

Abstract
The major nonspecific porin of Pseudomonas aeruginosa, OprF, produces a large channel yet allows only a slow diffusion of various solutes. Here we provide an explanation of this apparent paradox. We first show, by introduction of tobacco etch virus protease cleavage site in the middle of OprF protein, that most of OprF population folds as a two-domain protein with an N-terminal β-barrel domain and a C-terminal periplasmic domain rich in α-helices. However, sedimentation of unilamellar proteoliposomes through an iso-osmotic gradient showed that only about 5% of the OprF population produced open channels. Gel filtration showed that the open channel conformers tended to occur in oligomeric associations. Because the open channel conformer is likely to fold as a single domain protein with a large β-barrel, we reasoned that residues near the C terminus may be exposed on cell surface in this conformer. Introduction of a cysteine residue at position 312 produced a functional mutant protein. By using bulky biotinylation reagents on intact cells, we showed that this cysteine residue was not exposed on cell surface in most of the OprF population. However, the minority OprF population that was biotinylated in such experiments was enriched for the conformer with pore-forming activity and had a 10-fold higher pore-forming specific activity than the bulk OprF population. Finally trypsin treatment, which preferentially cleaves the C-terminal domain of the two-domain conformer, did not affect the pore-forming activity of OprF nor did it digest the minority conformer whose residue 312 is exposed on cell surface.
Structure and functions of the now classical, trimeric bacterial porins, such as OmpF, PhoE, and OmpC of Escherichia coli, are well understood. Each monomer forms a β-barrel composed of 16 membrane-traversing β-strands, and the central channel in the barrel is narrowed by the infolding of an external loop, L3 (for reviews, see Refs. 1–4). The crystal structure of OmpF and PhoE shows that the channel is open (2), and measurement of permeability of the outer membrane in intact cells shows that at least a majority of the porin channel is open at any given time (5).
In contrast, much controversy still surrounds the properties of the major outer membrane protein OprF of Pseudomonas aeruginosa, first identified as the major porin of this species by reconstitution studies in 1979 (6). The specific pore-forming activity of this porin was very low in the reconstitution assay (7), and this was not due to inactivation of the protein during purification because the intact outer membrane of P. aeruginosa also showed permeability coefficients about 2 orders of magnitude lower than the E. coli outer membrane (8, 9). The difficulty in explaining this low specific activity was magnified by the paradoxical observation that the OprF channel is wider than the E. coli OmpF channel, which has a much higher specific activity (6, 7). To explain these observations in a simplistic manner, it was claimed that purified OprF has no porin activity (10) and that the outer membrane channels in P. aeruginosa are very narrow, consistent with their low permeability (10–12). The finding that OprF is a homolog of E. coli OmpA (13), which is not a classical trimeric porin, seemed to add further support to these claims. However, we have shown that OmpA also produces very slow permeation of solutes, yet its pore size is comparable to that of OmpF, both properties reminiscent of those of OprF (14). Furthermore careful repetition of the OprF purification procedure of Yoshihara and Nakae (10) has shown that the purified product has distinct pore-forming activity, that the OprF contributes the major nonspecific porin activity to the P. aeruginosa outer membrane, and that the channel is indeed significantly larger than that of OmpF (15). Finally Bellido et al. (16) have shown that in intact cells OprF channel has a large size. Nevertheless the question remained on how the porin with large channel size could produce such a low permeability, that is, catalyze such slow permeation of solutes. Furthermore the accepted folding pattern of the OprF homolog, OmpA, consisting of an N-terminal eight-stranded β-barrel and a C-terminal, α-helix-rich, periplasmic domain (17) has been confirmed by the x-ray crystallographic structure of the N-terminal domain (18), and the absence of sizable channel in this narrow barrel is a contradiction with the assumed porin function of this protein.
We have shown earlier (19) that OmpA protein population consists of two conformers, a majority (about 98%) containing “closed” channels and a minority (about 2%) containing open channels. The closed conformer presumably folds as a two-domain protein (17) with the N-terminal eight-stranded β-barrel and the C-terminal periplasmic globular domain. That most of the OprF protein also folds in this manner was suggested by our circular dichroism studies (20). However, another laboratory has shown evidence that the folding pattern of OprF is OmpF-like with up to 18, rather than eight, transmembrane β-strands (21–23). In this study, we showed that OprF, like OmpA, becomes folded into two distinct conformers, the abundant, closed channel conformer and the rare, open channel conformer, and that the latter tends to occur as oligomers at high protein concentrations. We further present data demonstrating that the closed conformer folds in two domains, the N-terminal eight-stranded β-barrel and the C-terminal periplasmic globular domain, whereas the minority, open conformer folds as a single domain protein with a larger number of transmembrane β-strands, similar to the folding pattern of OmpF.
Experimental Procedures
Bacterial Strains and Their Cultivation
P. aeruginosa PAO1 was used as the source of OprF. To assess the degree of possible contamination of other proteins, we also used oprF null mutant H636 (15), which has an “omega” insertion in the middle of the gene. The oprF gene was also cloned by PCR amplification and inserted between the BamHI and PstI sites of low copy number plasmid pHSG576 (24), producing plasmid pHSG-oprF, which was used as the starting material for the production of the other plasmids described below. The cloned DNA included a 108-bp sequence upstream of the ATG initiation codon, and it was sequenced to ascertain that there was no errors introduced by PCR. Bacteria were grown at 37 °C in Luria-Bertani medium containing 0.5% glucose (and 30 μg/ml chloramphenicol when needed for plasmid maintenance) with aeration by rotary shaking.
Cleavage of OprF into N-terminal and C-terminal Domains
Most of the OprF population appears to fold into a two-domain conformation with the N-terminal β-barrel domain and the C-terminal globular domain (see “Results”). Digestion of unmodified OprF by common proteases, however, results in the nearly complete digestion of the C-terminal domain and does not produce any information on the conformation of this domain in intact OprF. We therefore introduced a cleavage site for a highly specific protease in OprF. Inspection of OprF primary sequence and especially comparison with its homolog OmpA show that the N-terminal β-barrel of OprF is likely to end at Phe-160 followed by a short linker sequence rich in proline and alanine and finally by the C-terminal domain. Thus an 8-residue sequence specifically cleaved by tobacco etch virus protease (25), SENLYFQS, was introduced between residues 162 and 163 of OprF. In addition, for purification, a decahistidine tag was introduced at the N terminus of the mature sequence.
First, an upstream primer containing an EcoRI site and a downstream primer containing a PstI site as well as the sequence coding for the decahistidine tag were used to amplify the modified signal sequence of E. coli ompA gene. After restriction enzyme treatment, the amplicon was cloned in between the EcoRI and PstI sites of vector pBCKS(+) (Stratagene) whose multiple cloning sequence contains restriction sites EcoRI, PstI, BamHI, XbaI, and NotI in this order. Second, the sequence coding for the N-terminal half of the mature OprF (residues 1 through 162 of the mature sequence or residues 25 through 186 of the precursor OprF) was amplified with pHSG-oprF as the template by using an upstream primer containing a PstI site and a downstream primer containing a BamHI site as well as the sequence coding for the tobacco etch virus protease site (see above). The amplicon, after restriction enzyme treatment, was cloned into the recombinant plasmid described above that was cut with PstI and BamHI. Finally the oprF sequence coding for the C-terminal half, or amino acid residues 163 through 326 of the mature OprF, was PCR-amplified by using an upstream primer with a BamHI site and a downstream primer with a XbaI site, and the amplicon, after restriction enzyme treatment, was inserted into the recombinant plasmid obtained in the previous step cut with BamHI and XbaI.
The complete modified oprF gene was sequenced to ascertain the absence of any PCR-induced alterations. It was excised from the pBCKS(+)-based construct with EcoRI and NotI and was inserted into vector pKY9790 (obtained from K. Yoshida) cut with the same two enzymes. This is a 5.1-kb plasmid with the pBR322 origin, chloramphenicol marker, lacI gene, and ptac promoter. Use of a gene that was cut with EcoRI at its upstream end places the Shine-Dalgarno sequence and the ptac promoter at optimal distances from the initiation codon. The recombinant plasmid, pKY-oprFTev, was transformed into a recA E. coli strain, BLR (Novagen). Overnight culture of this strain (10 ml) was diluted into 1 liter of fresh LB broth containing 30 μg/ml chloramphenicol, and the suspension was incubated at 37 °C with aeration until the A600 reached 0.6. At this time, 0.1 mm isopropyl 1-thio-β-d-galactopyranoside was added to initiate protein expression, and the culture was incubated at 30 °C for 3 h more. Crude envelope fraction was prepared by French pressure cell disruption, and it was then extracted with 0.12% octyl polyoxyethylene in 20 mm Tris-HCl, pH 8.0 to remove cytoplasmic membrane proteins. Outer membrane proteins were solubilized with a solution containing 3% octyl polyoxyethylene, 0.5 m NaCl, and 20 mm Tris-HCl, pH 8.0. Decahistidine-tagged OprF with the internal cleavage site was purified by using nickel-nitrilotriacetic acid Superflow column (Qiagen). Two hundred micrograms of the purified OprF was treated with 10 units of AcTEV protease (an enhanced form of tobacco etch virus protease, Invitrogen) in a total volume of 0.5 ml at room temperature overnight, and the mixture was loaded onto a nickel column, as above, to separate the C-terminal fragment from the decahistidine-tagged N-terminal fragment. The flow-through fraction containing the C-terminal fragment was concentrated.
CD Spectra
CD spectra were recorded on an Aviv model 62DS circular dichroism spectrometer (Aviv Associates, Lakewood, NJ). All spectra were corrected for light scattering by subtracting spectra of samples not containing protein.
Preparation of Unmodified OprF Protein
OprF protein was purified from the outer membrane fraction of P. aeruginosa strain PAO1 by following essentially the procedure published earlier (20). In brief contaminating proteins (mostly from the cytoplasmic membrane) were first removed from the crude outer membrane fraction with dilute non-ionic detergents, and then the membranes were extracted with 68 mm octyl β-d-glucoside in the presence of 5 mm EDTA and 10 mm Tris-HCl buffer, pH 8.0. The extract was fractionated by ion-exchange chromatography on a Macro-Prep 50Q column (10 × 50 mm) (Bio-Rad) in 50 mm Tris-Cl, pH 8.0, 1 mm EDTA, 0.3% (w/v) dodecyl β-d-maltoside (buffer A) and with a gradient of NaCl. The second peak containing mostly OprF and a small amount of OprG was combined from three successive runs, and the combined sample was fractionated on the same column with a shallower NaCl gradient, and the main peak was finally purified by gel filtration through a 1.5 × 90-cm column of Toyo Pearl HW-50F. Elution was by gravity using a buffer containing 20 mm Tris-HCl, pH 8.0, 1 mm dithiothreitol, 3 mm NaN3, 0.4 m NaCl, 10% (v/v) glycerol, and 0.1% (w/v) dodecyl β-d-maltoside. The OprF was eluted in the first large peak, which slightly overlapped with the second peak containing OprG. To assess the degree of contamimnation by other pore-forming proteins, the crude outer membrane fraction of the mutant H636 was treated in exactly the same manner.
Functional Fractionation of OprF
These experiments were carried out as described for OmpA (19). Purified OprF was mixed with the extensively sonicated egg phosphatidylcholine, octyl β-d-glucoside was added to a final concentration of 1.1%, and unilamellar, OprF-containing proteoliposomes were produced by dilution of this solution. The amount of OprF was chosen so that each vesicle would contain less than about 10 molecules of this protein. The uniform size distribution of these vesicles was ascertained in an earlier study (19). The vesicles, made in a buffer containing 0.3 m urea, 10 mm KCl, 10 mm HEPES-NaOH, pH 7.0, and 3 mm NaN3, were sedimented through a linear gradient that contained 0.3 m urea at the top and a mixture of 0.15 m urea and 0.15 m sucrose at the bottom in the same KCl-HEPES-NaN3 buffer at 300,000 × g for 16 h in a Beckman SW65 swinging bucket rotor at 10 °C. Fractions were collected, and the lipids in proteoliposomes were quantitated by the total phosphate assay (26). They were also analyzed for protein composition by SDS-PAGE followed by silver staining (27). Vesicles were also collected and used for the production of multilamellar vesicles for use in the osmotic swelling assay as described earlier for OmpA (19).
High Resolution Size Fractionation of OprF
OprF samples were fractionated by gel filtration with a 1.5 × 90-cm column of high resolution medium with narrow particle size distribution (Toyo Pearl HW-50F, Tosoh Biosep, Montgomeryville, PA). The buffer used contained 0.1% (w/v) dodecyl maltoside, 0.4 m NaCl, 20 mm Tris-HCl, pH 8.0, 1 mm DTT, and 3 mm NaN3. Fractions of 0.5 ml each were collected and analyzed for protein with A280 or BCA protein assay reagent (Pierce) and for pore-forming activity with the liposome swelling assay described below.
Determination of Pore-forming Activity
Pore-forming activity was routinely assayed by the osmotic swelling rates of proteoliposomes containing different amounts of OprF (15). Proteoliposomes were made in 15% Dextran T-40 (Pharmacia Corporation). As a routine measure of pore-forming activity, swelling rates in iso-osmotic l-arabinose were used because influx of this small, pentose sugar was rapid, and therefore the swelling rates could be determined more accurately. The pore size was inferred from the dependence of swelling rates on the size of the permeating solutes as described earlier (15).
Channel Reconstitution into Planar Lipid Bilayers
The channel-forming activity of open channel-enriched, unfractionated, and open channel-depleted OprF fractions has been probed with bilayer lipid membrane technique (see also Ref. 28). For multichannel experiments reported here, “solvent-free” bilayer lipid membranes were formed on a 100-μm aperture in the 15-μm-thick Teflon film that separated two compartments as described in Ref. 28. The channel measurements were carried out at room temperature of 23 ± 2 °C in planar diphytanoyl phosphatidylcholine lipid membrane; the aqueous phase of the membrane chamber contained 1 m KCl in 5 mm Tris at pH 7.42. OprF was added from 0.6–2 mg/ml stock solutions. In contrast, for the single channel experiments in Ref. 28 we used OprF stock solutions of 30 μg/ml concentration.
Labeling of Intact Cells and Outer Membrane Preparations with Biotin-Maleimide
A mutant OprF protein, whose Ala-312 (the numbering is that of the mature protein) was changed into cysteine, was used for this purpose. Site-directed mutagenesis was achieved by the PCR method (29) starting from pHSG576-oprF. Briefly an upstream primer containing the BamHI site and a downstream primer covering amino acid residue 312 and containing the desired mutant sequence were used in the first step, and the amplicon was used as the upstream megaprimer together with the downstream primer outside the coding sequence in the second PCR. The purified product was digested with BamHI and was ligated into pHSG576 to yield plasmid pHSG-A312C. The PCR-amplified segment was sequenced to confirm the absence of unwanted mutations.
E. coli DH5α containing the plasmid pHSG-FA312C was grown at 37 °C in LB broth containing 30 μg/ml chloramphenicol up to the midexponential phase (A600 = 1.0). Cells from the four sets of 20-ml cultures, grown in parallel, were collected by centrifugation, and two sets each were used either for intact cell labeling or for preparation of the cell envelope fractions. The sets for intact cell labeling were washed twice with 20 ml of M63 medium (0.1 m potassium phosphate buffer, pH 7.4, 15 mm (NH4)2SO4, 1 mm MgSO4), and one set was treated with 2% (v/v) 2-mercaptoethanol for 30 min at room temperature followed by washing in M63 medium. Cells of the other sets were resuspended each in 0.5 ml of 20 mm Hepes/NaOH buffer, pH 8.0, and were disintegrated by using a Gallenkamp Soniprep 150 sonicator. After the removal of unbroken cells by centrifugation at 5,000 rpm for 10 min, the envelope fractions were sedimented by centrifugation at 150,000 rpm for 15 min in a Beckman TA ultracentrifuge and were resuspended in M63 medium. One set of the envelope fractions was incubated with 2% (v/v) mercaptoethanol for 30 min at room temperature and was then washed three times with M63 medium by ultracentrifugation as above.
These four samples were labeled with 0.1 mm biotin-maleimide (N-biotinoyl-N′-(6-maleimidohexanoyl)hydrazide (Sigma)) for 5 min, the reaction was stopped by the addition of 1% (v/v) 2-mercaptoethanol, and the samples were washed twice by centrifugation in M63 medium. All samples were resuspended in 0.1 ml of lysis buffer (4% SDS, 10% 2-mercaptoethanol, 20% glycerol, 0.01% bromphenol blue in 0.125 m Tris-HCl, pH 6.8) and were solubilized by heating for 5 min in a boiling water bath. One-tenth aliquots of the samples were separated by SDS-PAGE, and the proteins were transferred onto a nitrocellulose membrane by electrophoretic blotting. The membrane was blocked for 1 h with 5% dry milk in TNT (20 mm Tris-HCl, pH 7.5, 0.5 m NaCl, and 0.05% Tween 80) followed by treatment for 30 min with horseradish peroxidase-conjugated streptavidin (Pierce) (0.06 μg in TNT containing 1% (w/v) dry milk). After washing the membrane three times with TNT, the biotinylated proteins were visualized with chemiluminescence detection (Renaissance Western blot ECL kit, DuPont).
Purification of the OprF Conformer with Its C-terminal Domain Exposed on Cell Surface
N-[6-(Biotinamido)hexyl]-3′-(2′-pyridyldithio)propionamide (biotin-HPDP2; Pierce), a cleavable biotinylation reagent, was used for this purpose. E. coli DH5α containing pHSG-OprFA312C was grown in 1 liter of LB broth containing 30 μg/ml chloramphenicol at 37 °C. Cells harvested by centrifugation were washed once with M63 medium and were resuspended in 20 ml of M63 medium. Cells were labeled with 0.1 mm biotin-HPDP for 5 min at room temperature and were then immediately centrifuged. Cells were washed quickly three times with M63 medium, and crude cell envelope fraction was prepared by French press treatment (14,000 p.s.i., repeated three times). Biotinylated OprFA312C was trapped onto an immobilized monomeric avidin column (Pierce) and was eluted by reducing the disulfide linkage in the biotin linker with a solution containing 10 mm 2-mercaptoethanol and 0.1% dodecyl maltoside in phosphate-buffered saline. As a control, labeling was done on crude envelope fraction, and the OprFA312C proteins that were biotinylated under these conditions were isolated by the same procedure as above.
Trypsin Digestion Kinetics of OprF
Intact cells of E. coli DH5α containing pHSG-OprFA312C were labeled with biotin-maleimide as described above. Cells were washed three times with M63 medium. Crude membrane fraction was isolated by French pressure cell disruption (see above), and the inner membrane proteins were removed by extraction with 1% Sarkosyl. The outer membrane fraction (0.4 mg) was then incubated at 37 °C with trypsin (20 μg) in a total volume of 0.15 ml. Digestion was stopped by the addition of 0.1 mm phenylmethylsulfonyl fluoride, and the samples were analyzed by SDS-PAGE after solubilization in the sample buffer for 5 min at 100 °C. The time course of digestion of total OprF population was determined by the scanning of the OprF band in the Coomassie Blue-stained gel, whereas that of the surface-exposed conformer of OprF was assayed by treating the blot of the gel with peroxidase-conjugated streptavidin as described above.
Results
Conformation of the Majority of OprF Population
E. coli OmpA has long been believed to fold into a two-domain conformation (17) with its N-terminal half producing an eight-stranded β-barrel and its C-terminal half folding into a globular periplasmic domain that interacts with the underlying peptidoglycan (30, 31). In fact, the eight-stranded β-barrel structure was confirmed by the crystallization of C-terminally truncated OmpA derivative (18). In contrast, P. aeruginosa OprF, a homolog of OmpA, has been described usually as a single domain protein that forms a 16-stranded β-barrel (21–23) just like the classical trimeric porin OmpF. Although this concept was challenged by our study of the secondary structures in OprF (20), direct proof for the OmpA-like, two-domain structure is lacking.
We wanted to isolate the two putative domains, the N-terminal β-barrel domain and the C-terminal globular domain, by cleaving the intact protein in the middle. However, treatment of OprF with general proteases such as trypsin or proteinase K results in the complete digestion of the C-terminal region. In this study, we introduced a peptide sequence coding for a cleavage site for a more specific protease, tobacco etch virus protease, between residues 162 and 163 of OprF within the region corresponding to the hinge between the two domains in OmpA (see “Experimental Procedures”). We also introduced a decahistidine tag at the N terminus of the protein. OprF, when produced in E. coli, is known to fold correctly and to become inserted in the outer membrane (22, 23, 32). The modified OprF was expressed in E. coli; it was found nearly exclusively in the outer membrane as long as its expression took place at 30 °C (results not shown). Cleavage with tobacco etch virus protease of the purified protein produced two fragments of about equal intensity on SDS-PAGE (Fig. 1A, left panel). These fragments were separated by a Ni2+ affinity column (Fig. 1A, right panel). Surprisingly the larger fragment, corresponding to the N-terminal half (187 residues, molecular weight 20,909 by electrospray ionization mass spectrometry) and containing the decahistidine tag added to the N terminus of the mature OprF sequence, turned out to correspond to the faster migrating band in Fig. 1A, right panel, lane 3. In SDS-PAGE, β-barrel proteins are often known to migrate faster than expected from their molecular weights (20). A smaller, C-terminal fragment (165 residues, molecular weight 17,598 by electrospray ionization mass spectrometry), not retained by the nickel column (Fig. 1A, right panel, lane 1), corresponded to the slower migrating band of the left panel. Because this fraction contained a few minor contaminants, it was purified further by gel filtration. The far-UV CD spectra clearly showed that the N-terminal fragment has essentially an all β-strand conformation with its single minimum at 217 nm (Fig. 1B) (33), whereas the C-terminal fragment has a conformation that is rich in α-helix with the characteristic negative peaks at about 207 and 223 nm (Fig. 1B) (33). This result is consistent with the notion that at least the majority of OprF folds as a two-domain protein, just like OmpA, as predicted earlier (20). (Although the N-terminal fragment eluted with imidazole contained small amounts of larger fragments, this was presumably due to the cleavage by contaminating protease(s) during the overnight treatment.)
FIGURE 1. Cleavage of His-tagged OprF-Tev into N-terminal and C-terminal domains by tobacco etch virus protease.

A (left panel) shows the time course of cleavage by SDS-PAGE analysis. The OprF protein modified by the insertion of the tobacco etch virus protease cleavage site in the middle and the addition of the decahistidine tag at the N terminus was cleaved as described under “Experimental Procedures.” Portions were taken at points indicated (ON, overnight) and analyzed by SDS-PAGE after heating at 100°C in the sample buffer followed by Coomassie Blue staining. The N-terminal and C-terminal fragments are indicated as N and C (see text). After overnight treatment with the protease, the sample was applied to the Ni2+ affinity column (right panel). Lanes 1, 2, and 3 show the flow-through fraction, the fraction obtained by washing with the wash buffer, and the eluate with 0.25 m imidazole, respectively. Samples were analyzed by SDS-PAGE as in the left panel. In both panels, the rightmost lane shows the molecular weight standards. B shows the far-UV CD spectra of the fragments isolated after overnight digestion. The N-terminal domain (●) shows a characteristic β-strand-rich spectrum with the single minimum occurring around 218 nm. In contrast, the C-terminal domain (▲) shows a spectrum suggesting the abundance of α-helices with characteristic double minima around 208 and 223 nm. The mean residue ellipticity values are in units of degrees times square centimeters divided by decimoles.
Purification of the Unmodified OprF
OprF protein used in many of the experiments here and also in the accompanying paper (28) was purified from the outer membrane fraction of P. aeruginosa by anion-exchange chromatography followed by gel filtration (see “Experimental Procedures”). The protein elution pattern in the anion-exchange step is shown in Fig. 2. Much of OprF appeared in peak 3, but this peak also contained many other proteins. We therefore collected peak 2, which contained mostly OprF contaminated with a small amount of what appeared to be OprG. After a second ion-exchange chromatography with a shallower gradient, the OprF was purified by gel filtration on a Toyo Pearl HW-50F column. The final preparation had a purity, by SDS-polyacrylamide electrophoresis, of about 97% (Fig. 3).
FIGURE 2. Purification of OprF by anion-exchange chromatography.
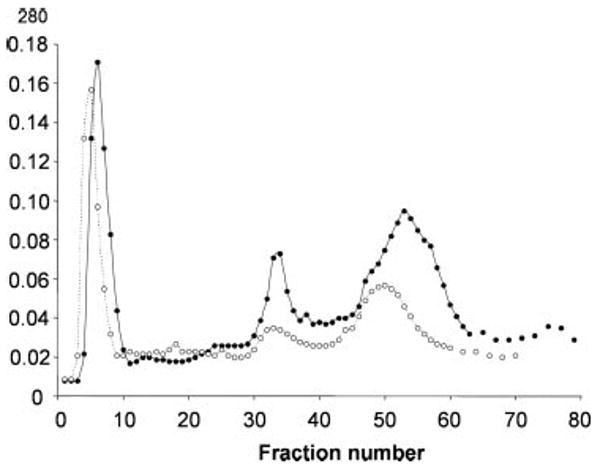
Crude outer membranes were prepared (20) from the wild-type strain PAO1 or its OprF-deficient mutant, H636, grown in LB broth. Octyl β-d-glucoside extracts of these fractions (2 mg of protein from each strain in each run) were loaded onto an anion-exchange column, Macro-Prep 50Q (10 × 50 mm), equilibrated with 50 mm Tris-Cl, pH 8.0, 1 mm EDTA, 0.3% dodecyl β-d-maltoside (buffer A), and the proteins were eluted, at a flow rate of 1 ml/min, with the following linear gradients of NaCl; 0–20 min, 0 m; 20–60 min, 0–0.2 m; and 60–80 min, 0.2–0.5 m. Fractions were collected every minute. SDS-PAGE showed that the major constituent of the first peak was OprE with small amounts of OprH and OprG, that the second peak contained mostly OprF and small amounts of OprG, and that the third peak contained a complex mixture of the proteins. The total amount of OprF present was larger in the third peak than in the second peak, but we retained the second peak because OprF was much less contaminated in that fraction. Filled circles and empty circles show absorbances of fractions from PAO1 and H636, respectively.
FIGURE 3. Purity of the OprF protein.
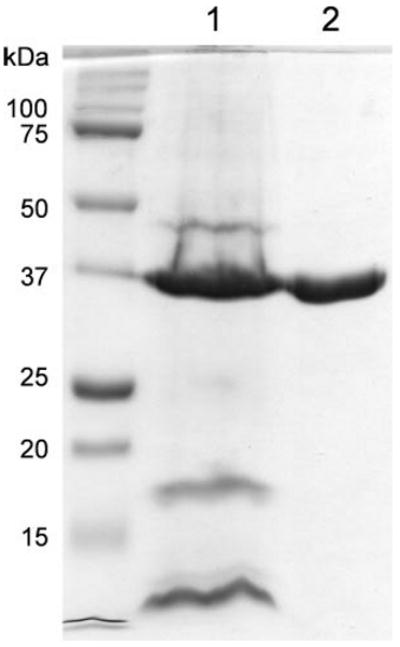
The starting material of the OprF purification (the crude outer membrane fraction) (lane 1) and the final OprF protein after fractionation on ion-exchange column and gel filtration (see “Experimental Procedures”) (lane 2) were separated by SDS-PAGE and stained with Coomassie Blue. Lane 1 contained molecular weight standards. The doublet pattern of OprF (lane 2) is characteristic of this protein and is caused partly by the difficulty in denaturing the N-terminal β-barrel structure (20) and perhaps also by the partial reduction of the two disulfide bonds present in OprF (see the legend for Fig. 5). We applied 5 μg of OprF preparation to show the absence of contaminating proteins. Densitometric scanning of gels to which less OprF was applied showed that this preparation of OprF was at least 97% pure.
Functional Fractionation of OprF Population
OprF, the major porin of P. aeruginosa (6), allows only a very slow penetration of solutes both in intact cells and in reconstituted proteoliposomes (7–9, 15). With OmpA of E. coli, we have previously shown that its low permeability was due to the small fraction of open channel conformers (19). We used the same functional fractionation technique to see whether the same explanation applies to the low permeability of OprF.
The OprF protein, purified from P. aeruginosa outer membrane as described above, was reconstituted into unilamellar liposomes with the octyl glucoside dilution method of Racker et al. (34) as described in detail in Ref. 19. The proteoliposomes originally contained 0.3 m urea inside, and they were centrifuged in an iso-osmotic gradient of urea/sucrose so that the sucrose concentration linearly increased from 0 m at the top to 0.15 m at the bottom and the urea concentration decreased from 0.3 to 0.15 m. Centrifugation is expected to separate vesicles that contain no open channels, which stay close to the top because sucrose cannot enter the vesicles, from those that contain open channels that become heavier because of the exchange of internal urea with sucrose outside. Fig. 4 shows the result of a typical experiment. As seen in Fig. 4A, the control vesicles containing no OprF remained entirely on top. In contrast, the OprF-containing vesicles were separated into two populations, the one remaining at the top (representing 59% of the entire vesicle population on the basis of phospholipid assay) and the one that sedimented close to the bottom (representing 41%) (Fig. 4B). Vesicles of both fractions contained OprF, but no other protein, when examined by SDS-PAGE followed by silver staining (not shown). Thus the channels in the sedimented vesicles were not due to contaminating proteins. We can calculate the fraction of open channel conformers by using Poisson statistics as follows (see also Ref. 19). If the average number of open OprF conformer per vesicle is n, the fraction of vesicles not containing any open OprF is predicted to be exp(−n). Because the fraction of vesicles remaining on top was 0.59 in Fig. 4B, n is −ln(0.59) = 0.528. In this particular experiment, 10 molecules of OprF, on average, were present in each vesicle. Thus the fraction of open channel conformer among the OprF population was 0.528/10 = 0.0528 or 5.28%. This experiment was repeated three more times, and the calculated fraction of open conformers was 4.2, 5.0, and 5.0%. The open and closed conformations of the OprF in vesicles sedimenting close to the bottom and remaining near the top, respectively, appeared to be stable because the OprF protein from sedimented vesicles, but not the OprF from vesicles remaining on top, produced multilamellar vesicles swelling rapidly in arabinose in the osmotic swelling assay (results not shown).
FIGURE 4. Functional fractionation of OprF population.

Unilamellar liposomes containing no protein (A) or 10 OprF molecules on average per vesicle (B) were centrifuged in an iso-osmotic gradient of urea and sucrose as described under “Experimental Procedures,” and fractions were collected from the bottom. Vesicles were quantitated by assaying for total phospholipid by the total phosphorus assay (26). Vesicles with open pores can take up sucrose and thus sediment through the gradient and appear in bottom fractions 1 and 2 in B.
Open Conformers Tend to Be Oligomeric
With OprF as well as OmpA, we found that the leading edges of the protein peaks eluted from a gel filtration column had a much higher pore-forming specific activity than the bulk of the peak (this was reported in a preliminary form in 1997 (35)). We tried such a fractionation in 1983 using Sephacryl S-200 but saw similar pore-forming specific activity across the OprF peak (7). In this study, we used a matrix with much more uniform size, Toyo Pearl HW-50F, and this resulted in a clear demonstration that the leading edge had a higher specific activity. Fig. 5A shows that the leading edge fraction had a specific activity of pore formation (as measured by reconstitution into proteoliposomes followed by osmotic swelling in iso-osmotic l-arabinose) more than 3 times higher than the peak fraction for OprF protein. In repeated experiments, this observation was confirmed, and pore-forming specific activity even 10 times higher than that of the bulk OprF was encountered at the leading edge of the peak. This was not due to the contamination of a different porin because the preparation obtained from the outer membrane of an OprF deletion strain, H636, by the same purification procedure produced no significant pore-forming activity (Fig. 5A). Interestingly SDS-PAGE of unheated leading edge fractions showed higher molecular weight protein bands (Fig. 5B, left half), which collapsed into the OprF (monomer) band upon heating (Fig. 5B, right half). The presence of such oligomeric complexes at the leading edge, showing higher pore-forming activity, is consistent with the hypothesis that the open conformers tend to exist as oligomers. The higher channel-forming activity of the leading edge fractions was also confirmed in numerous experiments with the planar bilayer reconstitution approach (Fig. 6) (for details, see Ref. 28).
FIGURE 5. Fractionation of P. aeruginosa OprF by gel filtration on Toyo Pearl HW-50F.

A, OprF preparation from P. aeruginosa PAO1 was fractionated on a high resolution column in the presence of 0.1% dodecyl maltoside and 0.4 m NaCl as described under “Experimental Procedures.” The leading edge of the OprF peak (seen from protein concentration in mg/ml (diamonds)) was seen to show a much higher specific poreforming activity (determined by the swelling rate of proteoliposomes upon dilution into iso-osmotic l-arabinose in the unit of ΔA400/min/10 μg (triangles)) than the bulk of the peak. The total pore-forming activity per 10 μl of each fraction (in ΔA400/min) is also shown (squares). The fact that the pore-forming activity of the leading edge is not due to other contaminating porin(s) is seen by the essentially undetectable pore-forming activity seen after the identical treatment of the fractions obtained from the outer membrane of an OprF-deleted strain, H636 (inverted triangles). B, portions (10 μl, after 5-fold dilution in sample buffer) of each fraction were separated by SDS-PAGE before or after heating at 100 °C in SDS-containing sample buffer of the samples, and the gel was stained with silver. Fraction 1 (the leading edge) is seen to contain significant portions of OprF behaving as oligomers (left panel, some of the bands are highlighted with arrowheads), which collapsed into a monomeric form after heat denaturation (right). The bands highlighted with arrowheads roughly correspond, in mobility, to 150, 70, and 55 kDa, respectively. Although several closely located bands are seen in most lanes, this is apparently due to the partial reduction of the two, closely spaced disulfide bonds in mercaptoethanol-containing running buffer as matrix-assisted laser desorption ionization time-of-flight analysis shows only OprF in each band.
FIGURE 6. Channel-forming activity of fractionated OprF assessed by planar bilayer assay.

Unfractionated, open channel-enriched (from the leading edge of gel filtration peak similar to that shown in Fig. 5), and open channel-depleted (from the trailing edge of gel filtration peak) preparations of the OprF were examined by planar bilayer reconstitution under the conditions described under “Experimental Procedures” and in the accompanying paper (28). It is seen (upper track) that at 0.5 μg/ml concentration of the open channel-enriched sample in the membrane-bathing solution, OprF induces ionic permeability developing in time. This permeability corresponds to a multichannel system where large amplitude steps probably represent insertion of OprF aggregates (see Ref. 28 for details). The unfractionated OprF sample was inactive up to 5 μg/ml protein concentration (middle track). None of our experiments showed any channel activity for the open channel-depleted OprF fraction (lower track).
Fractionation similar to that shown on Fig. 5 was repeated several times and always produced open form-enriched, leading edge fraction as well as the open form-depleted, trailing edge fraction. In one such trial, the unfractionated OprF protein showed specific pore-forming activity of 4.7 ± 0.2 A400/min/mg (n = 6) with l-arabinose as the permeating solute, and the open form-enriched and open form-depleted fractions showed specific activity of 24 ± 5 (n = 6) and 0.7 ± 0.1 (n = 6), respectively. If the open conformers represent 5% of the total OprF protein in the unfractionated sample, as described above, then 100% pure open conformers should show a specific activity of 4.7/0.05 = 94, and the specific activity of the open form-enriched fraction suggests that it contains (24/94) × 100 = 25% open conformers. These two fractions were examined by electrospray ionization mass spectrometry and exhibited main peaks corresponding to 35,241 and 35,242 m/e units. If the leading edge fraction contained 25% open conformers, these are likely to have been detected as a separate peak if there were covalent modifications in such conformers. We therefore conclude that there is probably no difference in the primary structure between the open and closed conformers.
We tried to produce, by repeated gel filtration, a preparation that is predominantly composed of open conformers. Although a single run of gel filtration leads to a preparation in which the estimated fraction of the open conformer was as high as 25% as described above, we were not able to get preparations of pure open conformers in this way. Repeated gel filtration runs decreased the total amount of proteins, and at low protein concentrations the oligomers tended to dissociate into monomers as the oligomeric form was apparently not completely stable. On the other hand, attempts to start purification from very large amounts of OprF resulted in poor resolution presumably because of the high viscosity of the sample.
Open Conformers Are Trypsin-resistant
OprF purified from P. aeruginosa PAO1 without the use of denaturing detergents was treated with trypsin at the weight ratio of 200:1. After 1 h at 37 °C, most of the OprF was converted into a smaller fragment of about 23 kDa (Fig. 7). This is a behavior similar to that found with the majority conformer of OmpA in which trypsin completely digests the C-terminal domain, leaving behind the trypsin-resistant N-terminal β-barrel domain (36). However, the pore-forming activity of this trypsin-treated OprF preparation was hardly diminished as seen in Table 1, although the N-terminal domain of OprF does not allow the permeation of even a small sugar, l-arabinose, as described in the accompanying paper (28). Furthermore there was no difference on the apparent pore size between the untreated and trypsin-treated preparations as seen with the effect of solute size on permeation rate. Thus N-acetylglucosamine (221 daltons) and sucrose (342 daltons) penetrated the untreated OprF channel at rates corresponding to 34 and 12% of the rate for l-arabinose (150 daltons), and N-acetylglucosamine and sucrose diffused through the trypsin-treated OprF at relative rates of 27 and 12%, respectively. These results indicate that the pore-forming fraction is not the majority conformer, which is digested away by trypsin, but a trypsin-resistant species.
FIGURE 7. Trypsin sensitivity of OprF.

OprF isolated from P. aeruginosa PAO1 was treated with trypsin (weight ratio, 200:1) at 37° C in a buffer containing 10 mm Tris-HCl, pH 8.0, 0.5 m NaCl, 1 mm EDTA, and 0.1% (w/v) dodecyl β-d-maltoside. The final concentration of OprF was 1.0 mg/ml. At the indicated times, portions of the samples were mixed with equal volumes of sample buffer, and the mixture was heated at 100 °C for 10 min. Portions were analyzed by SDS-PAGE, and the gel was stained with Coomassie Blue. Portions of the sample were also used for proteoliposome reconstitution for assay of permeability; these results are shown in Table 1.
TABLE 1. Pore-forming activity of OprF preparation before and after overnight trypsinization.
OprF purified from P. aeruginosa PAO1 was treated with trypsin as described in the legend for Fig. 5 at 37 °C for 1 h. The preparations were used immediately for reconstitution of proteoliposomes for the assay of pore-forming activity.
| Amount of OprF in proteoliposome | Proteoliposome swelling rate with l-arabinose | |
|---|---|---|
| Before trypsinization | After trypsinization | |
| μg | ΔA400/min | |
| 5 | 0.064 | 0.071 |
| 10 | 0.102 | 0.148 |
The open channel conformers, however, are apparently not completely trypsin-resistant, and after prolonged trypsin treatment, the pore-forming activity became diminished. This explains why we could not isolate pure populations of open conformers by the simple trypsinization of bulk OprF preparation.
C-terminal Domain of OprF Majority Conformer Is Not Exposed on Cell Surface
A cysteine residue was introduced by site-directed mutagenesis at a position 15 residues from the C terminus. This mutant porin (A312C) had pore-forming activity in the same range as the wild-type OprF and thus appeared to fold normally. However, in the experiments described below, its correct insertion into the outer membrane was a crucial issue. Thus E. coli DH5α cells containing no plasmid, pHSG-oprF, or pHSG-A312C were fractionated, and the outer membrane fraction was analyzed by SDS-PAGE. Coomassie Blue staining clearly showed that OprF was present at a level slightly lower than that of OmpA in the outer membrane regardless of the presence or absence of A312C mutation (Fig. 8). OprF was not present at a significant level in the cytoplasmic membrane fraction or cytosolic fraction (not shown).
FIGURE 8. Both unaltered OprF and OprFA312C are present in comparable amounts in the outer membrane of DH5α cells.
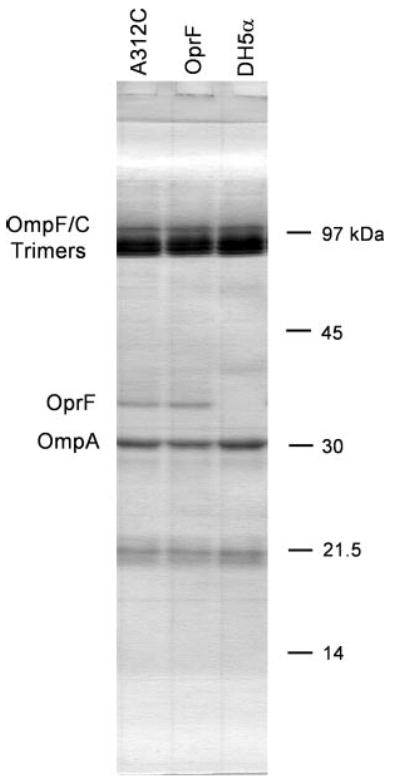
DH5α strain containing no plasmid, pHSG-oprF, or pHSG-A312C was grown in LB broth, and the cells were disrupted by French pressure cell treatment. Outer membrane fractions obtained from equal number of cells were separated by SDS-PAGE, and the gel was stained with Coomassie Blue.
E. coli DH5α cells expressing OprF A312C from the plasmid pHSG-A312C were labeled with a large (452-dalton) biotinylation reagent, biotin-maleimide (N-biotinoyl-N′-(6-maleimidohexanoyl)hydrazide), for 5 min at room temperature. Because the biotinylation reagent is both large and quite lipophilic, it is expected to permeate through the narrow E. coli porin channels only with marginal efficiency (37, 38). In fact, the only protein labeled to a significant extent was the A312C OprF regardless of whether the cells were pretreated with β-mercaptoethanol or not (Fig. 9, lanes 1 and 2). When the same experiment was carried out with the E. coli strain expressing an unmodified OprF, no labeling of this protein occurred (not shown). These experiments indicate (i) that the reagent is to a large extent impermeable through the cell envelope so that sulfhydryl-containing cytosolic proteins were not labeled and (ii) that the disulfide bonds present in some outer membrane proteins, including OprF, are so stable that it is hardly labeled by the biotinylation procedure (even after the attempted reduction of these bonds with β-mercaptoethanol).
FIGURE 9. C-terminal domain of OprF majority conformer is not exposed on cell surface.
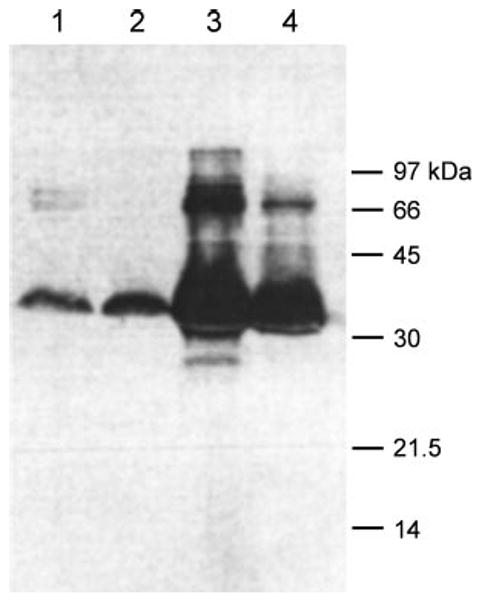
E. coli DH5α cells expressing the A312C mutant OprF were labeled with biotin-maleimide either as intact cells (lanes 1 and 2) or as crude envelope fraction after cell disruption by sonication (lanes 3 and 4). Labeling was carried out either after 2-mercaptoethanol treatment (lanes 1 and 3) or without such treatment. Fractions each containing 50 μg of protein were separated by SDS-PAGE, and the blot was stained with streptavidin reagent for biotin as described under “Experimental Procedures.”
Intact cells as well as isolated membrane fractions from an identical amount of cells were labeled in parallel under the same conditions. After solubilization of membranes the proteins were separated by SDS-PAGE, and the biotinylated proteins were visualized by streptavidin-based reagent as described under “Experimental Procedures.” Fig. 9 shows that in the isolated cell envelope fraction from comparable numbers of cells the A312C OprF was labeled far more strongly than in the intact cells (compare lanes 3 and 4 with lanes 1 and 2). Because outer membrane “vesicles” in the isolated crude envelope fractions are leaky and allow rapid influx of rather large agents (39), these results are consistent with the conclusion that the C-terminal domain of OprF, containing the residue 312, is largely located on the periplasmic side of the outer membrane and reacts with the biotinylation reagent in the isolated fragments of the outer membrane but not in intact cells.
Enrichment for the Less Frequent OprF Conformer Whose C-terminal Domain Is Exposed on Cell Surface
In the preceding section, we showed that the cysteine residue at position 312 of the majority conformer of OprF is probably hidden in the periplasmic domain and is largely inaccessible from the outside in intact cells. This localization reflects the two-domain conformation of the majority conformer. In contrast, the minority conformer, which forms open channels, is likely to traverse the membrane many more times as β-strands. In such a conformer the cysteine residue near the C terminus may have a possibility of being exposed on the cell surface (see Fig. 10). If so, we should be able to label this minority conformer by treating intact cells with a biotinylation reagent and isolate the labeled protein by utilizing the affinity of biotin group to streptavidin, thereby achieving enrichment for this conformer. This was carried out as detailed under “Experimental Procedures” following a brief (5-min) treatment of intact cells of E. coli expressing OprFA312C with a cleavable labeling agent, biotin-HPDP (Pierce). The large size (540 daltons) and the lipophilic nature of this compound is expected to hinder its diffusion through the E. coli porin channel (38), and thus the labeling will be largely limited to the conformer with surface-exposed Cys-312 residue. Membrane proteins were solubilized and then passed through an avidin affinity column. The biotinylated protein was then eluted by cleaving the disulfide bond in the biotin linker with 10 mm 2-mercaptoethanol.
FIGURE 10. Presumed folding patterns in the two conformers of OprF.

Our results are consistent with the hypothesis that the closed channel conformers consist of OprF folded as a two-domain protein (left), whereas the open channel conformers correspond to OprF folded as a single domain, large β-barrel. The gel filtration data (Fig. 3) indicates that the latter conformers tend to exist as unstable oligomers, most likely as trimers.
The C-terminal surface-exposed conformer of OprFA312C, isolated in this manner, produced a proteoliposome swelling rate (with l-arabinose) of 0.289 ΔA400/20 μg. This was nearly 10 times higher than the swelling rate (0.031 ΔA400/20 μg) produced by OprFA312C isolated by biotinylation in the crude envelope fraction where both conformers of OprF are expected to be accessible for biotin-HPDP as seen in Figs. 9 and 10. These results strongly suggest that the surface-exposed minority conformer, but not the majority conformer, contains open channels.
Another property we tested was the susceptibility of OprF to trypsin in the crude envelope fraction. In the majority conformer, the linker region that connects the N-terminal β-barrel to the C-terminal globular domain and the C-terminal domain itself are susceptible for trypsin attack. When OprFA312C was labeled with biotin-maleimide in intact E. coli cells and then the crude membrane fraction prepared from these cells was treated with trypsin, digestion of the (minority) conformer of OprF with its C-terminal domain exposed on the cell surface (followed by streptavidin staining for biotin after SDS-PAGE) was distinctly slower than the digestion of the bulk OprF (followed by the density of OprF band in SDS-PAGE) (Fig. 11). This result is consistent with the data that the minority conformer of OprF, with its putative single domain, β-barrel structure (Fig. 10, right), is relatively resistant to trypsin digestion, a property shared by other single domain, β-barrel proteins such as porins OmpF and OmpC (37).
FIGURE 11. Trypsin sensitivity of OprF conformers.

Intact cells of E. coli DH5α expressing OprFA312C mutant were first labeled with biotin-maleimide for 5 min. The cells were then disrupted with a French press, and the isolated crude outer membrane fraction was then treated with trypsin as described under “Experimental Procedures.” The envelope was analyzed by SDS-PAGE at 0, 1.5, and 3 h after the start of trypsin treatment, and the degradation of bulk OprF (▲) and OmpF (●) (as a control) was followed by staining and scanning of the gel. The degradation of the C-terminal domain-exposed conformer of OprF (■), which should have been labeled with biotin, was followed by streptavidin binding to a blot as described under “Experimental Procedures.”
Discussion
Our results argue that OprF folds into one of the two alternative conformations and that the majority conformation is a two-domain structure resembling that of E. coli OmpA. In addition to the circular dichroism data obtained with the whole OprF protein (19), we have now been able to cleave the majority OprF conformer into its N-terminal and C-terminal domains and show that they have, respectively, predominantly β-stranded and α-helical (presumably globular) structure (Fig. 1; see also Fig. 10, left). The N-terminal eight-stranded β-barrel structure is unlikely to allow the diffusion of organic molecules of even a small size, and indeed the N-terminal domain showed no evidence of allowing the passage of a pentose, l-arabinose, or monovalent ions (28). The two-domain, OmpA-like conformation of the majority conformer is at variance with earlier proposals of one-domain conformation for P. aeruginosa (21–23) and Pseudomonas fluorescens (40) OprF. The P. fluorescens study used digestion of OprF in intact cells with an extraordinarily high concentration (0.5 mg/ml) of papain, and perhaps we cannot assume the total absence of permeation of the enzyme under such conditions. The P. aeruginosa data are discussed below. The α-helix-rich conformation of the C-terminal domain of OprF is also consistent with the x-ray crystal structure of this domain from an OprF/OmpA homolog of Neisseria (41).
The unfractionated OprF protein has been shown to allow the slow permeation of sugars, including at least trisaccharides (which have difficulty in diffusing through the E. coli OmpF channel (3)) (7, 15, 16). Because the N-terminal domain of the majority conformer of OprF does not allow the diffusion of even l-arabinose as stated above, the likely explanation for the presence of these large pores is the alternative folding of the OprF channel. In this study, we showed that the population of OprF protein can be fractionated into a majority fraction that does not allow the diffusion of sucrose and a minority fraction that allows it by an iso-osmotic gradient centrifugation of unilamellar proteoliposomes (Fig. 4).
To permit the diffusion of large organic molecules, the β-barrel of the minority conformer of OprF must contain many more strands than the eight strands seen in the N-terminal domain of the majority conformer (Fig. 10). This large β-barrel structure was supported by the observation that the fraction of OprF with such a conformation, with residue 312 exposed on the external surface of the outer membrane (Figs. 9 and 10), had a nearly 10-fold higher channel-forming activity than the bulk OprF population (see “Results”). Interestingly this open channel conformer also appeared to form a metastable oligomeric complex (Fig. 5B). Finally this large β-barrel conformation of the minority conformer is also supported by its resistance to trypsin digestion as detected by the functional porin assay (Table 1) as well as by SDS-PAGE analysis (Fig. 11): classical, all β-barrel porins such as OmpF are usually trypsin-resistant, whereas the majority conformers of OmpA or OprF are easily cleaved in the exposed “linker” sequence between the N-terminal and C-terminal domains.
The concept of two alternative conformers also explains a long standing puzzle about the folding pattern of OprF. Thus extensive studies mostly in the laboratory of Hancock (22, 23) using epitope mapping or antigen insertion have shown that the C-terminal region of OprF is exposed on cell surface, and this led to the idea that OprF folds always as a 16-stranded β-barrel. In addition, an epitope in the C-terminal domain has been shown to be a strong immunogen that generates antibodies reacting with whole cells of P. aeruginosa (42–44). Again under the assumption that OprF folds into one unique conformation, these data were taken as a proof that it is a one-domain, large β-barrel protein just like the classical porin. However, this hypothesis is in obvious conflict with the circular dichroism data with the intact OprF (20) and the tobacco etch virus protease cleavage of the engineered OprF into the N-terminal β-barrel and the C-terminal globular domains (Fig. 1). There is no conflict among data, however, once we accept that OprF folds mostly as a two-domain protein but less frequently as a one-domain β-barrel protein. Because there are close to 105 molecules of OprF on the surface of a single cell, there will be thousands of one-domain conformers with the exposed C-terminal epitopes in each cell. In fact in a study in which malaria antigen epitopes were inserted at different positions of OprF sequence (22), there was some indication that the epitopes inserted into the C-terminal domain were less accessible in intact cells than those inserted into the external loops of N-terminal β-barrel. In this study, we showed clearly that a bulky labeling agent, biotin-maleimide, had a ready access to the C-terminal domain of the minority conformer of OprF and that its reaction with the C-terminal domain of the majority conformer occurred efficiently only after the disruption of the outer membrane barrier (Fig. 9), showing that the C-terminal domain is exposed on cell surface only in the minority conformer. A similar study, using monoclonal antibody directed to the C-terminal domain of OmpA of Salmonella enterica, an OprF homolog, showed again that this domain is exposed on cell surface only in the minority conformer of OmpA (45).
We should emphasize that the presence of the N-terminal eight-stranded β-barrel in the majority OprF conformer does not validate the earlier claim by Nakae and associates (10–12) of the “small” channels present in P. aeruginosa outer membrane as the N-terminal eight-stranded barrel does not allow the permeation of even a pentose or monovalent ions (28). Although Yoneyama and Nakae (11) reported that glucose, but not disaccharides, can permeate through the outer membrane, this experiment was done with intact cells, and the contribution of sugar-specific channel OprB (46, 47), for example, cannot be excluded. In addition, OprF, according to Yoshihara and Nakae (10), does not form channels of any size.
If OprF is a porin, as we argue, then it becomes a question of why only a small fraction of this protein folds into a conformation producing channels. The likely answer is that the major function of proteins of OmpA/OprF family is to connect together the components of cell envelope by using the N-terminal β-barrel domain that inserts into the outer membrane and the C-terminal periplasmic globular domain that interacts with peptidoglycan (30, 31). In fact, OprF has been shown to play a role in the stabilization of the cell envelope structure (48, 49). In organisms producing classical trimeric porins of high permeability, the contribution of OmpA/OprF family protein to permeability is negligible. However, in fluorescent pseudomonads that do not produce classical porins, OprF becomes the major nonspecific porin, and its low permeability contributes to their high levels of intrinsic resistance to noxious agents, a property presumably beneficial to these inhabitants of soil. In one strain of P. fluorescens, the inactivation of oprF gene led to the suppressor mutations causing the overexpression of another channel protein of OprD family (50). This is indeed a strong piece of genetic evidence suggesting that the major function of OprF protein is the porin function at least in this strain. In addition, OprF facilitates binding of bacteria to plant roots or animal cell surfaces (51–53).
Finally if the low permeability of OmpA/OprF depends on the alternative folding pathways of these proteins, it should be possible to influence the folding to alter the permeability. A closely related P. fluorescens OprF (54) and more recently P. aeruginosa OprF (55) were shown to exhibit single channel conductivity of different size depending on the growth temperature. Such a result is most easily explained by the temperature effect on protein folding.
Acknowledgments
We thank David King for mass spectrometric analysis of proteins, Kaoru Yoshida for the gift of the vector plasmid pKY9790, and Susan Marqusee for access to the circular dichroism spectrometer.
Footnotes
This work was supported by National Institutes of Health Grant AI-09644 (to H. N.).
The abbreviation used is: biotin-HPDP, N-[6-(biotinamido)hexyl]-3′-(2′-pyridyldithio)propionamide.
References
- 1.Schulz GE. Curr Opin Cell Biol. 1993;5:701–707. doi: 10.1016/0955-0674(93)90143-e. [DOI] [PubMed] [Google Scholar]
- 2.Cowan SW. Curr Opin Struct Biol. 1993;3:501–507. [Google Scholar]
- 3.Nikaido H. J Biol Chem. 1994;269:3905–3908. [PubMed] [Google Scholar]
- 4.Jap BK, Walian PJ. Physiol Rev. 1996;76:1073–1088. doi: 10.1152/physrev.1996.76.4.1073. [DOI] [PubMed] [Google Scholar]
- 5.Nikaido H, Rosenberg EY. J Gen Physiol. 1981;77:121–135. doi: 10.1085/jgp.77.2.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hancock REW, Decad GM, Nikaido H. Biochim Biophys Acta. 1979;554:323–331. doi: 10.1016/0005-2736(79)90373-0. [DOI] [PubMed] [Google Scholar]
- 7.Yoshimura F, Zalman LS, Nikaido H. J Biol Chem. 1983;258:2308–2314. [PubMed] [Google Scholar]
- 8.Yoshimura F, Nikaido H. J Bacteriol. 1982;152:636–642. doi: 10.1128/jb.152.2.636-642.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Angus BL, Carey AM, Caron DA, Kropinski AMB, Hancock REW. Antimicrob Agents Chemother. 1982;21:299–309. doi: 10.1128/aac.21.2.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yoshihara E, Nakae T. J Biol Chem. 1989;264:6297–6301. [PubMed] [Google Scholar]
- 11.Yoneyama H, Nakae T. Eur J Biochem. 1986;157:33–38. doi: 10.1111/j.1432-1033.1986.tb09634.x. [DOI] [PubMed] [Google Scholar]
- 12.Yoshihara E, Gotoh N, Nakae T. Biochem Biophys Res Commun. 1988;156:470–476. doi: 10.1016/s0006-291x(88)80865-9. [DOI] [PubMed] [Google Scholar]
- 13.Duchene M, Schweizer A, Lottspeieh F, Grauss G, Marget M, Vogel K, von Specht BU, Domdey H. J Bacteriol. 1988;171:4130–4137. doi: 10.1128/jb.171.8.4130-4137.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sugawara E, Nikaido H. J Biol Chem. 1992;267:2507–2511. [PubMed] [Google Scholar]
- 15.Nikaido H, Nikaido K, Harayama S. J Biol Chem. 1991;266:770–779. [PubMed] [Google Scholar]
- 16.Bellido F, Martin NL, Siehnel RJ, Hancock REW. J Bacteriol. 1992;174:5196–5203. doi: 10.1128/jb.174.16.5196-5203.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ried G, Koebnik R, Hindennach I, Mutscher B, Henning U. Mol Gen Genet. 1994;243:127–135. doi: 10.1007/BF00280309. [DOI] [PubMed] [Google Scholar]
- 18.Pautsch A, Schulz GE. Nat Struct Biol. 1998;5:1013–1017. doi: 10.1038/2983. [DOI] [PubMed] [Google Scholar]
- 19.Sugawara E, Nikaido H. J Biol Chem. 1994;269:17981–17987. [PubMed] [Google Scholar]
- 20.Sugawara E, Steiert M, Rouhani S, Nikaido H. J Bacteriol. 1996;178:6067–6069. doi: 10.1128/jb.178.20.6067-6069.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wong RS, Jost H, Hancock REW. Mol Microbiol. 1993;10:283–292. doi: 10.1111/j.1365-2958.1993.tb01954.x. [DOI] [PubMed] [Google Scholar]
- 22.Wong RS, Wirtz RA, Hancock REW. Gene (Amst) 1995;158:55–60. doi: 10.1016/0378-1119(95)00155-y. [DOI] [PubMed] [Google Scholar]
- 23.Rawling EG, Martin NL, Hancock REW. Infect Immun. 1995;63:38–42. doi: 10.1128/iai.63.1.38-42.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takeshita S, Sato M, Toba M, Masahashi W, Hashimoto-Gotoh T. Gene (Amst) 1987;61:63–74. doi: 10.1016/0378-1119(87)90365-9. [DOI] [PubMed] [Google Scholar]
- 25.Carrington JC, Dougherty WG. Proc Natl Acad Sci U S A. 1988;85:3391–3395. doi: 10.1073/pnas.85.10.3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ames BN, Dubin DT. J Biol Chem. 1960;235:769–775. [PubMed] [Google Scholar]
- 27.Henkeshoven J, Dernick R. Electrophoresis. 1988;9:28–32. doi: 10.1002/elps.1150090106. [DOI] [PubMed] [Google Scholar]
- 28.Nestorovich EM, Sugawara E, Nikaido H, Bezrukov SM. J Biol Chem. 2006;281:16230–16237. doi: 10.1074/jbc.M600650200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu J, Rutz JM, Klebba PE, Feix JB. Biochemistry. 1994;33:13274–13283. doi: 10.1021/bi00249a014. [DOI] [PubMed] [Google Scholar]
- 30.De Mot R, Vanderleyden J. Mol Microbiol. 1994;12:333–334. doi: 10.1111/j.1365-2958.1994.tb01021.x. [DOI] [PubMed] [Google Scholar]
- 31.Koebnik R. Mol Microbiol. 1995;16:1269–1270. doi: 10.1111/j.1365-2958.1995.tb02348.x. [DOI] [PubMed] [Google Scholar]
- 32.Woodruff WA, Parr TR, Jr, Hancock REW, Hanne LF, Nicas TI, Iglewski BH. J Bacteriol. 1986;167:473–479. doi: 10.1128/jb.167.2.473-479.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Greenfield N, Fasman GD. Biochemistry. 1969;8:4108–4116. doi: 10.1021/bi00838a031. [DOI] [PubMed] [Google Scholar]
- 34.Racker E, Violand B, O'Neal S, Alfonzo M, Telford J. Arch Biochem Biophys. 1979;198:470–477. doi: 10.1016/0003-9861(79)90521-6. [DOI] [PubMed] [Google Scholar]
- 35.Sugawara E, Nikaido H. Biophys J. 1997;72:A138. abstr. [Google Scholar]
- 36.Chen R, Schmidmayr W, Krämer C, Chen-Schmeisser U, Henning U. Proc Natl Acad Sci U S A. 1980;77:4592–4596. doi: 10.1073/pnas.77.8.4592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nikaido H, Vaara M. Microbiol Rev. 1985;49:1–32. doi: 10.1128/mr.49.1.1-32.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nikaido H. Microbiol Mol Biol Rev. 2003;67:593–656. doi: 10.1128/MMBR.67.4.593-656.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nakae T, Nikaido H. J Biol Chem. 1975;250:7359–7365. [PubMed] [Google Scholar]
- 40.De Mot R, Schoofs G, Roelandt A, Declerck P, Proost P, Van Damme J, Vanderleyden J. Microbiology. 1994;140:1377–1387. doi: 10.1099/00221287-140-6-1377. [DOI] [PubMed] [Google Scholar]
- 41.Grizot S, Buchanan SK. Mol Microbiol. 2004;51:1027–1037. doi: 10.1111/j.1365-2958.2003.03903.x. [DOI] [PubMed] [Google Scholar]
- 42.Hughes EE, Gilleland LB, Gilleland HE., Jr Infect Immun. 1992;60:3497–3503. doi: 10.1128/iai.60.9.3497-3503.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gilleland LB, Gilleland HE., Jr Infect Immun. 1995;63:2347–2351. doi: 10.1128/iai.63.6.2347-2351.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gilleland HE, Jr, Hughes EE, Gilleland LB, Matthews-Greer JM, Staczek J. Curr Microbiol. 1995;31:279–285. doi: 10.1007/BF00314580. [DOI] [PubMed] [Google Scholar]
- 45.Singh SP, Williams YU, Miller S, Nikaido H. Infect Immun. 2003;71:3937–3946. doi: 10.1128/IAI.71.7.3937-3946.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Trias J, Rosenberg EY, Nikaido H. Biochim Biophys Acta. 1988;938:493–496. doi: 10.1016/0005-2736(88)90148-4. [DOI] [PubMed] [Google Scholar]
- 47.Wylie JL, Worobec EA. J Bacteriol. 1995;177:3021–3026. doi: 10.1128/jb.177.11.3021-3026.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Woodruff WA, Hancock REW. J Bacteriol. 1989;171:3304–3309. doi: 10.1128/jb.171.6.3304-3309.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rawling EG, Brinkman FSL, Hancock REW. J Bacteriol. 1998;180:3556–3562. doi: 10.1128/jb.180.14.3556-3562.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chevalier S, Burini JF, Freulet-Marriere M, Regeard C, Schoofs G, Guespin-Michel J, De Mot R, Orange N. Res Micriobiol. 2000;151:619–627. doi: 10.1016/s0923-2508(00)90128-1. [DOI] [PubMed] [Google Scholar]
- 51.De Mot R, Proost P, Van Damme J, Vanderleyden J. Mol Gen Genet. 1992;231:489–493. doi: 10.1007/BF00292721. [DOI] [PubMed] [Google Scholar]
- 52.Azghani AO, Idell S, Bains M, Hancock REW. Microb Pathog. 2002;33:109–114. doi: 10.1006/mpat.2002.0514. [DOI] [PubMed] [Google Scholar]
- 53.Rebière-Huët J, Guérillon J, Pimenta AL, Di Martino P, Orange N, Hulen C. FEMS Microbiol Lett. 2002;215:121–126. doi: 10.1111/j.1574-6968.2002.tb11380.x. [DOI] [PubMed] [Google Scholar]
- 54.De E, Orange N, Saint N, Guerillon J, De Mot R, Molle G. Microbiology. 1997;143:1029–1035. doi: 10.1099/00221287-143-3-1029. [DOI] [PubMed] [Google Scholar]
- 55.Jaouen T, De E, Chevalier S, Orange N. Appl Environ Microbiol. 2004;70:6665–6669. doi: 10.1128/AEM.70.11.6665-6669.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]