

Abstract
A boron-pendant ruthenium species forms a unique agostic methyl bridge between the boron and ruthenium atoms in the presence of a ligating solvent, acetonitrile. NMR inversion-recovery experiments enable measurement of the activation and equilibrium thermochemistry for formation of the agostic bridge. The mechanism for bridge formation involves displacement of an acetonitrile ligand; thus, this is a rare example of a case where an agostic C—H ligand competitively displaces another tight-binding ligand from a coordinatively saturated complex. Characterization of this complex gives unique insights into development of C—H activation catalysis based on this ligand-metal bifunctional motif.
Hydrocarbon functionalization is an important reaction class in organic synthesis and has a central role in applications ranging from utilization of hydrocarbon feedstocks to fine chemical synthesis.i In this area, our group is developing ligand-metal bifunctional catalysts that manipulate hydrides. Our general approach to C—H functionalization involves cooperative effects between a transition metal and a ligand-centered coordination-directing element. This is analogous to hydrogen bond-directed oxidation catalysts such as Noyori's (Ts-DPEN)(cym)RuCl systemsii and cyclopentadienone-ligated systems from Shvoiii and Caseyiv (figure 1A), except that a Lewis acid directing group (such as boron, figure 1B) does not require a protic functionality in the substrate. In this communication, we report the synthesis, X-ray structure, and unique thermochemistry of this unusually strong C—H agostic methyl bridge between boron and ruthenium (figure 1C). This agostic complex has structural homology to a potential transition state for boron-directed C—H cleavage (figure 1B).
Figure 1.

An Agostic Boron Methyl has Structural Homology to Coordination-Directed C—H Activation Reactions.
Our design of a boron-pendant ruthenium-based oxidation catalyst requires a ligand that is robust under mild conditions. Along these lines, we became interested in the dimethyldi(2-pyridyl)borate (3) ligand as a platform from which to construct a heterobimetallic boron-transition metal hydride abstraction system.v Platinum(II) and (IV) complexes of this ligand are known, and are stable to dioxygen at elevated temperatures.vi A platinum(II) complex of 3 shows no reactivity between the borate and the metal, but the platinum(IV) congener participates in slow methyl transfer from boron to platinum with the intermediacy of an agostic bridge.
Analogous to the platinum(IV), we observe that a boron methyl on 3 forms an agostic interaction to ruthenium when ligated to ruthenium(II). This occurs with expulsion of an acetonitrile ligand from the metal (scheme 1). Unlike the analogous platinum system, this is a very rapid equilibrium between 6 and 7. To our knowledge, this is the first characterization of an alkyl group bridging boron and ruthenium in this way. Moreover, formation of the agostic bond occurs in the presence of acetonitrile, thus defining a very rare example of ligand displacement from a coordinatively saturated metal by a C—H bond.vii In fact, hydrolysis of this bridging methyl affords a μ-OH complex that has reactivity analogous to the Shvo catalyst.viii
Scheme 1.

Synthesis of dimethyl(2-pyridyl)borate ligated ruthenium complexes.
The syntheses of agostic complex 7 and precatalyst 1 are outlined in scheme 1. The route initiates with the formation of chlororuthenium adduct 4 from 3v and 2. Treatment of 4 with AgOTf in acetonitrile-d3 in a J. Young NMR tube affects rapid formation of 5 (1H δ (B-Me) = +0.09, +0.04); solvent then displaces cymene in minutes at elevated temperature to give an equilibrating mixture of 6 and 7 (1:1 at 90 °C), which can be hydrolyzed to give 1 (1H δ (B-Me) = +0.29).
Spectral data for 7 features 1H resonances at +0.18 and -5.13 ppm (Δδ = 5.3 ppm). While the downfield shift is consistent with bonding only at boron, the peak at -5.13 ppm is more consistent with an agostic interaction. In an attempt to arrest the agostic interaction, solutions of 6 plus 7 were cooled to -40 °C in acetonitrile-d3 and -95 °C in dichloromethane-d2 respectively. The upfield signal remains a singlet in each case, which indicates rapid interconversion among the three hydrogens of the bridging methyl at these temperatures. A 25 °C HMBC spectrum of the equilibrating mixture elucidated the carbon chemical shifts of the boron-bound carbon atoms and illustrated that the upfield methyl group in 7 was bound both to boron (correlation to the other BMe 13C, 8 ppm) and ruthenium (correlation to acetonitrile ligands 13C, 125 ppm). Low 1JC-H values provide further evidence for an agostic interaction.ix Although 1JC-H values for the boron methyls of 6 and 7 could not be observed directly (see Supporting Information), each was measured in a 13C-coupled HSQC experiment: 1JC-H (agostic) = 100 Hz; 1JC-H (free) = 107 Hz in 7. For comparison, 6 has 1JC-H (free) = 109 Hz. Further comparison of spectral details for 6 and 7 is presented in the Supporting Information. The nearest examples of a metal-bound alkyl borate in the literature include a platinum(IV) casevi and a bis(pyrazolyl)diethylborate-ligated molybdenum(II) complex. x
Importantly, NMR data for 7 show that the lifetimes 6 and 7 are on the order of 1 s in solution, and [7] / [6] ∼ T-1. Nonetheless, low temperature vapor diffusion enabled crystallization of 7 in dichloromethane at low [acetonitrile]. Remarkably, these crystals enabled collection of an X-ray structure that clearly defines the atomic connectivity in 7 (figure 2). A direct comparison of X-ray structures of 1 and 7 illustrates the positions of the dipyridylborate ligand with an oxygen or agostic bridge (figure 2). The agostic C—H in 7 was positioned using the electron difference map. A Ru1-C1 bond distance of 2.45 Å in 7 is in contrast to the bond distance of 2.10 Å for Ru1-O1 in 1.
Figure 2.
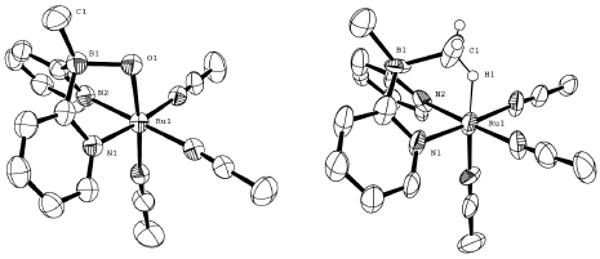
ORTEP diagrams of 1 (left), and 7 (right). Selected hydrogens and counter ion are omitted for clarity. Ellipsoids are drawn at the 50% level.xi For comparison sake, relative bond lengths (Å) for the agostic BMe-M interaction in 7 and [(3)PtIVMe3] are Ru1—H1, 1.72; B1—C1, 1.66; Ru1—C1, 2.53; Ru1—B1 2.89; and Pt1—H1, 2.02; B1—C1, 1.68; Pt1—C1, 2.76; Pt1—B1 3.08 respectively.
The thermochemistry for the equilibration of 6 and 7 was measured by NMR spectroscopy. This equilibrium has a linear van't Hoff plot from 20 to 80 °C with ΔH = 5.5(2) kcal/mol and ΔS = 20.1(5) eu. The conversion of 7 to 6 is conveniently studied by NMR magnetization transfer: at 85 °C, bridge cleavage has k-1 obs ∼ 1 s-1. The mechanism for conversion of 7 to 6 has kinetic order on [MeCN], which is consistent with a rapid pre-equilibrium followed by rate determining acetonitrile association or concerted displacement of the agostic bond from 7. The microscopic reverse of this reaction involves the very rareviib situation that an agostic bond displaces a ligand from an 18-electron metal center. Determination of second-order rate constants k-1 by inversion-recovery enables determination of the activation parameters for cleavage of the agostic interaction, ΔH‡ = 13.3(6) kcal/mol, and ΔS‡ = -27.5(43) eu, by Eyring analysis over a range of 41 °C.
Complex 1 shows catalytic reactivity in the oxidation of p-methoxyphenethylalcohol (scheme 2). Heating 10 mol% 1 with alcohol and 40 mol% t-BuOK in acetone resulted in > 95% yield of the corresponding ketone. The mechanism of this reaction is unknown.
Scheme 2.

Catalytic Reactivity of 1.
In summary, we report here the first example of an agostic bridge between boron and ruthenium atoms. The structure of this agostic bridge is established by a combination of NMR and X-ray diffraction methods. Cleavage of the bridge has kinetic order on [acetonitrile], which indicates that bridge dissociation is either a rapid pre-equilibrium or concerted displacement. Thus, this is a very rare situation in which an agostic interaction is in equilibrium with a tight-binding ligand. A hydrolyzed form of this ligand-metal bifunctional complex is a catalyst for transfer dehydrogenation of alcohols, although the mechanism of this reaction is not established. We are currently investigating the mechanism of this oxidation and optimizing the heterobifunctional motif for general hydride manipulation reactions.
Supplementary Material
Acknowledgments
We thank the University of Southern California, Loker Hydrocarbon Research Institute, Hydrocarbon Research Foundation, the Anton Burg Foundation, and the ACS Petroleum Research Fund (47987-G1) for research support and the NSF (DBI-0821671) and NIH (S10-RR25432) for NMR spectrometers. We are grateful to G. K. S. Prakash, T. Flood, A. Kershaw, S. Lynch, and C. Cares for valuable discussions and to T. Stewart and R. Haiges for assistance with X-ray crystallography. We thank Denver Guess for assistance with synthetic scale-up.
Footnotes
Supporting Information Available: Experimental procedures, kinetics data, graphical spectra, and crystallographic data (CIF) for compounds 1, 4, and 7. This materials is available free of charge via the Internet at http://pubs.acs.org/.
References
- i.(a) Deno NC, Peterson HJ, Saines GS. Chem Rev. 1960;60:7–14. [Google Scholar]; (b) Bäckvall JE. J Organomet Chem. 2002;652:105–111. [Google Scholar]; (c) Li Z, Bohle DS, Li CJ. Proc Natl Acad Sci (USA) 2006;103:8928–8933. doi: 10.1073/pnas.0601687103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ii.Noyori R, Sandoval CA, Muñiz K, Ohkuma T. Phil Trans R Soc A. 2005;363:901–912. doi: 10.1098/rsta.2004.1536. [DOI] [PubMed] [Google Scholar]
- iii.Conley BL, Pennington-Boggio M, Boz E, Williams TJ. Chem Rev. 2010;110 doi: 10.1021/cr9003133. in press. [DOI] [PubMed] [Google Scholar]
- iv.Casey CP, Guan H. J Am Chem Soc. 2009;131:2499–2507. doi: 10.1021/ja808683z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- v.Hodgkins TG, Powell DR. Inorg Chem. 1996;35:2140–2148. [Google Scholar]
- vi.(a) Khaskin E, Zavalij PY, Vedernikov AN. J Am Chem Soc. 2008;130:10088–10089. doi: 10.1021/ja804222c. [DOI] [PubMed] [Google Scholar]; (b) Khaskin E, Zavalij PY, Vedernikov AN. Angew Chem Int Ed. 2007;46:6309–6312. doi: 10.1002/anie.200701257. [DOI] [PubMed] [Google Scholar]; (c) Khaskin E, Zavalij PY, Vedernikov AN. J Am Chem Soc. 2006;128:13054–13055. doi: 10.1021/ja0643570. [DOI] [PubMed] [Google Scholar]
- vii.For a discussion of agostic bonding, see Brookhart M, Green MLH, Parkin G. Proc Natl Acad Sci (USA) 2007;104:6908–6914. doi: 10.1073/pnas.0610747104.Crabtree RH. Chem Rev. 1985;85:245–269.
- viii.For an example of hydroboration catalyzed by an Ru-B bifunctional species, see Koren-Selfridge L, Londino HN, Vellucci JK, Simmons BJ, Casey CP, Clark TB. Organometallics. 2009;28:2085–2090.
- ix.Brookhart M, Green ML, Wong LL. Prog Inorg Chem. 1988;36:1–124. [Google Scholar]
- x.Trofimenko S. J Am Chem Soc. 1968;90:4754–4755. [Google Scholar]
- xi.CCDC 738031, 738030, and 755444 contain supplementary crystallographic data for compounds 1, 4, and 7 respectively. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.