

Oxindoles with a Twist
Transfer hydrogenation of substituted isatins in the presence of allyl acetate, α-methyl allyl acetate or 1,1,-dimethylallene employing an ortho-cyclometallated iridium catalyst modified by CTH-(R)-P-PHOS provides products of carbonyl allylation, crotylation and reverse prenylation, respectively, in highly enantiomerically enriched form. These studies represent the first use of activated ketones as electrophilic partners in asymmetric C-C bond forming transfer hydrogenation.

Keywords: Iridium, Allylation, Crotylation, Prenylation, Transfer Hydrogenation, Catalytic, Enantioselective
3-Substituted-3-hydroxy-oxindoles appear as substructures within a fascinating array of natural products, including the convulutamydines,[1a,b] maremycins,[1c,d] donaxaridines,[1e,f] dioxibrassinins,[1g,h,i] celogentin K,[1j] hydroxyglucoisatisins[1k] and TMC-95A–D (Figure 1).[1l] While catalytic asymmetric additions to isatins are known,[2–6] highly enantioselective catalytic allylation, crotylation and reverse prenylation of isatins has remained elusive. In the course developing hydrogen-mediated C-C couplings beyond hydroformylation,[7–15] chiral ortho-cyclometallated iridium C,O-benzoates were found to catalyze highly enantioselective carbonyl allylation,[14a,b] crotylation[14c] and reverse prenylation[12d] under transfer hydrogenation conditions. In contrast to classical allylation procedures that employ stoichiometric organometallic reagents,[16] transfer hydrogenation protocols exploit allyl acetate, α-methyl allyl acetate and 1,1-dimethylallene as precursors to transient allyl-, crotyl- and prenylmetal intermediates, respectively.[12,14a–c] To further evaluate the scope of this emergent methodology, catalytic enantioselective additions to ketones were explored.[17,18] In this account, we report that activated ketones in the form of substituted isatins are subject to highly enantioselective carbonyl allylation, crotylation and reverse prenylation, constituting a convenient synthesis of optically enriched 3-substituted-3-hydroxy-oxindoles.
Figure 1.

Examples of naturally occurring 3-substituted-3-hydroxy-oxindoles.
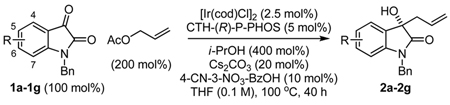
Our initial studies focused on the asymmetric allylation of N-benzyl isatin 1a. Using the cyclometallated C,O-benzoate generated in situ from [Ir(cod)Cl]2, BIPHEP and 4-chloro-3-nitrobenzoic acid,[14b] the coupling of allyl acetate (1000 mol%) to 1a at 100 °C in THF (0.2 M) delivered the tertiary homoallyl alcohol 2a in 42% isolated yield. Under otherwise identical conditions, but with a lower loading of allyl acetate (200 mol%) and optimization of reaction temperature, reaction time, and concentration, the isolated yield of homoallyl alcohol 2a was increased to 77%. An assay of chelating chiral phosphine ligands was undertaken, which revealed dramatic enhancement in the level of asymmetric induction at lower reaction temperatures. However, lower temperatures also diminished conversion. This impasse was resolved by increasing the loading of isopropanol from 200 mol% to 400 mol%, which enabled conversion of N-benzyl isatin 1a to homoallyl alcohol 2a in 73% isolated yield and 91% enantiomeric excess using CTH-(R)-P-PHOS as ligand. Notably, under analogous conditions employing our initially disclosed iridium catalyst modified by 3-nitrobenzoic acid,[14a,b] 2a is obtained in 61% isolated yield and 90% enantiomeric excess. These data further illustrate how catalyst performance is enhanced through structural variation of the C,O-benzoate moiety. Data pertaining to the optimization of the catalytic enantioselective allylation of N-benzyl isatin 1a is tabulated in the supporting information.
Optimal conditions identified for the conversion of N-benzyl isatin 1a to the hydroxy-oxindole 2a were applied to substituted isatins 1a–1g (Table 1). To our delight, the products of ketone allylation 2a–2g were produced in moderate to excellent isolated yield (65–92% yield) with uniformly high levels of optical enrichment (91–96% ee). The absolute stereochemical assignment of adducts 2a–2g are based upon that determined for the 5-bromo-dervative 2b via single crystal X-ray diffraction analysis using the anomalous dispersion method.
Table 1.
Catalytic enantioselective allylation N-benzyl isatins 1a–1g via iridium catalyzed C-C bond forming transfer hydrogenation.
 | ||||
|---|---|---|---|---|
| Entry | Isatins 1a–1g | Products | Yield (%) | ee (%) |
| 1 | 1a, N-benzyl isatin | 2a | 73 | 91 |
| 2b | 1b, 5-bromo-N-benzyl isatin | 2b | 83 | 94 |
| 3 | 1c, 5-methyl-N-benzyl isatin | 2c | 89 | 92 |
| 4 | 1d, 5-methoxy-N-benzyl isatin | 2d | 92 | 94 |
| 5 | 1e, 6-chloro-N-benzyl isatin | 2e | 73 | 96 |
| 6b | 1f, 6-bromo-N-benzyl isatin | 2f | 80 | 94 |
| 7c | 1g, 7-fluoro-N-benzyl isatin | 2g | 65 | 93 |
All reactions were performed in 13 × 100 mm pressure tubes. Cited yields are of material isolated by silica gel chromatography. Enantiomeric excess was determined by chiral stationary phase HPLC analysis. See supporting information for further details.
10 mol% loading of Cs2CO3 was used and the reaction was conducted for 72 hours.
400 mol% loading of allyl acetate was used.
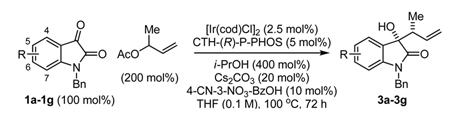
Given these favorable results, the crotylation of substituted isatins 1a–1g was attempted under identical conditions employing α-methyl allyl acetate as the crotyl donor (Table 2). The products of ketone crotylation 3a–3g were produced in moderate to excellent isolated yield (64–87% yield) with moderate to excellent levels of optical enrichment (80–92% ee). In general, crotylation required longer reaction times (Table 2, entries 1, 2, 5–7). Additionally, it was found that lower loadings of Cs2CO3 increased conversion in certain cases. The absolute stereochemical assignment of adducts 3a–3g are based upon that determined for the 5-bromo-dervative 3b via single crystal X-ray diffraction analysis using the anomalous dispersion method.
Table 2.
Catalytic enantioselective crotylation of N-benzyl isatins 1a–1g via iridium catalyzed C-C bond forming transfer hydrogenation.
 | ||||
|---|---|---|---|---|
| Entry | Isatins 1a–1g | Products | Yield (%) | ee (%), dr |
| 1b | 1a, N-benzyl isatin | 3a | 83 | 80, 13:1 |
| 2f | 1b, 5-bromo-N-benzyl isatin | 3b | 72 | 86, 16:1 |
| 3d,e | 1c, 5-methyl-N-benzyl isatin | 3c | 81 | 89, 18:1 |
| 4d,e | 1d, 5-methoxy-N-benzyl isatin | 3d | 87 | 92, 29:1 |
| 5b | 1e, 6-chloro-N-benzyl isatin | 3e | 70 | 91, 19:1 |
| 6f | 1f, 6-bromo-N-benzyl isatin | 3f | 81 | 89, 15:1 |
| 7b,c | 1g, 7-fluoro-N-benzyl isatin | 3g | 64 | 85, 19:1 |
As described in Table 2 footnotes.
10 mol% loading of Cs2CO3 was used.
400 mol% loading of allyl acetate was used.
Me-THF was used as solvent.
The reaction was run for 40 hours.
5 mol% loading of [Ir(cod)Cl]2, 10 mol% loading of CTH-(R)-P-PHOS and 20 mol% loading of 4-CN-3-NO2-BzOH were used.
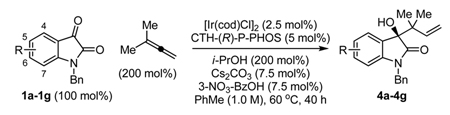
Finally, the reverse prenylation of substituted isatins 1a–1g was attempted (Table 3). To our delight, adducts 4a–4g were generated in uniformly high isolated yields (70–90% yield) and levels of optical enrichment (90–96 % ee) under mild conditions. Notably, this transformation enables creation of two contiguous quaternary carbon centers. The absolute stereochemical assignment of adducts 4a–4g are based upon that determined for the 5-bromo-dervative 4b via single crystal X-ray diffraction analysis using the anomalous dispersion method. Here, the enantiofacial selectivity of carbonyl addition is opposite to that observed in the case of allylation and crotylation.
Table 3.
Catalytic enantioselective prenylation of N-benzyl isatins 1a–1g via iridium catalyzed C-C bond forming transfer hydrogenation.
 | ||||
|---|---|---|---|---|
| Entry | Isatins 1a–1g | Products | Yield (%) | ee (%) |
| 1 | 1a, N-benzyl isatin | 4a | 90 | 96 |
| 2 | 1b, 5-bromo-N-benzyl isatin | 4b | 86 | 90 |
| 3 | 1c, 5-methyl-N-benzyl isatin | 4c | 79 | 93 |
| 4 | 1d, 5-methoxy-N-benzyl isatin | 4d | 81 | 96 |
| 5 | 1e, 6-chloro-N-benzyl isatin | 4e | 80 | 93 |
| 6b | 1f, 6-bromo-N-benzyl isatin | 4f | 70 | 93 |
| 7b | 1g, 7-fluoro-N-benzyl isatin | 4g | 79 | 94 |
As described in Table 2 footnotes.
The reaction was run for 72 hours.
The inversion in absolute stereochemistry observed in isatin reverse prenylation merits further explanation. The catalytic mechanism for carbonyl prenylation employing 1,1-dimethylallene is analogous to that previously reported for corresponding allylations and crotylations (Scheme 1, left).14b,c Assuming isatin crotylation occurs through a chair-like transition structure and an (E)-σ-crotyl iridium intermediate, previously proposed absolute stereochemical models agrees with the observed π-facial selectivity with respect to the crotyl partner.14c The latter observation suggests that isatin crotylation occurs by way of transition structure A, whereas isatin prenylation occurs by way of transition structures B. The basis of this partitioning may arise from non-bonded interactions of the axial methyl group of the σ-prenyl iridium intermediate with the amide π-bond of isatin, which is presumably more destabilizing than non-bonded interactions of the axial methyl group with the electron-deficient rim of the arene (Scheme 1, right).
Scheme 1.

A simplified catalytic mechanism depicting isatin prenylation via transfer hydrogenation (left) and a plausible stereochemical model accounting for the observed inversion in absolute stereochemistry in the prenylation of isatins (right).a
aLn = CTH-(R)-P-PHOS (omitted for clarity).
In summary, we report the first enantioselective allylations, crotylations and prenylations of isatin, which are achieved via isopropanol-mediated transfer hydrogenation. Unlike conventional allylation methodologies that employ stoichiometric quantities of allylmetal reagents, the present method exploits allyl acetate, α-methyl allyl acetate and 1,1-dimethylallene as precursors to transient allyl-, crotyl- and prenylmetal intermediates, respetively.[12,14a–c] To our knowledge, these studies represent the first examples of catalytic enantioselective ketone allylation, crotylation and prenylation in the absence of stoichiometric allylmetal reagents. Future studies will focus on the development of related C-C bond forming transfer hydrogenations and synthetic applications of the methods reported herein.
Footnotes
Acknowledgment is made to Merck, the Robert A. Welch Foundation, the American Chemical Society Green Chemistry Institute Pharmaceutical Roundtable and the NIH (RO1-GM069445) for partial support of this research. Dr. Oliver Briel of Umicore is thanked for the generous donation of [Ir(cod)Cl]2.
References
- 1.For natural products that possess 3-substituted-3-hydroxy-oxindole substructures, see: Kamano Y, Zhang H-P, Ichihara Y, Kizu H, Komiyama K, Petit GR. Tetrahedron Lett. 1995;36:2783–2784. Zhang H-P, Kamano Y, Ichihara Y, Kizu H, Komiyama K, Itokawa H, Pettit GR. Tetrahedron. 1995;51:5523–5528. Balk-Bindseil W, Helmke E, Weyland H, Laatsch H. Liebigs Ann. 1995:1291–1294. Tang Y-Q, Sattler I, Thiericke R, Grabley S. Eur. J. Org. Chem. 2001:261–267. Ubaidullaev KA, Shakirov R, Yunosov SY. Khim. Prir. Soedin. 1976;12:553–554. Rasmussen HB, MacLeod JK. J. Nat. Prod. 1997;60:1152–1154. doi: 10.1021/np970006a. Monde K, Sasaki K, Shirata A, Takasugi M. Phytochemistry. 1991;30:2915–2917. Monde K, Osawa S, Harada N, Takasugi M, Suchy M, Kutschy P, Dzurilla M, Balentova E. Chem. Lett. 2000;8:886–887. Monde K, Taniguchi T, Miura N, Nishimura S-I, Harada N, Dukor RK, Nafie LA. Tetrahedron Lett. 2003;44:6017–6020. Suzuki H, Morita H, Shiro M, Kobayashi J-i. Tetrahedron. 2004;60:2489–2495. Fréchard A, Fabre N, Péan C, Montaut S, Fauvel M-T, Rollin P, Fourasté I. Tetrahedron Lett. 2001;42:9015–9017. Kohno J, Koguchi Y, Nishio M, Nakao K, Kuroda M, Shimizu R, Ohnuki T, Komatsubara S. J. Org. Chem. 2000;65:990–995. doi: 10.1021/jo991375+.
- 2.For enantioselective catalytic aldol addition to isatins, see: Luppi G, Cozzi PG, Monari M, Kaptein B, Broxterman QB, Tomasini C. J. Org. Chem. 2005;70:7418–7421. doi: 10.1021/jo050257l. Chen G, Wang Y, He H, Gao S, Yang X, Hao X. Heterocycles. 2006;68:2327–2333. Luppi G, Monari M, Corrêa RJ, Violante FA, Pinto AC, Kaptein B, Broxterman QB, Garden SJ, Tomasini C. Tetrahedron. 2006;62:12017–12024. Malkov AV, Kabeshov MA, Bella M, Kysilka O, Malyshev DA, Pluháčková K, Kočovskŷ P. Org. Lett. 2007;9:5473–5476. doi: 10.1021/ol7023983. Chen J-R, Liu X-P, Zhu X-Y, Liang L, Qiao Y-F, Zhang J-M, Xiao W-J. Tetrahedron. 2007;63:10437–10444. Corrêa RJ, Garden SJ, Angelici G, Tomasini C. Eur. J. Chem. 2008:736–744. Nakamura S, Hara N, Nakashima H, Kubo K, Shibata N, Toru T. Chem. Eur. J. 2008;14:8079–8081. doi: 10.1002/chem.200800981.
- 3.For enantioselective catalytic addition of arylboronic acids to isatins, see: Toullec PY, Jagt RBC, de Vries JG, Feringa BL, Minnaard AL. Org. Lett. 2006;8:2715–2718. doi: 10.1021/ol0608101. Shintani R, Inoue M, Hayashi T. Angew. Chem. 2006;118:3431–3434. doi: 10.1002/anie.200600392. Angew. Chem. Int. Ed. 2006;45:3353–3356. doi: 10.1002/anie.200600392. Lai H, Huang Z, Wu Q, Qin Y. J. Org. Chem. 2009;74:283–288. doi: 10.1021/jo802036m.
- 4.For enantioselective catalytic hydrogenation of isatins, see: Carpentier J-F, Morteux A. Tetrahedron: Asymmetry. 1997;8:1083–1099.
- 5.For enantioselective catalytic addition of organozinc reagents to isatins, see: Funabashi K, Jachmann M, Kanai M, Shibasaki M. Angew. Chem. 2003;115:5647–5650. doi: 10.1002/anie.200351650. Angew. Chem. Int. Ed. 2003;42:5489–5492. doi: 10.1002/anie.200351650.
- 6.For enantioselective catalytic allylation of organozinc reagents to isatins, see: Kitajima M, Mori I, Arai K, Kogure N, Takayama H. Tetrahedron Lett. 2006;47:3199–3202.
- 7.For selected reviews of C-C bond forming hydrogenation and transfer hydrogenation, see: Ngai M-Y, Kong J-R, Krische MJ. J. Org. Chem. 2007;72:1063–1072. doi: 10.1021/jo061895m. Iida H, Krische MJ. Top. Curr. Chem. 2007;279:77–104. Skucas E, Ngai M-Y, Komanduri V, Krische MJ. Acc. Chem. Res. 2007;40:1394–1401. doi: 10.1021/ar7001123. Shibahara F, Krische MJ. Chem. Lett. 2008;37:1102–1107. doi: 10.1246/cl.2008.1102. Bower JF, Kim IS, Patman RL, Krische MJ. Angew. Chem. 2009;121:36–44. doi: 10.1002/anie.200802938. Angew. Chem. Int. Ed. 2009;48:34–46.
- 8.For carbonyl and imine vinylation employing 1,3-enynes and 1,3-diynes as vinyl donors under hydrogenation conditions, see: Jang H-Y, Huddleston RR, Krische MJ. J. Am. Chem. Soc. 2004;126:4664–4665. doi: 10.1021/ja0316566. Kong J-R, Cho C-W, Krische MJ. J. Am. Chem. Soc. 2005;127:11269–11276. doi: 10.1021/ja051104i. Kong J-R, Ngai M-Y, Krische MJ. J. Am. Chem. Soc. 2006;128:718–719. doi: 10.1021/ja056474l. Komanduri V, Krische MJ. J. Am. Chem. Soc. 2006;128:16448–16449. doi: 10.1021/ja0673027. Hong Y-T, Cho C-W, Skucas E, Krische MJ. Org. Lett. 2007;9:3745–3748. doi: 10.1021/ol7015548. Cho C-W, Krische MJ. Org. Lett. 2006;8:3873–3876. doi: 10.1021/ol061485k.
- 9.For carbonyl and imine vinylation employing non-conjugated alkynes as vinyl donors under hydrogenation and transfer hydrogenation conditions, see: Rhee J-U, Krische MJ. J. Am. Chem. Soc. 2006;128:10674–10675. doi: 10.1021/ja0637954. Kong J-R, Krische MJ. J. Am. Chem. Soc. 2006;128:16040–16041. doi: 10.1021/ja0664786. Ngai M-Y, Barchuk A, Krische MJ. J. Am. Chem. Soc. 2007;129:280–281. doi: 10.1021/ja0670815. Skucas E, Kong J-R, Krische MJ. J. Am. Chem. Soc. 2007;129:7242–7243. doi: 10.1021/ja0715896. Barchuk A, Ngai M-Y, Krische MJ. J. Am. Chem. Soc. 2007;129:8432–8433. doi: 10.1021/ja073018j. Ngai M-Y, Barchuk A, Krische MJ. J. Am. Chem. Soc. 2007;129:12644–12645. doi: 10.1021/ja075438e. Han SB, Kong JR, Krische MJ. Org. Lett. 2008;10:4133–4135. doi: 10.1021/ol8018874. Patman RL, Chaulagain MR, Williams VM, Krische MJ. J. Am. Chem. Soc. 2009;131:2066–2067. doi: 10.1021/ja809456u.
- 10.For a review of catalytic reductive aldol and Mannich additions employing gaseous hydrogen as terminal reductant, see: Han SB, Hassan A, Krische MJ. Synthesis. 2008:2669–2679. doi: 10.1055/s-2008-1067220.
- 11.For olefin reductive hydroacylation employing anhydrides as acyl donors under hydrogenation conditions, see: Hong Y-T, Barchuk A, Krische MJ. Angew. Chem. 2006;118:7039–7042. Angew. Chem. Int. Ed. 2006;128:6885–6888. Kokubo K, Miura M, Nomura M. Organometallics. 1995;14:4521–4524.
- 12.For carbonyl allylation employing allenes as allyl donors under hydrogenation and transfer hydrogenation conditions, see: Skucas E, Bower JF, Krische MJ. J. Am. Chem. Soc. 2007;129:12678–12679. doi: 10.1021/ja075971u. Bower JF, Skucas E, Patman RL, Krische MJ. J. Am. Chem. Soc. 2007;129:15134–15135. doi: 10.1021/ja077389b. Ngai M-Y, Skucas E, Krische MJ. Org. Lett. 2008;10:2705–2708. doi: 10.1021/ol800836v. Skucas E, Zbieg JR, Krische MJ. J. Am. Chem. Soc. 2009;131:5054–5055. doi: 10.1021/ja900827p. Han SB, Kim I-S, Han H, Krische MJ. J. Am. Chem. Soc. 2009;131:6916–6917. doi: 10.1021/ja902437k.
- 13.For carbonyl allylation employing 1,3-dienes as allyl donors under hydrogenation and transfer hydrogenation conditions, see: Jang H-Y, Huddleston RR, Krische MJ. Angew. Chem. 2003;115:4208–4211. doi: 10.1002/anie.200351986. Angew. Chem. Int. Ed. 2003;42:4074–4077. doi: 10.1002/anie.200351986. Bower JF, Patman RL, Krische MJ. Org. Lett. 2008;10:1033–1035. doi: 10.1021/ol800159w. Shibahara F, Bower JF, Krische MJ. J. Am. Chem. Soc. 2008;130:6338–6339. doi: 10.1021/ja801213x. Shibahara F, Bower JF, Krische MJ. J. Am. Chem. Soc. 2008;130:14120–14122. doi: 10.1021/ja805356j. 2008.
- 14.For carbonyl allylation employing allylic acetates as allyl donors under transfer hydrogenation conditions, see: Kim IS, Ngai M-Y, Krische MJ. J. Am. Chem. Soc. 2008;130:6340–6341. doi: 10.1021/ja802001b. Kim IS, Ngai M-Y, Krische MJ. J. Am. Chem. Soc. 2008;130:14891–14899. doi: 10.1021/ja805722e. Kim IS, Han SB, Krische MJ. J. Am. Chem. Soc. 2009;131:2514–2520. doi: 10.1021/ja808857w.
- 15.For carbonyl propargylation employing 1,3-enynes as propargyl donors under transfer hydrogenation conditions, see: Patman RL, Williams VM, Bower JF, Krische MJ. Angew. Chem. 2008;120:5298–5301. doi: 10.1002/anie.200801359. Angew. Chem. Int. Ed. 2008;47:5220–5223. doi: 10.1002/anie.200801359.
- 16.For selected reviews on conventional enantioselective carbonyl allylation, crotylation and prenylation employing stoichiometric organometallic reagents, see: Hoffmann RW. Angew. Chem. 1982;94:569–580. Angew. Chem., Int. Ed. Engl. 1982;21:555–566. Yamamoto Y, Asao N. Chem. Rev. 1993;93:2207–2293. Ramachandran PV. Aldrichim. Acta. 2002;35:23–35. Kennedy JWJ, Hall DG. Angew. Chem. 2003;115:4880–4887. Angew. Chem., Int. Ed. 2003;42:4732–4739. doi: 10.1002/anie.200301632. Denmark SE, Fu J. Chem. Rev. 2003;103:2763–2793. doi: 10.1021/cr020050h. Yu C-M, Youn J, Jung H-K. Bull. Korean Chem. Soc. 2006;27:463–472. Marek I, Sklute G. Chem. Commun. 2007:1683–1691. doi: 10.1039/b615042j. Hall DG. Synlett. 2007:1644–1655.
- 17.For reviews encompassing catalytic asymmetric ketone addition, see: Hatano M, Ishihara K. Synthesis. 2008:1647–1675. Shibasaki M, Kanai M. Chem. Rev. 2008;108:2853–2873. doi: 10.1021/cr078340r. Cozzi PG, Hilgraf R, Zimmermann N. Eur. J. Org. Chem. 2007:5969–5994. Riant O, Hannedouche J. Org. Biomol. Chem. 2007;5:873–888. doi: 10.1039/b617746h. Garcia C, Martin VS. Curr. Org. Chem. 2006;10:1849–1889. Ramon DJ, Yus M. Angew. Chem. 2004;116:286–289. Angew. Chem., Int. Ed. 2004;43:284–287.
- 18.For selected examples of catalytic enantioselective ketone allylation, see: Allylboration. Lou S, Moquist PN, Schaus SE. J. Am. Chem. Soc. 2006;128:12660–12661. doi: 10.1021/ja0651308. Wada R, Oisaki K, Kanai M, Shibasaki M. J. Am. Chem. Soc. 2004;126:8910–8911. doi: 10.1021/ja047200l. Allylsilation. Wadamoto M, Yamamoto H. J. Am. Chem. Soc. 2005;127:14556–14557. doi: 10.1021/ja0553351. Allylsilation. Kim JG, Waltz KM, Kwiatkowski D, Walsh PJ. J. Am. Chem. Soc. 2004;126:12580–12585. doi: 10.1021/ja047758t. Casolari S, D’Addario D, Tagliavini E. Org. Lett. 1999;1:1061–1063.