

Abstract
Synaptotagmin-IV (syt-IV) is a membrane trafficking protein that influences learning and memory, but its localization and role in synaptic function remain unclear. Here we discovered that syt-IV localizes to BDNF-containing vesicles in hippocampal neurons. Syt-IV/BDNF-harboring vesicles undergo exocytosis in both axons and dendrites, and syt-IV inhibits BDNF release at both sites. Knockout of syt-IV increases, and over-expression decreases, the rate of FM dye destaining from presynaptic terminals indirectly via changes in post-synaptic release of BDNF. Hence, post-synaptic syt-IV regulates the trans-synaptic action of BDNF to control presynaptic vesicle dynamics. Furthermore, selective loss of presynaptic syt-IV increased spontaneous quantal release, while loss of post-synaptic syt-IV increased quantal amplitude. Finally, syt-IV knockout mice exhibit enhanced LTP, which depends entirely on disinhibition of BDNF release. Thus, regulation of BDNF secretion by syt-IV emerges as a mechanism to maintain synaptic strength within a useful range during long-term potentiation.
Introduction
Synaptic plasticity relies on highly regulated membrane fusion events on both sides of the synapse, including presynaptic neurotransmitter release, neuropeptide release and post-synaptic receptor trafficking. Regulation of these fusion events allows synapses to adjust their strength in response to stimulation. Many of the seventeen members of the synaptotagmin family of proteins promote membrane fusion by penetrating lipid bilayers and binding to SNARE proteins in a Ca2+-promoted manner1, 2. The most well-studied isoform is syt-I, which plays an essential role in rapid synaptic vesicle exocytosis1, 2. Interestingly, the calcium sensitivity and kinetics of syt interactions with SNAREs and phospholipids vary among the different isoforms3, 4, and some syt isoforms do not appear to sense Ca2+ at all. These isoforms might regulate membrane traffic by inhibiting fusion5. Thus, different syt isoforms can potentially confer distinct functional properties on synapses6.
Within the syt family of proteins, syt-IV has attracted particular interest as an immediate early gene7 that is upregulated by seizures and activity7, 8. Syt-IV harbors an aspartate to serine substitution in a calcium coordination site, and can bind SNARE proteins in the absence of calcium, but fails to bind more avidly to SNAREs, or to penetrate membranes, in response to calcium9, 10. Thus, syt-IV can join the fusion complex, but prevents an essential fusion step3. In PC12 cells, syt-IV reduces the frequency of fusion events11 and gives rise to small conductance, long-lived fusion pores10. However, syt-IV has also been reported to play a positive role in an earlier step in secretory granule biogenesis in PC12 cells12, and in glutamate release from astrocytes13.
In neurons, the sub-cellular localization and function of syt-IV remain unresolved. Conflicting reports have found syt-IV on synaptic vesicles14, 15, or absent from synaptic vesicles16, 17, and an investigation of synaptic transmission revealed no effect of syt-IV over-expression15. Drosophila syt-IV, which retains some ability to bind phospholipids in response to calcium18, has also been found on synaptic vesicles in some studies, but tests of function yielded disparate results; expression of syt-IV was reported to decrease exocytosis18, rescue synaptic transmission in syt-I nulls19, or modify fusion pores20. A recent re-examination of syt isoform distribution in Drosophila localized syt-IV to postsynaptic muscle cells at the neuromuscular junction and proposed that syt-IV mediated the release of an unidentified retrograde messenger to enhance presynaptic function21. In summary, the localization and function of syt-IV, and of mammalian syt-IV in particular, remains unclear.
Syt-IV is clearly involved in learning and memory22, 23, as well as adaptive responses to pathological states such as epilepsy7, 8 and exposure to certain drugs24, 25. To bridge the gap between the putative regulation of exocytosis by syt-IV and changes in memory, behavior, and pathology governed by this molecule, we examined where syt-IV is targeted within neurons, and how it might affect synaptic function and plasticity. We found that syt-IV is localized to BDNF-containing vesicles in hippocampal neurons, where it negatively regulates BDNF release. During evoked activity, loss of post-synaptic syt-IV enhances synaptic vesicle recycling trans-synaptically via BDNF. In addition, syt-IV limits spontaneous quantal transmission pre and post-synaptically, in a cell autonomous manner. Finally, we found that loss of syt-IV enhances both the magnitude and spatial spread of LTP, and this enhancement is BDNF-dependent.
Results
Syt-IV localizes to BDNF-containing vesicles
To investigate the function of syt-IV, we first examined its subcellular localization in cultured hippocampal neurons, where syt-IV knockout neurons were used to control for the specificity of the syt-IV antibody (Supplemental Fig. 1a). We tested colocalization of syt-IV with a series of antibodies and GFP-tagged fusion proteins known to localize to distinct organelles and subcellular locations. We found that syt-IV is prominent in the Golgi, as previously reported8 (Supplemental Fig. 1b) and colocalized significantly with BDNF-GFP (Fig. 1a), and with endogenous BDNF (Fig. 1b), but not with synaptic vesicles marked with synaptophysin (Fig. 1c). We also performed immuno-organelle isolation experiments from hippocampal culture lysates using an antibody to syt-IV. We detected a small amount of BDNF associated with syt-IV vesicles, but not with synaptic vesicles pulled down by an antibody to synaptophysin. Only trace amounts of synaptophysin were found in the syt-IV immunoprecipitate, corresponding to ~2.5% of total synaptophysin suggesting that little, if any, syt-IV is present on synaptic vesicles (Supplemental Fig. 1c). These experiments do not, however, exclude the possibility that some BDNF-containing vesicles lack syt-IV, or that syt-IV is localized to other, as yet unidentified vesicles, in addition to those containing BDNF.
Figure 1. Syt-IV is targeted to BDNF-containing vesicles.

(a) Hippocampal neurons transfected with BDNF-GFP and immunostained for syt-IV. Endogenous syt-IV colocalizes with BDNF-GFP in transfected neurons. (b) Syt-IV colocalizes with endogenous BDNF in immunostained hippocampal neuron cultures. 66 ± 3% of detectable endogenous BDNF signal colocalized with syt-IV, and 68 ± 4% of syt-IV signal colocalized with BDNF. (c) High magnification images of syt-IV/BDNF and syt-IV/synaptophysin immunostained neurons, for comparison of colocalization of syt-IV with BDNF-containing vesicles versus synaptic vesicles, marked with synaptophysin. (d) Axon (top panel), axonal growth cone (arrow in middle panel) and dendrites (bottom panel) of neurons co-transfected with mCherry-syt-IV and BDNF-GFP. Scale bar is 10 μm in all panels.
We also constructed a fluorescently-tagged syt-IV fusion protein and found that mCherry-syt-IV targeted equally well to both axons and dendrites where it colocalized extensively with BDNF-GFP in both structures (Fig. 1d) similar to the endogenous proteins. In contrast, mCherry-syt-IV did not colocalize significantly with synaptophysin-GFP at presynaptic sites or with the post-synaptic marker PSD95-GFP (Supplemental Fig. 1d). To investigate the colocalization of syt-IV and BDNF in more detail, we performed time-lapse experiments on hippocampal neurons co-transfected with mCherry-syt-IV and BDNF-GFP and found that these fusion proteins traffic rapidly together (Supplemental Fig. 2).
To examine the trafficking of endogenous syt-IV and BDNF we took advantage of the fact that both are induced by activity7, 26. We increased activity in isolated hippocampal neurons by transfection with the TRPV1 channel, which is activated by addition of exogenous capsaicin, thereby selectively depolarizing the transfected cell27. In the absence of capsaicin, TRPV1 expression had no effect on syt-IV protein levels (Fig. 2a). Activation of the channel with 50 nM capsaicin for 16 hours, caused a marked increase in syt-IV levels in both axons and dendrites of transfected cells (Fig. 2b). BDNF was concomitantly increased specifically in capsaicin-treated TRPV1-expressing cells (Fig. 2c), and colocalized with syt-IV in both axons (Fig. 2d) and dendrites.
Figure 2. Activity-induced syt-IV and BDNF colocalize and traffic together.

(a) Syt-IV signal in TRPV1/GAP-43 transfected hippocampal neurons, identified by GFP fluorescence, without capsaicin treatment. (b) Following treatment with 50 nM capsaicin for 16 hours, the syt-IV signal in axons (arrow) and dendrites of transfected cells increased; note that gain settings for this image were set slightly lower than for controls in panel a, due to saturating syt-IV signal. (c) BDNF is concomitantly upregulated in capsaicin treated TRPV1-transfected cells (dendrites of a transfected cell are shown). (d) Activity-induced syt-IV and BDNF traffic together and colocalize in neuronal processes of TRPV1-transfected, capsaicin-treated cells. Syt-IV and BDNF vesicles in an axon of a capsaicin treated TRPV1 expressing cell are shown. Syt-IV and BDNF were increased 4.96 ± 0.18 fold and 3.81 ± 0.15 fold, respectively, in capsaicin treated TRPV1-expressing cells. In these cells 75 ± 4% of detectable BDNF signal colocalized with syt-IV, and 82 ± 5% of syt-IV signal colocalized with BDNF. Scale bar is 10 μm in all panels.
Syt-IV regulates the secretion of BDNF
The localization of syt-IV to BDNF-containing vesicles prompts the interesting hypothesis that syt-IV itself might regulate the secretion of BDNF. To test this, we used an in situ ELISA28. Over-expression of syt-IV decreased BDNF release, compared to GFP-expressing controls. Conversely, syt-IV knockouts released higher amounts of BDNF compared to wild-type littermate cultures (Fig. 3). Thus, syt-IV acts to negatively regulate BDNF release.
Figure 3. Syt-IV regulates BDNF release.
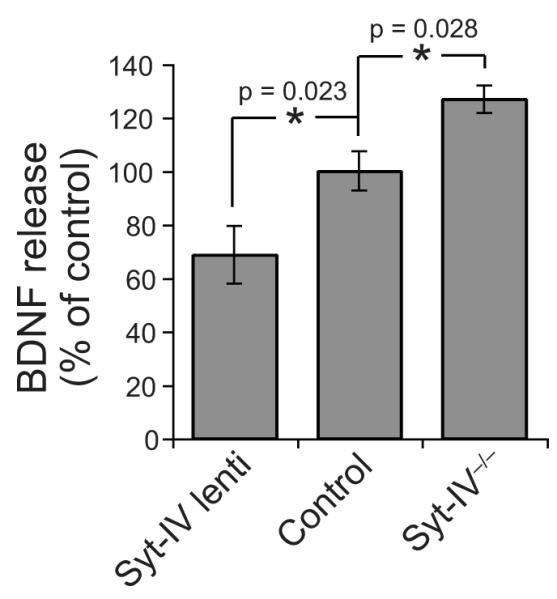
BDNF release, assayed by in situ ELISA, from hippocampal neurons over-expressing syt-IV, and from syt-IV knockout neurons compared to controls. For comparison of conditions, BDNF release was expressed relative to control (either GFP-expressing, or wild-type), which was normalized to 100%. Over-expression of syt-IV decreased BDNF release, and knockout of syt-IV increased BDNF release, compared to controls (n = 5 different neuronal cultures for each condition, 3-4 duplicate wells for each culture; error bars indicate SEM. Significance was determined by paired Student’s t-tests where * = p < 0.05, and one-way ANOVA where p = 0.003).
Total BDNF was not significantly different between syt-IV over-expressing, control, and syt-IV knockout cultures. However, we observed a trend toward an increase in total BDNF in syt-IV over-expressing neurons, which may result from an accumulation of intracellular BDNF due to inhibition of release by excess syt-IV (Supplemental Fig. 3).
Depolarization induces syt-IV vesicle recycling
BDNF can be secreted both constitutively, and via regulated release in response to depolarization26. To distinguish between these two possibilities we first tested whether syt-IV-containing vesicles recycle in response to depolarization, using an antibody uptake assay. Neurons transfected with GFP-syt-IV were depolarized in the presence of extracellular antibodies to GFP. If GFP-syt-IV-containing vesicles fuse with the plasma membrane in response to activity, their lumenal GFP tags will be exposed to the cell surface, allowing extracellular GFP antibodies to bind, thus marking sites of exocytosis. Using this assay, we observed numerous sites of syt-IV-vesicle exocytosis. These exocytotic events were activity-dependent and calcium-dependent, with no significant uptake in non-depolarizing or calcium-free conditions (Fig. 4a-c). To mark recycling synaptic vesicles, we included an antibody to the lumenal domain of syt-I in the uptake experiments. Syt-I uptake was also activity and calcium-dependent, as expected (Fig. 4b). Syt-IV vesicle recycling sites were distinct from sites of syt-I antibody uptake (Fig. 4d), but were often located perisynaptically along neuronal processes.
Figure 4. Depolarization induces syt-IV vesicle recycling in axons and dendrites.

(a) Immunofluorescence image of GFP antibodies taken up during depolarization with 45 mM KCl for 5 minutes, marking sites of syt-IV vesicle recycling. Neurons treated with 5 mM KCl, and with 5 mM KCl in the absence of calcium failed to take up antibody and served as controls. Scale bar is 10 μm. (b) Immunofluorescence image of antibodies to the lumenal domain of syt-I that have been taken up during activity. (c) Total GFP fluorescence in transfected cells, pseudocolored cyan. (d) Enlarged images of the indicated rectangular area in panels a-c showing sites of syt-IV vesicle recycling, syt-I vesicle recycling, and merged images. (e) pHluorin-syt-IV assay schematic. During exocytosis, the vesicle lumen pH is neutralized, resulting in an increase in GFP fluorescence. Vesicles are then re-acidified following endocytosis, which results in a concomitant decrease in GFP fluorescence. (f) pHluorin-syt-IV exocytotic events (white arrows) in a dendrite and an axon following depolarization at time zero. Scale bar is 5 μm. (g) Sample traces and average fluorescence change of small, fast pHluorin events in dendrites. (h) Fluorescence traces of large pHluorin events in dendrites. (i) Fluorescence traces of axonal pHluorin events (n=35 events of each type; 10 different cells from 3 different cultures/transfections for each event type; error bars indicate SEM).
To examine syt-IV vesicle exocytosis in real-time, we constructed a pHluorin-syt-IV expression construct (Fig. 4e). Exocytotic events were detected in both dendrites and axons upon depolarization (Fig. 4f). Interestingly, the kinetics of fluorescence changes differed markedly between axons and dendrites. In dendrites, puncta were visible before depolarization, and two types of events were observed: small, fast increases in puncta fluorescence followed by a decay back to baseline within seconds (Fig. 4g), and larger slower increases (Fig. 4h) which remained elevated in the presence of 45 mM KCl but recovered back to baseline when 45 mM KCl was replaced with 5 mM KCl. This recovery was prevented by treatment with bafilomycin, which inhibits the vesicular H+ ATPase and blocks re-acidification of newly endocytosed vesicles (Supplemental Fig. 4a), indicating that the drop in fluorescence following addition of 5 mM KCl is due to endocytosis of syt-IV vesicles. Both types of events in dendrites were tightly correlated in time with the onset of depolarization.
Depolarization-induced fusion events were also observed in axons, as apparent from the average overall rise in fluorescence following addition of 45 mM KCl (Fig. 4i). But unlike dendritic events, puncta that were not visible before stimulation appeared after depolarization (hence, the pH of syt-IV-containing vesicles appears to be higher in dendrites than in axons). The initial rises in fluorescence of axonal events were not as tightly synchronized in time with the onset of depolarization and were slower than dendritic events.
Syt-IV inhibits depolarization-induced secretion of BDNF
The experiments above demonstrate that syt-IV containing vesicles undergo exocytosis in both axons and dendrites in response to depolarization. However, they do not assess the effect of syt-IV on regulated release of BDNF. To test this, we used a BDNF-pHluorin fusion protein to visualize BDNF release events induced by depolarization. BDNF-pHluorin was expressed in a punctate pattern in axons and dendrites of transfected neurons, with no detectable changes in fluorescence over periods of minutes in the absence of depolarization.
In dendrites, puncta that were visible prior to depolarization exhibited decreases in fluorescence upon addition of 45 mM KCl (Fig. 5a white arrows, b); these events were occasionally preceded by brief spikes of increased fluorescence (Fig. 5c, asterisks). In axons, puncta that were not visible prior to depolarization appeared upon addition of 45 mM KCl (Fig. 5a, yellow arrows, b). These puncta appeared rapidly, followed by either an exponential decay to baseline fluorescence within 15 seconds (fast-decaying events) or a linear decay occurring over tens of seconds (slow-decaying events) (Fig. 5d). Co-expression of mCherry-syt-IV with BDNF-pHluorin (Supplemental Fig. 4b) decreased the amplitude of both dendritic (Fig. 5c) and axonal (Fig. 5d) events.
Figure 5. Syt-IV inhibits regulated release of BDNF.

(a) BDNF-pHluorin fluorescence decreased in dendrites (white arrows), and increased in axons (yellow arrows) following depolarization at t = 30 seconds. Scale bar is 5 μm in all panels. (b) Individual events in dendrites and axons. (c) Sample and average traces of BDNF-pHluorin fluorescence decrease events in dendrites in control and syt-IV-over-expressing neurons. Arrows indicate addition of 45 mM KCl. These events were occasionally preceded by brief spikes of increased fluorescence (asterisks). Average traces were normalized to puncta fluorescence intensity prior to depolarization, which was not significantly different in control versus syt-IV over-expressing neurons (control = 537 ± 52 a.u., syt-IV over-expressing = 343 ± 29 a.u.). (d) Sample and average traces of BDNF-pHluorin fluorescence increase events in axons of control and syt-IV-over-expressing neurons. These events exhibited either fast-decaying (asterisks) or slow-decaying kinetics of fluorescence recovery to baseline. Average traces include all axonal events, and are normalized to background fluorescence prior to depolarization. n = 120 events of each type for axons and dendrites; 10 cells, 4 cultures/transfections; error bars indicate SEM. (e) BDNF-pHluorin fluorescence decrease events in dendrites during depolarization in the absence and presence of bafilomycin. (f) BDNF-pHluorin fluorescence increase events in axons during depolarization in the absence and presence of bafilomycin. (g) Quantitation of the fraction of events exhibiting no change in fluorescence, an increase in fluorescence (in axons), or a decrease in fluorescence (in dendrites) in BDNF-pHluorin and BDNF-pHluorin/mCherry-syt-IV transfected cells (n = 10 cells; 3 cultures/transfections; error bars indicate SEM).
The fluorescence drop in dendritic events could be due to 1) BDNF-pHluorin release, where a pH-dependent spike of increased fluorescence is too fast to resolve in the majority of events, or 2) endocytosis and re-acidification of BDNF-pHluorin containing vesicles. To discriminate between these two possibilities, we used bafilomycin to block re-acidification of newly endocytosed vesicles. Because each individual BDNF-pHluorin punctum exhibited the same characteristic fluorescence changes during successive rounds of depolarization, we compared individual puncta with and without bafilomycin. Dendritic events were unchanged by bafilomycin (Fig. 5e) and thus most likely correspond to fast and complete release of BDNF-pHluorin from vesicles.
In axons, the initial fluorescence increase (appearance of puncta) most likely corresponds to fusion pore opening, while the decay in fluorescence, as outlined above for dendritic events, could be due to loss of BDNF-pHluorin cargo from vesicles, or to reacidification of vesicles following fusion pore closure and endocytosis, without release of BDNF-pHluorin. Again, we used bafilomycin to distinguish these two possibilities. The fluorescence decrease of fast-decay axonal events was only partially blocked by bafilomycin, suggesting that the initial spike in fluorescence of these fast-decay events corresponds to partial release of BDNF-pHluorin contained in vesicles (Fig. 5f).
Over-expression of syt-IV decreased the proportion of fast-decay events (partial release) compared to slow-decay events (no release) in axons, but did not change the relative fraction of events showing no change in fluorescence, an increase in fluorescence (in axons), or a decrease in fluorescence (in dendrites) (Fig. 5g).
Together these experiments demonstrate that syt-IV regulates activity-driven exocytosis of BDNF in both axons and dendrites, where secretory events have distinct properties.
Syt-IV levels affect synaptic function
One of the most well-characterized effects of BDNF is to potentiate synaptic vesicle release from presynaptic nerve terminals26, 29-31. Since syt-IV negatively regulates BDNF release, we tested the effects of syt-IV on presynaptic function using FM dye destaining experiments.
To increase syt-IV, neurons were infected with syt-IV/GFP lentivirus under control of the synapsin promoter, which has the advantage of generating modest expression levels, similar to the physiological levels of syt-IV induced by activity (Fig. 6a). Synaptic vesicles were loaded with FM4-64 (Fig. 6b), and the rate of synaptic vesicle exocytosis was determined by loss of FM dye from presynaptic boutons following subsequent depolarization (Supplemental Fig. 5a, b). We found that the rate of FM dye destaining was slower in syt-IV over-expressing neurons compared to GFP-infected controls (Fig. 6c). Conversely, We syt-IV knockouts had a faster rate of destaining compared to wild-type (Fig. 6c). Thus, the level of syt-IV affects the rate of FM dye release from presynaptic boutons.
Figure 6. Syt-IV levels affect synaptic vesicle exocytosis.

(a) Western blots of syt-IV in control and activity-induced (treatment with 50 μm forskolin for four hours) conditions, and in control versus syt-IV over-expressing neurons. 12 μg of protein was loaded per lane for control versus forskolin, and 8 μg for control versus syt-IV over-expressing. Anti-tubulin served as a load control. (b) FM4-64 dye-loaded boutons in a culture over-expressing syt-IV/GFP via lentiviral infection. Scale bar is 5 μm. (c) Normalized FM dye destaining rates in control, syt-IV over-expressing and syt-IV-/- neurons. Arrow indicates addition of 45 mM KCl to depolarize neurons. Exponential fits yielded τ(sec) = 34.0 ± 2.6 (syt-IV lentivirus), 18.4 ± 1.4 (GFP lentivirus), 17.4 ± 0.8 (wt), and 9.8 ± 0.7 (syt-IV-/-). (d) Normalized destaining rates in syt-IV over-expressing and wt neurons treated with 25 nM BDNF for 1 hour, compared to traces from panel c. τ(sec) = 14.2 ± 3.0 (syt-IV lentivirus + 25 nM BDNF), 9.2 ± 0.8 (wt + 25 nM BDNF). (e) Normalized FM dye destaining rates in syt-IV over-expressing and wt neurons treated with 50 nM BDNF for 1 hour, compared to traces from panel c. τ(sec) = 10.4 ± 1.0 (syt-IV lentivirus + 50 nM BDNF), 8.7 ± 0.7 (wt + 50 nM BDNF). For all panels, n = 12-16 coverslips; 3-4 cultures, 10 boutons per coverslip for each condition; error bars indicate SEM.
If syt-IV acts to reduce BDNF release, which in turn slows the rate of FM dye efflux from nerve terminals, one would expect that the slower FM dye destaining rate in syt-IV over-expressing neurons could be rescued by exogenous BDNF. Indeed, we found that incubation with 25 nM BDNF for one hour prior to FM dye destaining experiments rescued the slow FM dye destaining rate in syt-IV overexpressing neurons, resulting in a destaining rate slightly faster than wild-type (Fig. 6d). Treatment of wild-type neurons with 25 nM BDNF also accelerated destaining, to a rate similar to that of syt-IV knockout neurons (Fig. 6d). Finally, we found that saturating amounts of BDNF (50 nM for 1 hour) resulted in virtually identical rapid destaining rates in wild-type and syt-IV over-expressing neurons (Fig. 6e). Together these experiments are consistent with the hypothesis that the decrease in FM dye destaining rate in syt-IV over-expressing neurons is caused by decreased secretion of BDNF.
Syt-IV affects presynaptic function trans-synaptically
Because syt-IV-containing vesicles undergo exocytosis in both axons and dendrites, and BDNF can act in an autocrine or a paracrine manner26, we next tested whether the increase in synaptic vesicle exocytosis caused by knockout of syt-IV is due to increased pre or post-synaptic release of BDNF using a co-culture assay. Two co-culture conditions were used; in the first, 95% of neurons were syt-IV knockout and 5% were wild-type neurons labeled with DiO. We then determined the rate of FM dye destaining in knockout presynaptic terminals contacting wild-type post-synaptic neurons, identified by DiO fluorescence (Fig. 7a). In the second condition, neurons were cultured in the reciprocal configuration; 95% of the neurons were wild-type and 5% were DiO labeled syt-IV knockout neurons, and the rate of FM dye destaining in wild-type terminals contacting DiO-labeled knockout neurons was determined (Fig. 7b).
Figure 7. Syt-IV modulates presynaptic function via regulation of post-synaptic BDNF release.

(a) Co-culture assay in which 5% of neurons are DiO-labelled wild-type, and 95% are unlabelled syt-IV-/- neurons. (b) Co-culture in which 5% of neurons are DiO-labelled syt-IV-/-, and 95% are wild-type. (c) Images of co-cultured wild-type and DiO-labelled syt-IV-/- neurons (left panel), and a DiO-labeled neuron stained with FM4-64, indicating boutons selected for analysis (right panel, arrowheads). Scale bar is 5 μm. (d) Normalized FM dye destaining rates in the indicated conditions. τ(sec) = 10.4 ± 0.7 (wt pre/syt-IV-/- post) and 16.0 ± 0.6 (syt-IV-/- pre/wt post). τ(sec) = 17.4 ± 0.8 (wt) and 9.8 ± 0.7 (syt-IV-/- ) as indicated in Figure 6. (e) Normalized destaining rates following 30-60 min incubation with 4 μg/ml TrkB-IgG scavenger. τ(sec) = 14.8 ± 1.4 (wt TrkB-IgG), 14.4 ± 1.3 (syt-IV-/- TrkB-IgG), 15.3 ± 1.4 (wt pre/syt-IV-/- post TrkB-IgG), and 14.7 ± 1.3 (syt-IV-/- pre/wt post TrkB-IgG), (n = 20 coverslips; 4 cultures, 2-5 boutons per coverslip for each condition; error bars indicate SEM). (f) mEPSCs recorded from syt-IV-/- and wt littermate neurons. (g) mEPSC frequency. (h) mEPSC amplitude (n = 20 cells; 4 cultures, 2-3 coverslips for each condition; error bars indicate SEM). Significance, determined by a Student’s t-test is shown relative to wild-type. One-way ANOVA p values for mEPSC frequency/amplitude were 1.2/ 0.006 (wt versus wt pre/sytIV-/- post), 0.043/ 0.702 (wt versus sytIV-/- pre/wt post), 0.606/ 0.004 (sytIV-/- versus sytIV-/- pre/wt post) and 0.033/ 0.178 (sytIV-/- versus wt pre/sytIV-/- post).
An example of a co-culture is shown in Figure 7c (left panel). FM dye-labeled boutons contacting DiO labeled dendrites were selected for analysis (Fig. 7c, right panel). Although we cannot be sure that all FM dye-labeled boutons on DiO labeled dendrites correspond to presynaptic knockout/post-synaptic wild-type synapses (or vice versa), the probability is high (~95%, assuming that neurons contact each other randomly and extend processes to similar distances) given the excess of knockout neurons in these cultures.
Upon depolarization, we observed a retrograde effect of syt-IV. The FM dye destaining rate from wild-type presynaptic terminals contacting post-synaptic syt-IV knockout neurons was increased. Conversely, the destaining rate from syt-IV knockout terminals contacting wild-type post-synaptic neurons remained “wild-type” (Fig. 7d). Thus, the phenotype of the post-synaptic side of the synapse determined the rate of FM dye destaining from the presynaptic side.
To test if this effect is due to increased BDNF release from syt-IV knockout neurons, we used a recombinant TrkB-IgG chimera, which acts as a scavenger by binding free BDNF32, 33. Pre-incubation of cultures with 4 μg/ml TrkB-IgG for 30-60 minutes largely reversed the enhanced rate of FM dye destaining mediated by syt-IV knockout neurons; the destaining rates of wild-type, syt-IV knockout, and the two co-culture configurations were indistinguishable (Fig. 7e). Thus, the enhancement of synaptic vesicle exocytosis in syt-IV knockout cultures is due to increased post-synaptic BDNF release. The destaining rate for all culture conditions following TrkB-IgG treatment was slightly faster than for wild-type (Supplemental Fig. 5c). This may be due to compensation for lack of BDNF signaling by separate slower mechanisms34, since we observe a gradual increase in wild-type destaining rate with increasing time of TrkB-IgG incubation, with maximal destaining rates following 24 hour incubation (data not shown).
We also investigated synaptic function by electrophysiological recording of mEPSCs in wild-type and syt-IV knockout cultures (Fig. 7g). Knockout neurons had an increase in both mEPSC frequency (Fig. 7h) and amplitude (Fig. 7i). The density of vGluT-positive terminals was similar in wild-type and syt-IV knockout cultures (Supplemental Fig. 1a, wild-type = 5.9 ± 0.2 puncta/ 10 μm, syt-IV knockout = 6.1 ± 0.2 puncta), suggesting that the increase in mEPSC frequency is due to an increase in spontaneous fusion rate, and not to an increase in the number of presynaptic terminals.
Using co-cultures, we found that the effect of syt-IV on mEPSC frequency was cell autonomous; the increase in frequency was due to a loss of presynaptic syt-IV, whereas loss of postsynaptic syt-IV had no effect on frequency. The increase in mEPSC amplitude was also cell autonomous, being due to the loss of post-synaptic syt-IV; loss of presynaptic syt-IV had no effect on the amplitude of quantal responses. These experiments indicate that syt-IV has both pre and postsynaptic cell autonomous effects, consistent with the autocrine actions of BDNF, in addition to the paracrine effects during depolarization described above (Supplemental Fig. 5d, e).
Syt-IV-/- mice have enhanced LTP due to excess BDNF release
Our finding that syt-IV regulates the release of BDNF to affect synaptic function, in conjunction with mounting evidence for a role of BDNF in LTP26, suggests a mechanism by which syt-IV might regulate the level of BDNF release during LTP and thus ultimately impact memory and learning22, 23. Syt-IV knockout mice would lack a “brake” on BDNF-containing vesicles, resulting in unchecked BDNF release during activity, and leading to changes in LTP. To test this hypothesis, we examined LTP in hippocampal slices from syt-IV knockout mice and wild-type littermates by simultaneous field potential recording and voltage imaging35.
Hippocampal slices were stained with the voltage-sensitive absorbance dye RH482, and Schaffer collaterals were stimulated (Fig. 8a) while recording in the CA1 region before and after induction of LTP by theta burst stimulation. The optical signal from the dye, and the fEPSP slope increased with increasing stimulus intensity (Fig. 8b), and both signals were potentiated following theta burst stimulation (Fig. 8c). The optical signal remained potentiated and reliably reported LTP for one hour or more after induction (Fig. 8d).
Figure 8. Syt-IV knockouts have enhanced LTP due to excess BDNF release.

(a) Hippocampal slice with voltage-sensitive dye optical signals from each of 464 photodiodes overlayed. CA1, CA3, subiculum (SB), entorhinal cortex (EC), and stimulating electrode (arrow) are indicated. A recording electrode was placed in the CA1 region. (b) Optical signal of the voltage sensitive dye and fEPSP slope versus stimulation intensity, showing an increase in both optical signal and fEPSP slope with increasing stimulation current. (c) fEPSP and optical signals before and after theta burst stimulation (TBS). (d) LTP measured as the increase in voltage-sensitive dye optical signal and fEPSP slope. The optical signal reliably reports LTP over the course of at least one hour. (e) Color-coded amplitude map of the optical signal detected in wild-type and syt-IV knockout hippocampal slices pre and one hour post TBS, normalized to maximum amplitude in each case. Right panels are LTP maps generated by subtracting the optical signal pre TBS from the optical signal one hour post TBS, and normalized to maximum amplitude increase for comparison between syt-IV knockout and wild-type. (f) Average LTP maps (n = 5 slices each for wild-type and syt-IV knockout). (g) Time course of LTP (n = 7 slices each for wild-type and syt-IV knockout, from 4 mice of each genotype; error bars indicate SEM). For BDNF blockade experiments, slices were incubated in 4 μg/ml TrkB-IgG scavenger for 2-3 hours prior to voltage imaging experiments. The stimulation current was 25 μA for all LTP experiments.
Using this technique, we found that syt-IV knockouts had increased magnitude and spatial spread of LTP following theta burst stimulation, compared to wild-type (Fig. 8e). This increase in LTP was most notable in the CA1 region and subiculum (Fig. 8f), and remained elevated over the course of at least one hour (Fig. 8g). To determine if the increase in LTP in syt-IV knockouts is due to increased BDNF release, we neutralized all free BDNF with the recombinant TrkB-IgG chimera. Incubation with 4 μg/ml TrkB-IgG scavenger for 2-3 hours decreased LTP in wild-type slices, as shown previously32, 33 and decreased LTP in syt-IV knockout slices to the same level as wild-type (Fig. 8g, Supplemental Fig. 6a). Incubation with a control Fc-IgG had no effect on LTP (Supplemental Fig. 6b). These data indicate that the enhanced LTP observed in syt-IV knockouts can be attributed to excess BDNF release, and further suggest that syt-IV acts to limit LTP by negatively regulating BDNF release.
Discussion
Our experiments reveal that syt-IV is targeted to BDNF-containing vesicles, and upregulated syt-IV traffics with BDNF to both axons and dendrites in response to activity. A key finding is that syt-IV negatively regulates the release of BDNF. Thus, the emerging view is that syt-IV dynamically controls BDNF release, possibly at specific sites along neuronal processes, to modify synapse and circuit function in response to alterations in network activity. Indeed, we found that regulation of BDNF release by syt-IV has significant effects on synaptic function and plasticity.
Using co-cultures of wild-type and syt-IV knockout neurons, we found that the loss of pre versus post-synaptic syt-IV affected distinct aspects of synaptic function. During evoked activity, loss of post-synaptic syt-IV, but not presynaptic syt-IV, enhanced the destaining rate of FM dye from presynaptic terminals via retrograde action of increased BDNF release. Our pHluorin experiments demonstrate that depolarization-induced dendritic BDNF release events are larger and faster than axonal events, suggesting that post-synaptic BDNF release dominates during evoked activity.
In contrast to the paracrine effects of syt-IV during evoked activity, we found cell-autonomous effects of syt-IV on spontaneous quantal transmission, consistent with an autocrine effect of BDNF26. Loss of presynaptic syt-IV increased the frequency of mEPSCs, without affecting amplitude, and loss of post-synaptic syt-IV increased amplitude without affecting frequency. At present, it is not clear whether these potentially autocrine effects are due to BDNF, or to the secretion of other factors from syt-IV containing vesicles. Nonetheless, the enhanced synaptic function observed in syt-IV knockouts is consistent with the effects of increased BDNF29, 36, 37.
We found that syt-IV knockouts have enhanced LTP, which is entirely due to excess BDNF. The observed increase in LTP is consistent with a previous study showing enhanced potentiation in syt-IV knockouts for several minutes following LTP induction with 100 Hz trains of action potentials23. The more dramatic effect seen in our study could be a result of induction protocols, since BDNF is more efficiently released by patterned stimuli such as the theta burst protocol used in our experiments, than by trains of action potentials28, 38. Syt-IV knockouts have enhanced LTP, yet these mice have deficits in learning and memory22, 23, implying that a certain range of potentiation is optimal. Syt-IV may act to maintain an upper limit of LTP by limiting neurotrophin release.
The BDNF-dependent enhancement of FM dye release that we observe in syt-IV knockouts could in part explain why knockouts have enhanced LTP. The rate of FM dye release from presynaptic terminals in hippocampal slices is enhanced following LTP induction, and this enhancement is absent in BDNF knockout mice, where normal LTP is also absent31. We note that the faster FM dye destaining rate in syt-IV knockout neurons could be due either to an increase in the number of synaptic vesicles that fuse, or to an alteration in the mode of synaptic vesicle exocytosis. If BDNF promotes full fusion events (complete release of FM dye from single vesicles) over kiss-and-run events (partial release of dye from a transient fusion pore39, 40), this could result in the increased destaining rate observed in syt-IV knockouts. We note that a recent study reported no effect of syt-IV over-expression on FM dye destaining kinetics in autapses15. A network of neurons may be required to see an effect of syt-IV, which is not evident in single autaptic neurons (Liu et al., submitted).
Our findings would seem to suggest that syt-IV affects LTP by modifying post-synaptic release of BDNF, which then acts as a retrograde signal to alter presynaptic vesicle release. However, BDNF-mediated LTP has been reported to be expressed exclusively postsynaptically41, exclusively presynaptically31, or at both locations42. A definitive demonstration of the effects of pre- versus post-synaptic loss of syt-IV on LTP will require selective pre- and postsynaptic knockdown studies. Nonetheless, our studies show that syt-IV limits the extent of LTP by limiting BDNF release.
The incorporation of syt-IV into BDNF-containing vesicles could inhibit release via a decrease in the frequency of fusion events owing to impaired membrane and SNARE binding activity and/or by favoring the formation of tiny fusion pores that are too small to allow exit of neuropeptides10, 43. The observed reduction in amplitude of BDNF-pHluorin events in dendrites and axons by over-expression of syt-IV could be due to fewer BDNF vesicles undergoing fusion, or to release of less BDNF-pHluorin cargo from individual vesicles. Our results, and those of a concurrent study (Matsuda et al. submitted), indicate that axons and dendrites exhibit distinct modes of BDNF release assayed by BDNF-pHluorin. In dendrites, release events are consistent with full fusion and complete release of BDNF-pHluorin. Thus, the reduction in amplitude of these events by syt-IV likely corresponds to a reduction in the number of vesicles that fuse. In axons, we observed events corresponding to partial release of BDNF-pHluorin from vesicles, where syt-IV inhibited secretion by increasing the proportion of “non-productive” fusion events corresponding to transient fusion pore opening without release of BDNF.
In addition to differences in the mode of BDNF release in axons versus dendrites, our results suggest that pH regulation of BDNF-containing vesicles may be distinct in these two compartments. Indeed, pHluorin-conjugated tetanus neurotoxin is transported in vesicles shared with BDNF, and these vesicles display pH values ranging from acidic to neutral44, 45. This raises the interesting possibility that distinct types of neurotrophin-containing vesicles may be differentially sorted to axons versus dendrites.
In co-cultures, in which only 5% of neurons lack syt-IV (and therefore have excess BDNF release) we observe an enhancement in FM dye destaining rate specifically in presynaptic terminals contacting these knockout neurons. This finding suggests that syt-IV/BDNF acts in a highly specific, targeted manner, rather than as a more general factor that acts diffusely. This is intriguing in light of the idea that BDNF-TrkB signaling may be involved in synaptic tagging, where proteins are selectively trafficked to tetanized synapses to modulate their function46. Given the high steady state levels of syt-IV in the Golgi, syt-IV vesicles could be rapidly exported from the Golgi in response to activity. However, lack of syt-IV affects LTP within minutes of induction; if syt-IV is recruited to specific sites during (and not prior to) LTP to inhibit potentiation, it must do so extremely rapidly. We found that syt-IV-containing vesicles are present throughout axons and dendrites in resting conditions, and these vesicles are highly dynamic. This constant transit of vesicles may insure that they can be rapidly recruited to active synapses without requiring new synthesis and trafficking from the cell body47. Studies are currently underway to determine whether and how syt-IV/BDNF vesicles are recruited to specific sites in response to neuronal activity.
Materials and Methods
Hippocampal Cultures, Co-cultures, Transfection and Lentiviral Infection
Syt-IV-/- mice were provided by H. Herschman (University of California, Los Angeles). All procedures involving animals were performed in accordance with NIH guidelines, as approved by the Animal Care and Use Committee of the University of Wisconsin, Madison. Hippocampi were isolated from P1-3 mice or E18-19 rats and plated at 25,000-50,000 cells/cm2 on poly-lysine coated coverslips (Carolina Biologicals) in Neurobasal supplemented with 2% B-27 and 2 mM Glutamax (Gibco/ Invitrogen). For co-cultures, equal numbers of syt-IV-/- and wild-type pups were dissected. 5% of neurons from each genotype were dye-labeled with 40 μg/ml DiO in Neurobasal for 20 min at 37° C, followed by centrifugation for 90 sec at 1000 rpm to remove excess dye48. Dye-labeled cells were then mixed with 95% unlabeled neurons of the opposite genotype. The two mixtures of cells (5% DiO-labeled syt-IV-/-/95% wild-type, and 5% DiO-labeled wild-type/95% syt-IV-/-) were plated at equal density.
Neurons in 24-well plates were transfected with Lipofectamine 2000 (Invitrogen) at 6-9 DIV, or with calcium phosphate at 3-4 DIV. For calcium phosphate transfection, medium was removed, saved, and replaced with Optimem (Life Technologies). 45 μl transfection buffer (274 mM NaCl, 10 mM KCl, 1.4 mM Na2HPO4, 15 mM glucose, 42 mM Hepes, pH 7.06) was added dropwise to 45 μl 250 mM CaCl2 plus 3 μg DNA, under gentle vortex, and 30 μl of this mixture was added per well for 60-90 min. Cells were then washed 3× in Neurobasal medium, and saved medium was replaced.
Syt-IV lentivirus was generated by co-transfecting a syt-IV/GFP double synapsin promoter lentiviral construct with two other packaging vectors encoding VSV-G, and gag and pol, into HEK293T cells. The supernatant was collected after 48-72 hrs, purified by filtration through a 0.45 μm filter, and centrifuged at 70,000g for 2 h to concentrate virus. Viral particles were resuspended in PBS overnight at 4° C and viral titer assessed by GFP fluorescence.
Antibodies, Reagents, and DNA Constructs
Antibodies used were: polyclonal syt-IV, VGAT (Synaptic Systems), VGluT1, BDNF (Promega), synaptophysin mAb 7.2, syt-I polyclonal 604.4 (provided by R. Jahn, Max Planck Institute for Biophysical Chemistry, Göttingen, Germany), monoclonal syt-IV, GFP (Abcam), beta-tubulin E7 (Developmental Studies Hybridoma Bank), Cy2 and Cy3 secondary antibodies (Jackson Immunologicals), and Alexa 647 secondary antibody (Molecular Probes). FM1-43 and FM4-64 lipophilic dyes were from Molecular Probes. Forskolin and capsaicin were from Sigma, and BDNF was from Promega.
Mammalian expression constructs used were: PSD95-GFP, synaptophysin-GFP (provided by T. Dresbach, University of Heidelberg, Germany), TRPV1/GAP43-GFP (provided by G. Miesenboeck, Yale University). pHluorin-syt-IV was constructed by replacing the syt-I in pHluorin-tagmin (provided by T. Ryan, Weill Medical College of Cornell University, New York, NY) between the MluI and NotI sites with PCR amplified syt-IV. mCherry-syt-IV and GFP-syt-IV were made by replacing the pHluorin with mCherry or GFP, respectively. BDNF-GFP was constructed by inserting PCR amplified BDNF cDNA (provided by P. Barker, McGill University, Montreal) between the BamHI and EcoRI sites of pEGFP-N1, as previously described49. BDNF-pHluorin was constructed by replacing the GFP with the pHluorin described above. The syt-IV lentivirus construct was made by inserting syt-IV into a double synapsin promoter lentiviral construct (provided by F. Gomez-Scholl, University of Seville, Spain) between the BamHI and NotI restriction sites.
Immunocytochemistry, Image Acquisition, Quantitation and Statistical Analysis
Cells were fixed with 4% paraformaldehyde, permeabilized and blocked in 10% goat serum and 0.1% Triton X-100, and immunostained at room temp for 2 h, or overnight at 4°C. Images were acquired on an Olympus FV1000 upright confocal microscope with a 60× 1.10 NA water immersion lens. For quantitation of percent colocalization images were acquired with identical laser and gain settings and imported into Metamorph (Improvision). Channels were thresholded separately to include all recognizable puncta. Percent colocalization was determined as percent of the total thresholded area of one channel that overlapped with thresholded area of the corresponding channel. Statistical significance for all quantitation was determined by a Student’s t-test where * = p < 0.05, ** = p < 0.01, and *** = p < 0.001, or by one-way ANOVA.
In Situ ELISAs
In situ ELISAs were performed as previously described28 using the BDNF Emax ImmunoAssay System (Promega). ELISA plates were coated with poly-lysine, and then with anti-BDNF monoclonal antibody at 4°C overnight. Plates were washed and blocked, followed by two 1 h incubations with culture medium to remove wash residue. Neurons were plated at high density (200,000 cells/well) in anti-BDNF-coated wells, and grown for 3-4 days, during which secreted BDNF was released onto the plate surface. Plates were then extensively washed to remove cells. Total BDNF (intra- and extracellular) was determined by lysing cells in duplicate wells with 1% Triton-X100 and 0.05% SDS prior to washing to release all intracellular BDNF. Anti-BDNF polyclonal antibody was then applied to plates, and subsequent steps performed according to the manufacturer’s protocol. Absorbance values were read at 450 nm in a plate reader. Positive (BDNF standards) and negative (culture medium) wells were included in all experiments. Neurons infected with GFP lentivirus served as controls for syt-IV lentiviral infection. Wild-type littermate neurons served as controls for syt-IV-/- neurons. The amount of secreted BDNF over 3-4 days corresponded to ~5-10% of the total BDNF determined by cell lysis.
Western Blots
For western blots of syt-IV, 12-14 DIV neurons in 10 cm dishes were washed in PBS. 1 ml hypotonic buffer (10 mM Tris-HCl pH 7.4) was added per plate for 5 min. Cells were harvested by scraping in 0.5 ml homogenization buffer (320 mM sucrose, 1 mM EDTA, 10 mM Tris-HCl pH 7.4) and passed through a 27-gauge needle 10x. A final concentration of 150 mM NaCl was added to maintain osmolarity. Lysates were centrifuged at 4000 rpm for 10 min to pellet nuclei and cellular debris. The supernatant was collected and protein concentration determined by BCA assay. Equal amounts of protein were loaded per lane for comparison of syt-IV levels between conditions, resolved by SDS-PAGE, and analyzed by immunoblotting.
Immuno-organelle Isolation
30 μL (50% slurry in TBS) of Protein A beads (Pierce) were blocked overnight with 10 μl control mouse ascites fluid, washed 3× in TBS, incubated with 10 μl rabbit polyclonal anti-syt-IV, anti-synaptophysin (mAb 7.2), or control ascites for 6 h and washed 3× with TBS. 12-14 DIV neurons in 10 cm dishes were washed twice in PBS, and 1 ml of TBS added per plate for 5 min. Cells were harvested by scraping and passed through a 27-gauge needle 10x. Cell lysate was centrifuged at 1000 × g for 15 min to pellet nuclei and cellular debris. The supernatant was collected and protein concentration determined by BCA assay. Lysates were incubated with beads at 4°C with shaking overnight. Samples were washed 3× with TBS, by centrifugation at 500 × g for 5 minutes, and bound protein was eluted from beads by boiling in SDS sample buffer. 5 or 2.5 μg of supernatant, and 90% or 30% of the pellet were resolved by SDS-PAGE and analyzed by immunoblotting.
Timelapse, FM Dye Destaining, and pHluorin Experiments
For timelapse experiments, neurons were transferred to a live imaging chamber (Warner Instruments) containing bathing saline solution (140 mM NaCl, 5 mM KCl, 2 mM CaCl2, 2 mM MgCl2, 5.5 mM glucose, 20 mM Hepes, pH = 7.3). Images were acquired at 1 sec intervals sequentially for BDNF-GFP (484ex/517em) and mCherry-syt-IV (555ex/605em) on a Nikon TE300 inverted microscope with a Roper Scientific Photometric Cascade IIB EMCCD camera, and Lambda DG-4 high-speed wavelength switcher interfaced with Metamorph software.
For FM dye experiments, neurons were loaded with 10 μM FM dye (FM1-43 or FM4-64) in depolarizing buffer (100 mM NaCl, 45 mM KCl, 2 mM CaCl2, 2 mM MgCl2, 5.5 mM glucose, 20 mM HEPES, ph=7.3) for 2 min, and washed in bathing solution for 5 min to remove extracellular dye. Images of dye-labeled boutons (484ex/755em for FM4-64, or 484ex/605em for FM1-43) were acquired at 1 sec intervals. A baseline of 10 images was collected before addition of depolarizing buffer to destain neurons. Dyelabeled boutons were selected as regions in Metamorph and fluorescence intensity plotted versus time. Destain traces were normalized by setting starting fluorescence to one and complete destain (disappearance of bouton to background levels) to zero, for comparison of rates. For BDNF scavenger experiments, 4 μg/ml TrkB-IgG (R & D Systems) was added to culture medium for 30-60 min prior to FM destaining.
For pHluorin experiments, transfected cells (identified by faint GFP fluorescence in non-depolarizing conditions) in which axons and dendrites were clearly discernible by morphology, were selected. Images were collected during depolarization, as described above, with 484ex/517em filter sets. Puncta that did not exhibit any lateral movement during image acquisition were chosen for analysis. For inhibition of the vesicular H+ ATPase, neurons were incubated with 1 μm bafilomycin for 2 min (Calbiochem) prior to imaging experiments.
Electrophysiology
Whole cell recordings of mEPSCs were made from 15-21 DIV neurons from syt-IV-/- and wild-type littermates. For co-cultures, neurons were identified by DiO fluorescence prior to recording. Neurons were voltage clamped at −70 mV in the presence of 1 μm TTX and currents recorded using an EPC-10/2 amplifier (HEKA Electronics, Germany) and Patchmaster software (HEKA). Cells were continuously perfused with extracellular solution consisting of (in mM): 125 NaCl, 4 KCl, 1.25 NaH2PO4, 26 NaHCO3, 2.5 CaCl2, 1.3 MgCl2, 10 glucose, bubbled with 95% O2 and 5% CO2 (pH 7.3, 310 mOsm). Intracellular solution consisted of, in mM: 130 K-gluconate, 10 EGTA,10 HEPES, 5 Phosphocreatine, 2 Mg-ATP, 0.3 Na-GTP, (pH 7.3, 310 mOsm). Recordings were filtered at 2.9 kHz and digitized at 5 kHz. Events were detected with MiniAnalysis software (Synaptosoft) using a threshold of five times the RMS noise.
Voltage Imaging
Detection of LTP by voltage imaging was performed as previously described35. Five-week old mice were euthanized with CO2. Brains were removed into ice-cold cutting solution consisting of, in mM: 124 NaCl, 3.2 KCl, 26 NaHCO3, 1.25 NaH2PO4, 1 CaCl2, 6 MgSO4,10 glucose, and bubbled with 95% O2/5% CO2. Horizontal 350 μm thick hippocampal slices were cut with a tissue slicer (HR2, Sigmann Elektronik, Germany). A cut at the CA2 or CA3 region was made to prevent epileptiform activity in LTP experiments. Slices were incubated in bubbled artificial cerebrospinal fluid (ACSF), identical to the cutting solution but containing 2.5 mM CaCl2 and 1.3 mM MgSO4, for 1 h, stained with bubbled ACSF containing 0.02 mg/ml of the voltage-sensitive absorbance dye RH482 (NK3630, Hayashibara Biochemical Laboratories, Okayama, Japan) for 1 hr, and then returned to ACSF. For BDNF scavenger experiments, slices were incubated with 4 μg/ml TrkB-IgG for 2-3 h prior to recording and compared to control slices incubated without TrkB-IgG, or with 4 μg/ml control Fc-IgG (R & D Systems), for the same amount of time (2-3 h). During recording, slices were continuously perfused with bubbled ACSF at 29-31°C. Slices were stimulated with 200 μs current pulses delivered by a stimulus isolator (Model A365, World Precision Instruments, Sarasota, FL) every 30 seconds. LTP was induced by three theta bursts (TB) separated by 10 seconds. Each TB consisted of 10 bursts at 5 Hz and each burst consisted of six pulses at 100 Hz. The stimulation current was 25 μA for all LTP experiments. Field potentials were recorded with an Axopatch 200B amplifier (Axon Instruments, Foster City, CA). The photodiode array instrumentation has been previously described35. Optical signals (average of four consecutive trials) are presented as the change in transmitted light divided by the resting light intensity (ΔI/I), and are proportional to the membrane potential change within the physiological range of voltages50. LTP was determined by the difference in optical signal amplitude pre and post TBS. To generate average LTP maps, images were overlaid according to morphological landmarks, and the average optical signal for each photodiode calculated. LTP time courses were generated from the average peak optical signal (determined by the average signal of six photodiodes in the middle of the CA1 region) at each time point.
Supplementary Material
Acknowledgements
We thank Mu-ming Poo and Naoto Matsuda for sharing their BDNF-pHluorin data with us and for comments on the manuscript, and members of the Chapman and Jackson labs for helpful discussions and comments on the manuscript. C.D. designed and performed experiments, analyzed data and wrote the paper, H.L. performed and analyzed mEPSC recordings, F.M.D constructed syt-IV fusion proteins, P.Y.C. wrote software for analysis of voltage imaging data, E.R.C., and M.B.J. supervised the project. This work was supported by an NIH NRSA to C.D. (NS049748) NIH grants to M.B.J. (NS30016 and NS44057), and NIH (NIGMS GM 56827 and NIMH MH61876) and AHA (0440168N) grants to E.R.C. E.R.C. is an Investigator of the Howard Hughes Medical Institute.
References
- 1.Chapman ER. How Does Synaptotagmin Trigger Neurotransmitter Release? Annu Rev Biochem. 2008;77:615–41. doi: 10.1146/annurev.biochem.77.062005.101135. [DOI] [PubMed] [Google Scholar]
- 2.Koh TW, Bellen HJ. Synaptotagmin I, a Ca2+ sensor for neurotransmitter release. Trends Neurosci. 2003;26:413–422. doi: 10.1016/S0166-2236(03)00195-4. [DOI] [PubMed] [Google Scholar]
- 3.Bhalla A, Tucker WC, Chapman ER. Synaptotagmin isoforms couple distinct ranges of Ca2+, Ba2+, and Sr2+ concentration to SNARE-mediated membrane fusion. Mol Biol Cell. 2005;16:4755–4764. doi: 10.1091/mbc.E05-04-0277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hui E, et al. Three distinct kinetic groupings of the synaptotagmin family: candidate sensors for rapid and delayed exocytosis. Proc Natl Acad Sci U S A. 2005;102:5210–5214. doi: 10.1073/pnas.0500941102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bhalla A, Chicka MC, Chapman ER. Analysis of the synaptotagmin family during reconstituted membrane fusion: Uncovering a class of inhibitory isoforms. J Biol Chem. 2008;283:21799–807. doi: 10.1074/jbc.M709628200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xu J, Mashimo T, Sudhof TC. Synaptotagmin-1, -2, and -9: Ca(2+) sensors for fast release that specify distinct presynaptic properties in subsets of neurons. Neuron. 2007;54:567–581. doi: 10.1016/j.neuron.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 7.Vician L, et al. Synaptotagmin IV is an immediate early gene induced by depolarization in PC12 cells and in brain. Proc Natl Acad Sci U S A. 1995;92:2164–2168. doi: 10.1073/pnas.92.6.2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ibata K, Fukuda M, Hamada T, Kabayama H, Mikoshiba K. Synaptotagmin IV is present at the Golgi and distal parts of neurites. J Neurochem. 2000;74:518–526. doi: 10.1046/j.1471-4159.2000.740518.x. [DOI] [PubMed] [Google Scholar]
- 9.Chapman ER, Desai RC, Davis AF, Tornehl CK. Delineation of the oligomerization, AP-2 binding, and synprint binding region of the C2B domain of synaptotagmin. J Biol Chem. 1998;273:32966–32972. doi: 10.1074/jbc.273.49.32966. [DOI] [PubMed] [Google Scholar]
- 10.Wang CT, et al. Different domains of synaptotagmin control the choice between kiss-and-run and full fusion. Nature. 2003;424:943–947. doi: 10.1038/nature01857. [DOI] [PubMed] [Google Scholar]
- 11.Wang CT, et al. Synaptotagmin modulation of fusion pore kinetics in regulated exocytosis of dense-core vesicles. Science. 2001;294:1111–1115. doi: 10.1126/science.1064002. [DOI] [PubMed] [Google Scholar]
- 12.Ahras M, Otto GP, Tooze SA. Synaptotagmin IV is necessary for the maturation of secretory granules in PC12 cells. J Cell Biol. 2006;173:241–251. doi: 10.1083/jcb.200506163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang Q, Fukuda M, Van Bockstaele E, Pascual O, Haydon PG. Synaptotagmin IV regulates glial glutamate release. Proc Natl Acad Sci U S A. 2004;101:9441–9446. doi: 10.1073/pnas.0401960101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Osborne SL, Herreros J, Bastiaens PI, Schiavo G. Calcium-dependent oligomerization of synaptotagmins I and II. Synaptotagmins I and II are localized on the same synaptic vesicle and heterodimerize in the presence of calcium. J Biol Chem. 1999;274:59–66. doi: 10.1074/jbc.274.1.59. [DOI] [PubMed] [Google Scholar]
- 15.Ting JT, Kelley BG, Sullivan JM. Synaptotagmin IV does not alter excitatory fast synaptic transmission or fusion pore kinetics in mammalian CNS neurons. J Neurosci. 2006;26:372–380. doi: 10.1523/JNEUROSCI.3997-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ibata K, et al. Non-polarized distribution of synaptotagmin IV in neurons: evidence that synaptotagmin IV is not a synaptic vesicle protein. Neurosci Res. 2002;43:401–406. doi: 10.1016/s0168-0102(02)00066-4. [DOI] [PubMed] [Google Scholar]
- 17.Berton F, et al. Synaptotagmin I and IV define distinct populations of neuronal transport vesicles. Eur J Neurosci. 2000;12:1294–1302. doi: 10.1046/j.1460-9568.2000.00013.x. [DOI] [PubMed] [Google Scholar]
- 18.Littleton JT, Serano TL, Rubin GM, Ganetzky B, Chapman ER. Synaptic function modulated by changes in the ratio of synaptotagmin I and IV. Nature. 1999;400:757–760. doi: 10.1038/23462. [DOI] [PubMed] [Google Scholar]
- 19.Robinson IM, Ranjan R, Schwarz TL. Synaptotagmins I and IV promote transmitter release independently of Ca(2+) binding in the C(2)A domain. Nature. 2002;418:336–340. doi: 10.1038/nature00915. [DOI] [PubMed] [Google Scholar]
- 20.Pawlu C, DiAntonio A, Heckmann M. Postfusional control of quantal current shape. Neuron. 2004;42:607–618. doi: 10.1016/s0896-6273(04)00269-7. [DOI] [PubMed] [Google Scholar]
- 21.Yoshihara M, Adolfsen B, Galle KT, Littleton JT. Retrograde signaling by Syt 4 induces presynaptic release and synapse-specific growth. Science. 2005;310:858–863. doi: 10.1126/science.1117541. [DOI] [PubMed] [Google Scholar]
- 22.Ferguson GD, Anagnostaras SG, Silva AJ, Herschman HR. Deficits in memory and motor performance in synaptotagmin IV mutant mice. Proc Natl Acad Sci U S A. 2000;97:5598–5603. doi: 10.1073/pnas.100104597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ferguson GD, Wang H, Herschman HR, Storm DR. Altered hippocampal short-term plasticity and associative memory in synaptotagmin IV (-/-) mice. Hippocampus. 2004;14:964–74. doi: 10.1002/hipo.20013. [DOI] [PubMed] [Google Scholar]
- 24.Denovan-Wright EM, Newton RA, Armstrong JN, Babity JM, Robertson HA. Acute administration of cocaine, but not amphetamine, increases the level of synaptotagmin IV mRNA in the dorsal striatum of rat. Brain Res Mol Brain Res. 1998;55:350–354. doi: 10.1016/s0169-328x(98)00042-4. [DOI] [PubMed] [Google Scholar]
- 25.Peng W, et al. Synaptotagmin I and IV are differentially regulated in the brain by the recreational drug 3,4-methylenedioxymethamphetamine (MDMA) Brain Res Mol Brain Res. 2002;108:94–101. doi: 10.1016/s0169-328x(02)00518-1. [DOI] [PubMed] [Google Scholar]
- 26.Poo MM. Neurotrophins as synaptic modulators. Nat Rev Neurosci. 2001;2:24–32. doi: 10.1038/35049004. [DOI] [PubMed] [Google Scholar]
- 27.Zemelman BV, Nesnas N, Lee GA, Miesenbock G. Photochemical gating of heterologous ion channels: remote control over genetically designated populations of neurons. Proc Natl Acad Sci U S A. 2003;100:1352–1357. doi: 10.1073/pnas.242738899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Balkowiec A, Katz DM. Cellular mechanisms regulating activity-dependent release of native brain-derived neurotrophic factor from hippocampal neurons. J Neurosci. 2002;22:10399–10407. doi: 10.1523/JNEUROSCI.22-23-10399.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tyler WJ, Pozzo-Miller LD. BDNF enhances quantal neurotransmitter release and increases the number of docked vesicles at the active zones of hippocampal excitatory synapses. J Neurosci. 2001;21:4249–4258. doi: 10.1523/JNEUROSCI.21-12-04249.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tyler WJ, et al. BDNF increases release probability and the size of a rapidly recycling vesicle pool within rat hippocampal excitatory synapses. J Physiol. 2006;574:787–803. doi: 10.1113/jphysiol.2006.111310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zakharenko SS, et al. Presynaptic BDNF required for a presynaptic but not postsynaptic component of LTP at hippocampal CA1-CA3 synapses. Neuron. 2003;39:975–990. doi: 10.1016/s0896-6273(03)00543-9. [DOI] [PubMed] [Google Scholar]
- 32.Chen G, Kolbeck R, Barde YA, Bonhoeffer T, Kossel A. Relative contribution of endogenous neurotrophins in hippocampal long-term potentiation. J Neurosci. 1999;19:7983–7990. doi: 10.1523/JNEUROSCI.19-18-07983.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kang H, Welcher AA, Shelton D, Schuman EM. Neurotrophins and time: different roles for TrkB signaling in hippocampal long-term potentiation. Neuron. 1997;19:653–664. doi: 10.1016/s0896-6273(00)80378-5. [DOI] [PubMed] [Google Scholar]
- 34.Rutherford LC, Nelson SB, Turrigiano GG. BDNF has opposite effects on the quantal amplitude of pyramidal neuron and interneuron excitatory synapses. Neuron. 1998;21:521–530. doi: 10.1016/s0896-6273(00)80563-2. [DOI] [PubMed] [Google Scholar]
- 35.Chang PY, Jackson MB. Heterogeneous Spatial Patterns of Long-Term Potentiation in Hippocampal Slices. J Physiol. 2006;576:427–43. doi: 10.1113/jphysiol.2006.112128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Levine ES, Crozier RA, Black IB, Plummer MR. Brain-derived neurotrophic factor modulates hippocampal synaptic transmission by increasing N-methyl-D-aspartic acid receptor activity. Proc Natl Acad Sci U S A. 1998;95:10235–10239. doi: 10.1073/pnas.95.17.10235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Caldeira MV, et al. BDNF regulates the expression and the synaptic delivery of AMPA receptor subunits in hippocampal neurons. J Biol Chem. 2007;282:12619–28. doi: 10.1074/jbc.M700607200. [DOI] [PubMed] [Google Scholar]
- 38.Aicardi G, et al. Induction of long-term potentiation and depression is reflected by corresponding changes in secretion of endogenous brain-derived neurotrophic factor. Proc Natl Acad Sci U S A. 2004;101:15788–15792. doi: 10.1073/pnas.0406960101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aravanis AM, Pyle JL, Tsien RW. Single synaptic vesicles fusing transiently and successively without loss of identity. Nature. 2003;423:643–647. doi: 10.1038/nature01686. [DOI] [PubMed] [Google Scholar]
- 40.Richards DA, Bai J, Chapman ER. Two modes of exocytosis at hippocampal synapses revealed by rate of FM1-43 efflux from individual vesicles. J Cell Biol. 2005;168:929–939. doi: 10.1083/jcb.200407148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kovalchuk Y, Hanse E, Kafitz KW, Konnerth A. Postsynaptic Induction of BDNF-Mediated Long-Term Potentiation. Science. 2002;295:1729–1734. doi: 10.1126/science.1067766. [DOI] [PubMed] [Google Scholar]
- 42.Gartner A, et al. Hippocampal long-term potentiation is supported by presynaptic and postsynaptic tyrosine receptor kinase B-mediated phospholipase Cgamma signaling. J Neurosci. 2006;26:3496–3504. doi: 10.1523/JNEUROSCI.3792-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Perrais D, Kleppe IC, Taraska JW, Almers W. Recapture after exocytosis causes differential retention of protein in granules of bovine chromaffin cells. J Physiol. 2004;560:413–428. doi: 10.1113/jphysiol.2004.064410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bohnert S, Schiavo G. Tetanus toxin is transported in a novel neuronal compartment characterized by a specialized pH regulation. J Biol Chem. 2005;280:42336–42344. doi: 10.1074/jbc.M506750200. [DOI] [PubMed] [Google Scholar]
- 45.Deinhardt K, et al. Rab5 and Rab7 control endocytic sorting along the axonal retrograde transport pathway. Neuron. 2006;52:293–305. doi: 10.1016/j.neuron.2006.08.018. [DOI] [PubMed] [Google Scholar]
- 46.Lu Y, Christian K, Lu B. BDNF: A key regulator for protein synthesis-dependent LTP and long-term memory? Neurobiol Learn Mem. 2007;89:312–23. doi: 10.1016/j.nlm.2007.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shakiryanova D, Tully A, Levitan ES. Activity-dependent synaptic capture of transiting peptidergic vesicles. Nat Neurosci. 2006;9:896–900. doi: 10.1038/nn1719. [DOI] [PubMed] [Google Scholar]
- 48.Tarsa L, Goda Y. Synaptophysin regulates activity-dependent synapse formation in cultured hippocampal neurons. Proc Natl Acad Sci U S A. 2002;99:1012–1016. doi: 10.1073/pnas.022575999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hartmann M, Heumann R, Lessmann V. Synaptic secretion of BDNF after high-frequency stimulation of glutamatergic synapses. Embo J. 2001;20:5887–5897. doi: 10.1093/emboj/20.21.5887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Waggoner AS, Grinvald A. Mechanisms of rapid optical changes of potential sensitive dyes. Ann N Y Acad Sci. 1977;303:217–241. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.