-derived tissues. Genes Dev. 1994;8:1019–1029. doi: 10.1101/gad.8.9.1019. [DOI] [PubMed] [Google Scholar]
- Cichowski K, Jacks T. NF1 tumor suppressor function: narrowing the GAP. Cell. 2001;104:593–604. doi: 10.1016/s0092-8674(01)00245-8. [DOI] [PubMed] [Google Scholar]
- Cichowski K, Santiago S, Jardim M, Johnson B, Jacks T. Dynamic regulation of the Ras pathway via proteolysis of the NF1 tumor suppressor. Genes Dev. 2003;17:449–454. doi: 10.1101/gad.1054703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cichowski K, Shih TS, Schmitt E, Santiago S, Reilly K, McLaughlin ME, Bronson RT, Jacks T. Mouse models of tumor development in neurofibromatosis type 1. Science. 1999;286:2172–2176. doi: 10.1126/science.286.5447.2172. [DOI] [PubMed] [Google Scholar]
- Courtois-Cox S, Genther Williams S, Reczek E, Johnson B, McGillicuddy L, Johannessen C, Hollstein P, MacCollin M, Cichowski K. A negative feedback signaling network underlies oncogene-induced senescence. Cancer Cell. 2006;10:459–472. doi: 10.1016/j.ccr.2006.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeClue JE, Papageorge AG, Fletcher JA, Diehl SR, Ratner N, Vass WC, Lowy DR. Abnormal regulation of mammalian p21ras contributes to malignant tumor growth in von Recklinghausen (Type 1) neurofibromatosis. Cell. 1992;69:265–273. doi: 10.1016/0092-8674(92)90407-4. [DOI] [PubMed] [Google Scholar]
- Dickersin G. The electron microscopic spectrum of nerve sheath tumors. Ultrastruct Pathol. 1987;11:103–146. doi: 10.3109/01913128709048319. [DOI] [PubMed] [Google Scholar]
- Feigenbaum L, Fujita K, Collins F, Jay G. Repression of the NF1 gene by Tax may expain the development of neurofibromas in human T-lymphotropic virus type 1 transgenic mice. J Virol. 1996;70:3280–3285. doi: 10.1128/jvi.70.5.3280-3285.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes K, McKenzie I, Mill P, Smith K, Akhavan M, Barnabé-Heider F, Biernaskie J, Junek A, Kobayashi N, Toma J, et al. A dermal niche for multipotent adult skin-derived precursor cells. Nat Cell Biol. 2004;6:1082–1093. doi: 10.1038/ncb1181. [DOI] [PubMed] [Google Scholar]
- Friedman J. Epidemiology of neurofibromatosis type 1. Am J Med Genet. 1999;89:1–6. [PubMed] [Google Scholar]
- Gitler A, Zhu Y, Ismat F, Lu M, Yamauchi Y, Parada L, JAE Nf1 has an essential role in endothelial cells. Nat Genet. 2003;33:75–79. doi: 10.1038/ng1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guha A, Lau N, Huvar I, Gutman D, Provias J, Pawson T, Boss G. Ras-GTP levels are elevated in human NF1 peripheral nerve tumors. Oncogene. 1996;12:507–513. [PubMed] [Google Scholar]
- Huson S. Neurofibromatosis type 1: Historical perspective and introductory overview. In: Upadhyaya M, Cooper DN, editors. Neurofibromatosis type 1: from genotype to phenotype. BIOS Sci Pub Ltd; Oxfor, U K: 1998. pp. 1–13. [Google Scholar]
- Jacks T, Shih TS, Schmitt EM, Bronson RT, Bernards A, Weinberg RA. Tumor predisposition in mice heterozygous for a targeted mutation in NF1. Nature Genetics. 1994;7:353–361. doi: 10.1038/ng0794-353. [DOI] [PubMed] [Google Scholar]
- Jaegle M, Ghazvini M, Mandemakers W, Piirsoo M, Driegen S, Levavasseur F, Raghoenath S, Grosveld F, Meijer D. The POU proteins Brn-2 and Oct-6 share important functions in Schwann cell development. Genes Dev. 2003;17:1380–1391. doi: 10.1101/gad.258203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessen K, Mirsky R. The origin and development of glial cells in peripheral nerves. Nat Rev Neurosci. 2005;6:671–682. doi: 10.1038/nrn1746. [DOI] [PubMed] [Google Scholar]
- Jippo T, Morii E, Tsujino K, Tsujimura T, Lee Y, Kim D, Matsuda H, Kim H, Kitamura Y. Involvement of transcription factor encoded by the mouse mi locus (MITF) in expression of p75 receptor of nerve growth factor in cultured mast cells of mice. Blood. 1997;90:2601–2608. [PubMed] [Google Scholar]
- Joseph N, Mukouyama Y, Mosher J, Jaegle M, Crone S, Dormand E, Lee K, Meijer D, Anderson D, Morrison S. Neural crest stem cells undergo multilineage differentiation in developing peripheral nerves to generate endoneurial fibroblasts in addition to Schwann cells. Development. 2004;131:5599–5612. doi: 10.1242/dev.01429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HA, Ling B, Ratner N. Nf1-deficient mouse Schwann cells are angiogenic and invasive and can be induced to hyperproliferate: reversion of some phenotypes by an inhibitor of farnesyl protein transferase. Molecular and Cellular Biology. 1997;17:862–872. doi: 10.1128/mcb.17.2.862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HA, Rosenbaum T, Marchionni MA, Ratner N, DeClue JE. Schwann cells from neurofibromin deficient mice exhibit activation of p21ras, inhibition of cell proliferation and morphological changes. Oncogene. 1995;11:325–335. [PubMed] [Google Scholar]
- Le Douarin N, Dupin E. Multipotentiality of the neural crest. Curr Opin Genet Dev. 2003;13:529–536. doi: 10.1016/j.gde.2003.08.002. [DOI] [PubMed] [Google Scholar]
- Ling B, Wu J, Miller S, Monk K, Shamekh R, Rizvi T, Decourten-Myers G, Vogel K, DeClue J, Ratner N. Role for the epidermal growth factor receptor in neurofibromatosis-related peripheral nerve tumorigenesis. Cancer Cell. 2005;7:65–75. doi: 10.1016/j.ccr.2004.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maertens O, De Schepper S, Vandesompele J, Brems H, Heyns IJ, Anssens S, Speleman F, Legius E, Messiaen L. Molecular dissection of isolated disease features in mosaic neurofibromatosis type 1. Am J Hum Genet. 2007;81:243–251. doi: 10.1086/519562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maro G, Vermeren M, Voiculescu O, Melton L, Cohen J, Charnay P, Topilko P. Neural crest boundary cap cells constitute a source of neuronal and glial cells of the PNS. Nat Neurosci. 2004;7:930–938. doi: 10.1038/nn1299. [DOI] [PubMed] [Google Scholar]
- Morrison S, White P, Zock C, Anderson D. Prospective identification, isolation by flow cytometry, and in vivo self-renewal of multipotent mammalian neural crest stem cells. Cell. 1999;96:737–749. doi: 10.1016/s0092-8674(00)80583-8. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Colbert M, Robbins J. Neural crest cells retain multipotential characteristics in the developing valves and label the cardiac conduction system. Circ Res. 2006;98:1547–1554. doi: 10.1161/01.RES.0000227505.19472.69. [DOI] [PubMed] [Google Scholar]
- Obata K, Katsura H, Sakurai J, Kobayashi K, Yamanaka H, Dai Y, Fukuoka T, Noguchi K. Suppression of the p75 neurotrophin receptor in uninjured sensory neurons reduces neuropathic pain after nerve injury. J Neurosci. 2006;26:11974–11986. doi: 10.1523/JNEUROSCI.3188-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parmantier E, Lynn B, Lawson D, Turmaine M, Namini S, Chakrabarti L, McMahon A, Jessen K, Mirsky R. Schwann cell-derived Desert hedgehog controls the development of peripheral nerve sheaths. Neuron. 1999;23:713–724. doi: 10.1016/s0896-6273(01)80030-1. [DOI] [PubMed] [Google Scholar]
- Ratner N, Williams JP, Kordich JJ, Kim HA. Schwann cell preparation from single mouse embryos: analyses of neurofibromin function in Schwann cells. Methods Enzymol. 2005;407:22–33. doi: 10.1016/S0076-6879(05)07003-5. [DOI] [PubMed] [Google Scholar]
- Rizvi T, Huang Y, Sidani A, Atit R, Largaespada D, Boissy R, Ratner N. A novel cytokine pathway suppresses glial cell melanogenesis after injury to adult nerve. J Neurosci. 2002;22:9831–9840. doi: 10.1523/JNEUROSCI.22-22-09831.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan J, Klein K, Neuberger T, Leftwich J, Westin E, Kauma S, Fletcher J, DeVries G, Huff T. Role for the stem cell factor/KIT complex in Schwann cell neoplasia and mast cell proliferation associated with neurofibromatosis. J Neurosci Res. 1994;37:415–432. doi: 10.1002/jnr.490370314. [DOI] [PubMed] [Google Scholar]
- Saito H, Yoshida T, Yamazaki H, Suzuki N. Conditional N-rasG12V expression promotes manifestations of neurofibromatosis in a mouse model. Oncogene. 2007;26:4714–4719. doi: 10.1038/sj.onc.1210250. [DOI] [PubMed] [Google Scholar]
- Serra E, Puig S, Otero D, Gaona A, Kruyer H, Ars E, Estivill X, Lazaro C. Confirmation of a double-hit model for the NF1 gene in benign neurofibromas. Am J Hum Genet. 1997;61:512–519. doi: 10.1086/515504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheela S, Riccardi V, Ratner N. Angiogenic and invasive properties of neurofibroma Schwann cells. J Cell Biol. 1990;111:645–653. doi: 10.1083/jcb.111.2.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman L, Atit R, Rosenbaum T, Cox A, Ratner N. Single cell Ras-GTP analysis reveals altered Ras activity in a subpopulation of neurofibroma Schwann cells but not fibroblasts. J Biol Chem. 2000;275:30740–30745. doi: 10.1074/jbc.M001702200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieber-Blum M, Grim M, Hu Y, Szeder V. Pluripotent neural crest stem cells in the adult hair follicle. Dev Dyn. 2004;231:258–269. doi: 10.1002/dvdy.20129. [DOI] [PubMed] [Google Scholar]
- Stemmer-Rachamimov A, Louis D, Nielsen G, Antonescu C, Borowsky A, Bronson R, Burns D, Cervera P, McLaughlin M, Reifenberger G, Schmale M, et al. Comparative pathology of nerve sheath tumors in mouse models and humans. Cancer Res. 2004;64:3718–3724. doi: 10.1158/0008-5472.CAN-03-4079. [DOI] [PubMed] [Google Scholar]
- Stemple D, Anderson D. Lineage diversification of the neural crest: in vitro investigations. Dev Biol. 1993;159:12–23. doi: 10.1006/dbio.1993.1218. [DOI] [PubMed] [Google Scholar]
- Vogel KS, Klesse LJ, Velasco-Miguel S, Meyers K, Rushing EJ, Parada LF. Mouse tumor model for neurofibromatosis type 1. Science. 1999;286:2176–2179. doi: 10.1126/science.286.5447.2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White P, Anderson D. In vivo transplantation of mammalian neural crest cells into chick hosts reveals a new autonomic sublineage restriction. Development. 1999;126:4351–4363. doi: 10.1242/dev.126.19.4351. [DOI] [PubMed] [Google Scholar]
- White P, Morrison S, Orimoto K, Kubu C, Verdi J, Anderson D. Neural crest stem cells undergo cell-intrinsic developmental changes in sensitivity to instructive differentiation signals. Neuron. 2001;29:57–71. doi: 10.1016/s0896-6273(01)00180-5. [DOI] [PubMed] [Google Scholar]
- Williams S, Mear J, Liang H, Potter S, Aronow B, Colbert M. Large-scale reprogramming of cranial neural crest gene expression by retinoic acid exposure. Physiol Genomics. 2004;19:184–197. doi: 10.1152/physiolgenomics.00136.2004. [DOI] [PubMed] [Google Scholar]
- Wong C, Paratore C, Dours-Zimmermann M, Rochat A, Pietri T, Suter U, Zimmermann D, Dufour S, Thiery J, Meijer D, et al. Neural crest-derived cells with stem cell features can be traced back to multiple lineages in the adult skin. J Cell Biol. 2006;175:1005–1015. doi: 10.1083/jcb.200606062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Crimmins J, Monk K, Williams J, Fitzgerald M, Tedesco S, Ratner N. Perinatal epidermal growth factor receptor blockade prevents peripheral nerve disruption in a mouse model reminiscent of benign world health organization grade I neurofibroma. Am J Pathol. 2006;168:1686–1696. doi: 10.2353/ajpath.2006.050859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, Ingram D, Chen S, Hingtgen C, Ratner N, Monk K, Clegg T, White H, Mead L, Wenning M, et al. Neurofibromin-deficient Schwann cells secrete a potent migratory stimulus for Nf1+/− mast cells. J Clin Invest. 2003;112:1851–1861. doi: 10.1172/JCI19195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Ghosh P, Charnay P, Burns D, Parada L. Neurofibromas in NF1: Schwann cell origin and role of tumor environment. Science. 2002;296:920–922. doi: 10.1126/science.1068452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorick T, Syroid D, Brown A, Gridley T, Lemke G. Krox-20 controls SCIP expression, cell cycle exit and susceptibility to apoptosis in developing myelinating Schwann cells. Development. 1999;126:1397–1406. doi: 10.1242/dev.126.7.1397. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Nf1flox/flox:DhhCre and Nf1flox/flox:Krox20-Cre mouse models were compared in strain background, promoter expression, mouse survival, environment, tumor type, location and size, and pigmentation.
Cell division varied greatly from region to region within GEM-neurofibromas, ranging from 1.4% to 29.9% of BrdU+ cells (black arrow), even within the same tumor. Sections were count staining with hematoxylin (blue). Representative images of high (A) and low (B) proliferation are shown. Bars=50μm.