

Abstract
Brain-derived neurotrophic factor (BDNF) and its receptor, TrkB, play prominent roles in food intake regulation through central mechanisms. However, the neural circuits underlying their anorexigenic effects remain largely unknown. We showed previously that selective BDNF depletion in the ventromedial hypothalamus (VMH) of mice resulted in hyperphagic behavior and obesity. Here, we sought to ascertain whether its regulatory effects involved the mesolimbic dopamine system, which mediates motivated and reward-seeking behaviors including consumption of palatable food. We found that expression of BDNF and TrkB mRNA in the ventral tegmental area (VTA) of wild-type mice was influenced by consumption of palatable, high-fat food (HFF). Moreover, amperometric recordings in brain slices of mice depleted of central BDNF uncovered marked deficits in evoked release of dopamine in the nucleus accumbens (NAc) shell and dorsal striatum but normal secretion in the NAc core. Mutant mice also exhibited dramatic increases in HFF consumption, which were exacerbated when access to HFF was restricted. However, mutants displayed enhanced responses to D1 receptor agonist administration, which normalized their intake of HFF in a 4 h food intake test. Finally, in contrast to deletion of Bdnf in the VMH of mice, which resulted in increased intake of standard chow, BDNF depletion in the VTA elicited excessive intake of HFF but not of standard chow and increased body weights under HFF conditions. Our findings indicate that the effects of BDNF on eating behavior are neural substrate-dependent and that BDNF influences hedonic feeding via positive modulation of the mesolimbic dopamine system.
Introduction
Brain-derived neurotrophic factor (BDNF) and its receptor, TrkB, mediate neuronal survival, differentiation, and plasticity and promote satiety through central mechanisms. Perturbed BDNF signaling triggers hyperphagia and dramatic obesity in mice (Lyons et al., 1999; Kernie et al., 2000; Rios et al., 2001; Xu et al., 2003). In humans, BDNF haploinsufficiency was linked to elevated food intake and obesity (Gray et al., 2006; Han et al., 2008). Even though the cumulative data support a pivotal role of BDNF in feeding control, the neural circuits underlying its regulatory effects remain largely unknown. Food intake is a complex behavior resulting from interactions between homeostatic and reward-related processes. Consistent with a role in homeostatic mechanisms acting in the hypothalamus and hindbrain, energy status influences levels of expression of BDNF and TrkB in these brain regions (Xu et al., 2003; Bariohay et al., 2005; Tran et al., 2006; Unger et al., 2007). Additionally, selective Bdnf deletion in the ventromedial hypothalamus (VMH) of mice elicited hyperphagia and obesity (Unger et al., 2007). BDNF and TrkB are also expressed in the mesolimbic dopamine system (Seroogy et al., 1994; Numan and Seroogy, 1999), which modulates motivated and reward-seeking behaviors, including drug and palatable food consumption. BDNF in this pathway has a demonstrated role in drug addiction and social defeat stress (Berton et al., 2006; Graham et al., 2007). The possibility that it influences eating behavior by acting here remains to be investigated.
Mesolimbic fibers originate in the ventral tegmental area (VTA) and terminate in the nucleus accumbens (NAc) and prefrontal cortex, where they release dopamine in response to drug and palatable food intake (Bassareo and Di Chiara, 1997; Ghiglieri et al., 1997; Bina and Cincotta, 2000; Lee and Clifton, 2002; Leddy et al., 2004). Appetite-regulating factors, including leptin, ghrelin, and melanin-concentrating hormone regulate mesolimbic dopaminergic activity and some of their effects on feeding involve this system (Georgescu et al., 2005; Abizaid et al., 2006; Fulton et al., 2006; Hommel et al., 2006). BDNF is expressed in dopamine-containing cells in the VTA and is anterogradely transported to the NAc, a region of minimal BDNF expression (Conner et al., 1997; Numan and Seroogy, 1999). TrkB, for its part, is localized to VTA dopamine cells and GABAergic medium spiny-projection neurons in the NAc (Yan et al., 1997; Numan and Seroogy, 1999; Freeman et al., 2003). While BDNF is not essential for survival of VTA dopamine neurons (Baquet et al., 2005), it is unclear whether dopamine secretion by these cells requires neurotrophin action.
We investigated whether BDNF in the mesolimbic pathway influenced feeding behavior. We found that mutant mice depleted of central BDNF exhibited marked decreases in evoked release of dopamine in the NAc and dorsal striatum. Furthermore, they displayed excessive intake of palatable food that was normalized by D1 receptor stimulation. Notably, VTA-specific deletion of Bdnf resulted in increased feeding and body weight when mutants received a palatable high-fat diet but minimal effects when administered standard chow. These findings indicate that BDNF regulates hedonic feeding by positively modulating the mesolimbic dopamine system.
Materials and Methods
Animals.
All of the following procedures were approved by the Institutional Animal Care and Use Committee at Tufts University and were in accordance with the National Institutes of Health guide for the Care and Use of Laboratory Animals. Every effort was made to minimize the number of animals used in these studies and their suffering. BDNF2L/2LCk-cre mice were generated as previously described and were in a hybrid background with C57BL/6 and 129 strain contributions (Rios et al., 2001; Chan et al., 2006; Rios et al., 2006). All animals were females between 10 and 12 weeks of age at the start of each experiment. They were fed ad libitum, unless otherwise noted, and housed individually in the Tufts University animal care unit on a 12 h light/dark cycle.
Food intake measurements.
Food intake of female BDNF2L/2LCk-cre and wild-type mice (10–12 weeks of age) that had unrestricted access to standard (SC; 5% Kcal Fat, Tekland Global Diet) or palatable high-fat chow (HFF; 45% Kcal Fat, Research Diets) was measured over 3 d of testing. Mice were individually housed and given premeasured amounts of food such that caloric intake could be calculated based on grams of food consumed measured at 2:00 P.M. daily (SC = 3.3 Kcal/g; HFF = 4.7 Kcal/g). Consumption of palatable high-fat chow was also measured when mice had restricted access to this diet. For this, fed female BDNF2L/2LCk-cre and wild-type mice (10–12 weeks of age) that were individually housed and maintained on a standard chow diet were presented with HFF for 1 h daily (2:00–3:00 P.M.) for three consecutive days. Daily intake of HFF during the restricted period was measured.
Quantitative reverse transcription-PCR analysis.
On day 1 of the experiment, SC or HFF was placed in the cages of naive, individually housed, fed wild-type mice (females, 10–12 weeks of age) for 60 min to minimize novelty effects. On day 2, following 30 or 60 min of SC or HFF exposure and consumption, mice were killed. Their brains were then rapidly extracted and tissue punches of NAc and VTA obtained from 500-μm-thick coronal sections prepared with a vibrating microtome (Leica VT100S). Samples were immediately frozen on dry ice and RNA extracted using Tri Reagent (Molecular Reasearch Center). RNA samples were treated with DNase and tested for genomic DNA contamination in PCRs. Reverse transcription to generate cDNA was conducted with 1 μg of RNA and using 200 units of Superscript II reverse transcriptase (Invitrogen) and 150 ng of random primers (Invitrogen) in a 20 μl reaction. Real time PCR amplification was performed using a MX-3000P Stratagene cycler and SYBR green PCR master mix (Qiagen). For each primer set, the specificity of the product amplification was confirmed by dissociation curve analysis and agarose gel electrophoresis. Furthermore, curves were created using serial dilutions and the efficiencies for each primer set was calculated. The amplification efficiency for all the primers used in this study was >90%. For each target primer set, a validation experiment was performed to demonstrate that the PCR efficiencies were approximately equal to those of the reference gene. A two-step protocol was used: 95°C for 10 min and 45 cycles with 95°C for 30 s, 55°C for 30 s, and 72°C for 30 s. Actin was used as a normalizer. The following primers were used: BDNF forward, GAAAGTCCCGGTATCCAAAG; BDNF reverse, CCAGCCAATTCTCTTTTT; TrkB forward, CCTCCACGGATGTTGCTGAC; TrkB reverse, GCAACATCACCAGCAGGCA; actin forward, GGCTGTATTCCCCTCCATCG; actin reverse, CCAGTTGGTAACAATGCCATGT. All samples were analyzed in triplicates, and nontemplate controls were included to ascertain any level of contamination. Data obtained were analyzed using the comparative Ct method. Every experiment was repeated at least once.
HPLC analysis.
Fed female BDNF2L/2LCk-cre and wild-type mice (10–12 weeks of age) maintained on a standard chow diet were killed and their brains rapidly extracted. NAc and dorsal striatum tissue punches were obtained from coronal sections (500 μm) prepared with a vibrating microtome (Leica VT100S). Samples were immediately frozen on dry ice. HPLC for biogenic amines was performed by the Neurochemistry Core Facility at Vanderbilt University.
Western blot analysis.
Protein was extracted from NAc and dorsal striatum tissue punches from wild-type and BDNF2L/2LCk-cre female mice (10–12 weeks of age) and 50 μg of each protein extract were separated in an 8% acrylamide gel and transferred to Immuno-blot PVDF membranes (Bio-Rad). Membranes were blocked in a 5% milk TBS-Tween solution and probed individually with antibodies against dopamine transporter and dopamine receptors 1 and 2 (1:1000, Millipore Bioscience Research Reagents) overnight at 4°C. Membranes were then washed, incubated with peroxidase-conjugated secondary antibodies, reacted in ECLplus solution (GE Healthcare) for 5 min, and exposed to Kodak maximum sensitivity film. Densitometry of blots was performed using the Kodak 1D Image analysis software program. Membranes were also hybridized to anti-β-tubulin (Sigma) to normalize the data for loading.
Amperometry.
Acute brain slices (300 μm thick) containing NAc and dorsal striatum were prepared from wild-type and BDNF2L/2LCk-cre mice (females 8–10 weeks of age) using a Leica vibratome. Disk carbon fiber electrodes of 5 μm in diameter with a freshly cut surface were placed ∼50 μm into the slice. Wild-type and mutant slices (n = 8) were electrically stimulated (400 μA, 1 ms) with a bipolar stimulating electrode (Plastics One) placed ∼100 μm from the recording electrode using an Iso-Flex stimulus isolator (A.M.P.I.) triggered by a pulse generator (A.M.P.I.). Background-subtracted cyclic voltammograms serve to calibrate the electrodes and to optimize the identification of the released substance as dopamine.
Pharmacology.
Food-nondeprived BDNF2L/2LCk-cre and wild-type mice were individually housed and maintained on standard chow. At the beginning of the experiment, 1:00 P.M., an intraperitoneal injection of R(+)Skf-38393 (20 mg/kg; Sigma-Aldrich) or saline (0.9% NaCl) was administered. Upon injection, mice were given premeasured amounts of HFF such that grams of food consumed and caloric intake (4.7 kcal/g) could be calculated at 1/2, 1, 2, 3, and 4 h after injection. Mice were habituated to experimental conditions by gentle handling and saline injections for 3 d preceding the experiment. One day before drug administration, mice were given 4 h of access to HFF to reduce novelty effects.
Deletion of Bdnf in the VTA.
AAV2/1 vectors were produced, purified, and titered as previously described (Broekman et al., 2006). Cre recombinase cDNA carrying an N-terminal nuclear localization signal was cloned into the AAV2-CBA-W vector plasmid. Adeno-associated virus (0.5 μl; 1 × 1013 infectious units per ml) encoding Cre recombinase (AAV2/1-Cre) or green fluorescence protein (AAV2/1-GFP) was delivered to the VTA of anesthetized female floxed Bdnf mice (10–12 weeks of age) using the coordinates: anteroposterior −3.2 mm, mediolateral, ±1.0 mm, and dorsoventral, −4.6 mm at 10 degrees from midline. The virus was administered bilaterally using a 10 μl Hamilton syringe with a 33-gauge needle attached to a digital stereotaxic apparatus (Benchmark) and an infusion microsyringe nanopump (KD Scientific) at a rate of 0.25 μl/min. To allow diffusion of the virus and minimize backflow after needle retraction, the needle was held in place for 10 min after injection. Mice were given a 1 week recovery from surgery before any experiments were conducted. Viral volume and coordinates for selectively targeting the VTA were optimized using the mouse atlas (Paxinos and Franklin, 2001) and by pilot experiments injecting AAV2/1-Cre into ROSA reporter mice (Soriano, 1999). Accurate targeting was confirmed in AAV2/1-GFP mice by analysis of GFP signal and in AAV2/1-Cre-injected mice by measurement of BDNF mRNA expression (in situ hybridization analysis).
In situ hybridization analysis.
Twelve-micrometer-thick sections containing VTA from wild-type, BDNF2L/2LCk-Cre mutant, and AAV2/1-GFP- and AAV2/1-Cre-injected mice were hybridized for 16 h at 60°C with a 35S-labeled, antisense riboprobe representing bases 507–833 of the BDNF cDNA. Specificity of this riboprobe was confirmed by lack of hybridization to tissue from BDNF2L/2LCk-Cre mutant mice (Rios et al., 2001). Following the hybridization step, sections were stringently washed and placed on x-ray film for 12 d. Densitometry of BDNF mRNA signal was conducted using the Kodak 1D Image Analysis software. Coronal level was confirmed by staining adjacent sections with cresyl violet.
Locomotor activity measurements.
Mice were housed individually in standard 15 × 24 cm plastic cages on a 12 h reverse light/dark cycle. Motor activity was monitored with the Smart Frame Activity System (Hamilton/Kinder), consisting of 12 PC-interfaced horizontal photobeam frames (8 cm length × 4 cm width spaced 1.5 cm apart) that surrounded the animal's home cage. At the start of the experiment, 10:00 A.M., an intraperitoneal injection either saline (0.9% NaCl), SK&F-81297 (5 mg/kg; Sigma-Aldrich), or Quinpirole (2.5 mg/kg; Sigma-Aldrich) was administered in a volume of 10 μl per gram body weight. Locomotor readouts (beam breaks) were subsequently recorded for 3 h using MotorMonitor software (Hamilton/Kinder). During the entire testing period, animals remained undisturbed. Mice were habituated to experimental conditions by gentle handling and saline injections for 3 d preceding the experiment.
Statistical analysis.
Food intake measurements for experiments involving BDNF2L/2LCk-Cre mice were analyzed using two-way ANOVA. Amperometric recordings were analyzed using a one-way ANOVA. Quantification of in situ hybridizations, quantitative reverse transcription (RT)-PCR, Western blot analysis and food intake and body weights of AAV2/1-GFP- and AAV2/1-Cre-injected mice were analyzed by unpaired t test. Data were considered statistically significant when p < 0.05 and all values represent mean ± SEM.
Results
BDNF conditional mutant mice exhibit increased intake of palatable food
The mesolimbic dopamine system is linked to appetitive motivation and consumption of highly palatable food (Berridge, 2009). As a first step to evaluate how BDNF activity in this pathway might influence eating behavior, we measured consumption of palatable HFF in mice with central depletion of BDNF (BDNF2L/2LCk-cre). Mutants were generated by crossing mice carrying floxed Bdnf alleles with transgenic mice expressing cre recombinase under the control of the α-calcium/calmodulin protein kinase II promoter and were described previously (Rios et al., 2001, 2006; Chan et al., 2006). In these animals, BDNF expression was terminated across the brain, the cerebellum exempted. Depletion of BDNF began during the first postnatal week and became maximal at 3 weeks after birth. To confirm that BDNF mRNA was depleted in the VTA of BDNF2L/2LCk-cre mutants, we conducted in situ hybridization analysis. As indicated in supplemental Figure 1 (available at www.jneurosci.org as supplemental material), BDNF mRNA was extensively depleted from the VTA of mutants.
We reported previously that when fed SC, BDNF2L/2LCk-cre mutants exhibited hyperphagic behavior and became dramatically obese (Rios et al., 2001). Here, we examined whether they also exhibited higher levels of consumption of palatable HFF. We also measured SC consumption for comparison. There was an effect of genotype on food intake (F(1,18) = 75.6, p = 0.000001). Whereas BDNF2L/2LCk-cre mice fed SC ad libitum consumed 83% more kilocalories than WT mice fed SC (p < 0.0001), their caloric intake was elevated by 111% (p < 0.0001) when they had unrestricted access to HFF compared with wild types fed a similar diet (Fig. 1A).
Figure 1.

BDNF2L/2LCk-cre mutant mice exhibit increased intake of palatable high-fat food. A, Caloric intake of wild-type (WT) and BDNF2L/2LCk-cre conditional mutant (CM) mice that were fed SC or palatable HFF ad libitum (n = 10; *p < 0.0001). B, Caloric intake of high-fat chow of wild-type and BDNF2L/2LCk-cre mutant mice during a 1 h restricted period over 3 consecutive days (n = 10; *p < 0.0001; **p = 0.0001).
Restricted access to palatable food elicits binge eating behavior in wild-type rodents (Corwin and Wojnicki, 2006; Davis et al., 2007; Berner et al., 2008). We asked whether this effect was exaggerated in BDNF2L/2LCK-cre mutant mice. Fed wild-type and BDNF mutant mice maintained on a SC diet had access to palatable HFF for 1 h/d for 3 consecutive days. Food intake during the restricted access period was measured. HFF caloric intake of BDNF mutant mice during the restricted period was increased by 214% (p < 0.0001), 209% (p < 0.0001), and 200% (p = 0.0001) on days 1, 2, and 3, respectively, compared with wild-type mice (Fig. 1B). The collective behavioral data indicate that central depletion of BDNF induces dramatic hyperphagic behavior when animals have free access to palatable HFF. Furthermore, limited exposure to HFF exacerbates the binge-eating behavior triggered by deficient BDNF signal.
Levels of TrkB and BDNF mRNA in the VTA are influenced by intake of palatable food
To investigate whether BDNF signaling in the mesolimbic system might influence feeding behavior, we examined the effect of palatable food ingestion on its expression and that of its receptor, TrkB, in the VTA and NAc of sated wild-type mice. Following 30 or 60 min of SC or HFF exposure and consumption, mice were killed and VTA and NAc tissue punches obtained for quantitative RT-PCR analysis. We found that TrkB mRNA expression in the NAc of SC and HFF-fed mice was similar at both time points (data not shown). In the VTA, there was a 46% increase (p = 0.04) in TrkB receptor transcript content following 30 min of HFF consumption and this increase returned to baseline levels after 1 h of palatable food intake (Fig. 2A). In contrast, BDNF expression in the VTA remained stable at 30 min but was decreased by 38% (p = 0.004) following 60 min of HFF consumption (Fig. 2B). These data indicate that expression of TrkB and BDNF mRNA in the VTA is influenced by consumption of palatable food and that BDNF signaling in this region is finely regulated during food intake-related processes.
Figure 2.

Intake of palatable high-fat food alters levels of BDNF and TrkB mRNA in the VTA of wild-type mice. A, Expression levels of TrkB mRNA in the ventral tegmental area of wild-type mice fed SC or HFF chow for 30 or 60 min as measured by quantitative RT-PCR analysis (n = 5; *p = 0.04). B, Relative expression of BDNF mRNA in the ventral tegmental area of wild-type mice fed standard or high-fat chow for 30 or 60 min (n = 5; *p = 0.004). Data are expressed as fold difference of HFF-fed relative to SC-fed wild-type animals. The mean value for SC-fed mice was set at 1. p values were calculated based on 2{Δ}{Δ}Ct values for each sample.
BDNF conditional mutants exhibit deficits in evoked release of dopamine in the nucleus accumbens shell and dorsal striatum
Because BDNF2L/2LCk-cre mutant mice exhibited increased intake of palatable HFF, which influenced expression of BDNF and TrkB mRNA in the VTA, we sought to ascertain whether alterations in the mesolimbic dopamine system might underlie their excessive eating. First, we evaluated dopamine synthesis and turnover in the nucleus accumbens of wild-type and BDNF mutant mice. The dorsal striatum was also examined for comparison. We found no significant changes in content of dopamine or its metabolite 3,4-dihydroxyphenylacetic acid (DOPAC) in the NAc of BDNF mutant mice (Fig. 3A). However, there was an 18% increase (p = 0.02) in the DOPAC/dopamine (DA) ratio in the mutants compared with wild types, indicating a mild elevation in dopamine turnover. In the dorsal striatum, BDNF2L/2LCk-cre mutant mice exhibited 22% (p = 0.002) and 40% (p = 0.007) increases in dopamine and DOPAC content, respectively, but normal DOPAC/DA ratios compared with wild-type mice (Fig. 3B).
Figure 3.

Dopamine content in nucleus accumbens and dorsal striatum of BDNF2L/2LCk-cre mutant mice. A, HPLC measurements of DA and DOPAC content and DOPAC/DA ratios in the nucleus accumbens of wild-type control (WT) and BDNF2L/2LCk-cre conditional mutant (CM) mice (n = 5; *p = 0.02). B, Dopamine and DOPAC content and DOPAC/DA ratios in the dorsal striatum of wild-type control and BDNF2L/2LCk-cre conditional mutant mice (n = 5; *p = 0.002; **p = 0.007).
Next, we performed amperometric recordings to determine whether BDNF influenced dopamine release through presynaptic mechanisms. We measured electrically evoked secretion of dopamine in acute brain slices obtained from wild-type and BDNF2L/2LCk-cre mutant mice. Amperometric recordings were obtained from dopamine terminals in the NAc shell and core that originated in the VTA. Deficient evoked release of dopamine was evident in the NAc shell but not in the NAc core of BDNF mutants (Fig. 4A–D). The mean evoked dopamine signal amplitude in the NAc shell of BDNF mutants was decreased by 39% (18.4 ± 1.5 pA; n = 68 stimulations in 14 slices) compared with wild types (30.1 ± 2.7 pA; n = 70 stimulations in 15 slices) (F(1,136) = 14.15, p < 0.01) (Fig. 4C). This difference persisted following addition of nomifensine, a dopamine transporter (DAT) inhibitor, indicating that the deficit was due to reduced secretion, not to increased reuptake of dopamine. When the number of dopamine molecules released was calculated based on spike amplitude (pA) and width (s), BDNF2L/2LCk-cre mutant mice exhibited a 43% decrease (F(1,136) = 17.33, p < 0.01) in dopamine release in the NAc shell compared with controls (Fig. 4B). This deficit persisted in the presence of nomifensine (F(1,117) = 14.39, p < 0.01). (Fig. 4B).
Figure 4.

Evoked release of dopamine in the nucleus accumbens shell is impaired in BDNF2L/2LCk-cre conditional mutant mice. A, Representative amperometric traces of electrical stimulation-evoked dopamine release in acute coronal accumbens slices of wild-type (WT) and BDNF2L/2LCk-cre conditional mutant (CM) mice. Stimulation electrodes and carbon fiber recording microelectrodes were positioned in the shell region of the nucleus accumbens, which receives the majority of the dopaminergic projections from the VTA. Data were acquired at 50 kHz and digitally postfiltered at 1 kHz. Arrow points to onset of electrical single pulse (2 ms of 0.5 mA). B, Mean evoked dopamine molecules in the nucleus accumbens core (NAc Core) and shell (NAc Shell) in brain slices from wild-type and BDNF2L/2LCk-cre conditional mutant mice in ACSF and ACSF with the dopamine reuptake inhibitor nomifensine (Nomifen.). *p < 0.01. C, Evoked dopamine signal amplitude in the NAc core and shell of wild-type and BDNF2L/2LCk-cre mutant mice. *p < 0.01. D, Evoked dopamine signal width in the NAc core and shell of wild-type and BDNF2L/2LCk-cre mutant mice. *p < 0.01.
Amperometric measurements in the dorsal striatum revealed additional deficits in BDNF mutant mice (Fig. 5A–D). BDNF mutant slices showed a 64% decrease in mean evoked dopamine molecules compared with wild types (BDNF mutants: 38.5 × 106 ± 4.0 × 106, n = 49 stimulations in 10 slices; wild types: 106.3 × 106 ± 15.6 × 106, n = 44 stimulations in 9 slices; F(1,135) = 30.60, P < 0.01) (Fig. 5B). As in the NAc shell, this difference persisted in the presence of nomifensine (Fig. 5B). Moreover, whereas mutants exhibited a mean signal amplitude of 15.9 ± 2.2 pA (n = 49 stimulations in 10 slices), wild types showed an amplitude of 36.0 ± 5.5 pA (n = 44 stimulations in 9 slices) (F(1,135) = 26.11, p < 0.01) (Fig. 5C). In artificial CSF (ACSF) with nomifensine, BDNF mutant slices also exhibited a significant decrease in mean signal amplitude, 20.7 ± 3.7 pA (n = 30 stimulations in 6 slices) versus 47.3 ± 4.9 (n = 20 stimulations in 4 slices) in wild-type slices (F(1,68) = 40.92, p < 0.01). These data demonstrate that lack of central BDNF triggers an increase in dopamine synthesis in the dorsal striatum and marked decreases in dopamine release in the NAc shell and dorsal striatum.
Figure 5.

BDNF2L/2LCk-cre conditional mutants exhibit deficits in evoked release of dopamine in the dorsal striatum. *p < 0.01. A, Representative amperometric traces of electrical stimulation-evoked dopamine release in acute coronal striatal slices from wild-type (WT) and BDNF2L/2LCk-cre conditional mutant (CM) mice. Stimulation electrodes and carbon fiber recording microelectrodes were positioned in the medial–dorsal region of the striatum. Data were acquired at 50 kHz and digitally postfiltered at 1 kHz. The arrow points to onset of electrical single pulse (2 ms of 0.5 mA). B, Mean evoked dopamine molecules in the dorsal striatum of wild-type and BDNF2L/2LCk-cre conditional mutant mice in ACSF and ACSF with the dopamine reuptake inhibitor nomifensine. *p < 0.01. C, Evoked dopamine signal amplitude in the dorsal striatum of wild-type and BDNF2L/2LCk-cre mutant mice. *p < 0.01. D, Evoked dopamine signal width in the dorsal striatum of wild-type and BDNF2L/2LCk-cre mutant mice. *p < 0.01.
Expression of dopamine receptors and transporter in the striatum of BDNF conditional mutant mice
To further evaluate dopamine transmission in the absence of central BDNF, we sought to ascertain whether the observed decreases in evoked release of dopamine in BDNF mutants were accompanied by changes in levels of expression of D1 (D1R) and D2 (D2R) receptors or dopamine transporter. We found that expression of D1R, D2R and DAT was normal in the NAc of BDNF2L/2LCk-cre mutant mice (data not shown). In the dorsal striatum, whereas expression of D1R and DAT was normal in the mutants, expression of D2R was decreased by 34% (p = 0.03) (Fig. 6A–C). These findings indicate that dopamine receptor and transporter content is normal in the NAc of BDNF mutants despite them having decreased release of dopamine in this region. However, diminished BDNF signal led to decreased D2R expression in the dorsal striatum, reminiscent of the D2R downregulation observed in dietary obese animals and humans (Pothos et al., 1998, Geiger et al., 2009, Wang et al., 2001).
Figure 6.

Expression of D2 receptor protein is reduced in the dorsal striatum of BDNF2L/2LCk-cre conditional mutants. A, Representative Western blot and densitometry analysis measuring D1R protein content in the dorsal striatum of wild-type (WT) and BDNF2L/2LCk-cre conditional mutant (CM) mice (n = 5). Data are expressed as expression of BDNF mutant mice relative to wild-type controls. B, Expression of D2R protein in the dorsal striatum of wild-type and BDNF conditional mutant mice (n = 5; p = 0.03). C, Expression of DAT in the dorsal striatum of wild-type and BDNF conditional mutant mice (n = 5).
Treatment with selective D1 receptor agonists normalized consumption of palatable high-fat food in BDNF2L/2LCk-cre mutant mice
In light of the deficient dopamine release but normal D1R expression observed in the NAc of BDNF mutant mice, we investigated whether D1 receptor stimulation normalized their eating behavior. To assess this, fed wild-type and BDNF2L/2LCk-cre mutant mice maintained on a SC diet received a peripheral injection of the selective D1R agonist, SKF38393 (20 mg/kg) and their intake of palatable HFF was monitored for the following 4 h. There was a significant effect of genotype (F(1,16) = 8.72, p < 0.01) and a significant interaction of genotype and drug treatment (F(1,16) = 5.72, p < 0.03) on caloric intake. Remarkably, SKF38393 treatment had a more profound and persistent effect in BDNF mutants compared with wild-type mice, completely normalizing their cumulative HFF intake (Fig. 7A,B). During the first hour following drug administration, wild types ate 80% less than vehicle-treated wild-type controls (p = 0.01) (Fig. 7A). During that same period of time, BDNF mutants treated with SKF38393 ate 95% less HFF than vehicle-treated BDNF2L/2LCk-cre mice (p = 0.01) (Fig. 7A). There were no significant differences in cumulative food intake after 2 h between vehicle and drug-treated wild types (Fig. 7A). In far contrast, the anorexigenic effect of SKF38393 persisted in BDNF mutants 2 h after drug treatment as indicated by a 94% reduction in cumulative caloric intake (p = 0.002) compared with their vehicle-treated counterparts (Fig. 7A). When total food consumption during the 4 h following D1R agonist administration was calculated, BDNF2L/2LCk-cre mice showed a 48% reduction in HFF caloric intake compared with vehicle-treated mutants (p = 0.04) (Fig. 7A,B). In contrast, SKF38393 treatment had no significant effect on the 4 h cumulative HFF intake of wild types compared with vehicle-treated wild-type controls. (Fig. 7A,B). It is important to note that the 4 h cumulative caloric intake of SKF38393-treated BDNF mutant mice was indistinguishable from that of SKF38393 or vehicle-treated wild-type mice (Fig. 7A,B). These results show that stimulation of D1 receptors normalizes consumption of palatable HFF in BDNF2L/2LCk-cre mice, which are hyper responsive to the effects of D1R agonists compared with wild types. These findings are consistent with deficient dopamine transmission contributing to the abnormal eating behavior triggered by depleted BDNF stores.
Figure 7.

Peripheral administration of the D1 receptor selective agonist SKF38393 reduced consumption of palatable high-fat food of BDNF2L/2LCk-cre mutant mice. A, Time course for HFF caloric intake of fed wild-type (WT) and BDNF2L/2LCk-cre conditional mutant (CM) mice following peripheral administration of saline vehicle or SKF38393 drug (n = 5 per group). B, Four-hour cumulative HFF intake of fed wild-type and BDNF mutant mice following saline vehicle (Sal.) or SKF38393 drug (D) treatment. *p = 0.04, n = 5 per group.
Site-specific deletion of Bdnf in the VTA of adult mice
To pinpoint the role of BDNF in the mesolimbic reward pathway, we examined the effect of selectively deleting Bdnf in the VTA, a principal source of BDNF for the nucleus accumbens (Guillin et al., 2001). VTA-specific mutant and control mice were generated by delivery of adeno-associated viral vectors encoding for cre recombinase (AAV2/1-Cre) or green fluorescent protein (AAV2/1-GFP), respectively, to the VTA of floxed Bdnf mice. Figure 8A illustrates a representative study entailing stereotaxic delivery of AAV2/1-Cre to the VTA of Rosa-β-gal reporter mice (Soriano, 1999). Blue cells represent sites where cre recombinase was expressed to mediate recombination of floxed sequences. Blue cells were evident in the VTA of Rosa-β-gal reporter mice injected with AAV2/1-Cre, indicating that floxed alleles were selectively and efficiently recombined in the VTA. Viral spread was restricted to the VTA with minimal infection of the adjacent substantia nigra, consistent with previous reports of contained infection within targeted brain areas due to large viral particle size and boundaries created by fiber bundles (Hommel et al., 2003, 2006; Berton et al., 2006; Graham et al., 2007). Densitometry of BDNF mRNA signal in the VTA showed that BDNF transcript levels were reduced by an average 68% in AAV2/8-Cre mice compared with AAV2/1-GFP controls (p = 0.004) (Fig. 8B). Figure 8, C and D, show representative coronal brain sections from floxed Bdnf mice injected with AAV2/1-GFP or AAV2/1-Cre, demonstrating selective depletion of BDNF mRNA in the VTA of the latter. Furthermore, expression of BDNF mRNA was intact in the hypothalamus of AAV2/1-Cre-injected mice (supplemental Fig. 2, available at www.jneurosci.org as supplemental material), ensuring that food intake behavior was not affected by hypothalamic depletion of this neurotrophin. Finally, examination of cresyl violet-stained sections containing VTA and obtained from AAV2/1-GFP and AAV2/1-Cre-injected mice failed to reveal any toxicity effects of the viral injection (data not shown).
Figure 8.
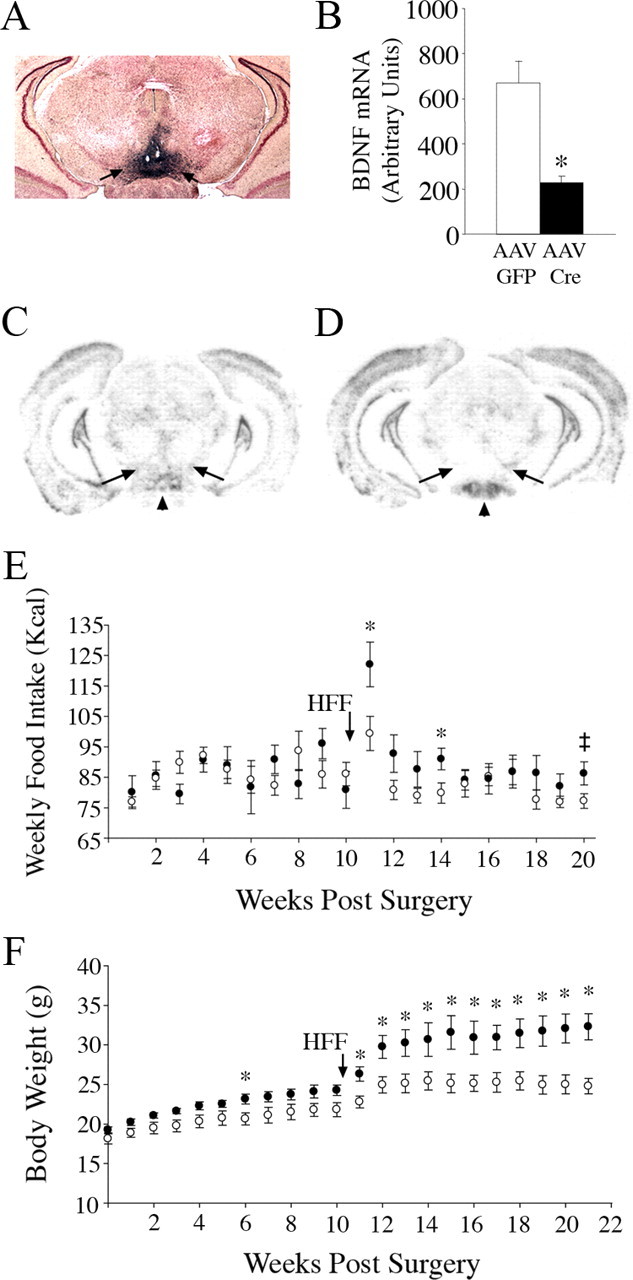
Selective targeting of Bdnf in the VTA results in increased intake of palatable HFF and higher body weights. A, Representative coronal brain section from a Rosa-β-gal reporter mouse that was delivered AAV2/1-Cre to the VTA. B, Densitometry of BDNF mRNA signal detected by in situ hybridization analysis in AAV2/1-GFP (n = 6) and AAV2/1-Cre (n = 5) injected mice. *p = 0.004. C, D, In situ hybridization analysis of BDNF mRNA expression in coronal brain sections from floxed Bdnf mice injected with AAV2/1-GFP (C) or AAV2/1-Cre (D) in the VTA. Arrows indicate the VTA and arrowhead indicates the mammillary nucleus. E, Weekly caloric intake of AAV2/1-GFP (open circles, n = 6) and AAV2/1-Cre (closed circles, n = 5) injected mice under standard and high-fat chow conditions (*p < 0.05; ‡, p = 0.06). Arrow indicates time point when mice were switched from a standard to a high-fat chow diet. F, Body weights of AAV2/1-GFP (open circles, n = 6) and AAV2/1-Cre (closed circles, n = 5) injected mice over 21 weeks following stereotaxic surgery and under standard and high-fat chow conditions (*p < 0.05). Arrow indicates time point when mice were switched from a standard to a high-fat chow diet.
AAV2/1-GFP and AAV2/1-Cre-injected mice identified through post hoc examination as having correct targeting of the VTA and AAV2/8-Cre-treated animals with BDNF mRNA depletion in the VTA ranging from 60 to 78% were included in our food intake and body weight analysis. Both sets of mice were fed SC ad libitum for 10 weeks following viral delivery and then switched to a palatable high-fat diet during the subsequent 10 weeks. AAV2/1-Cre-injected mice fed SC did not exhibit significant changes in food intake compared with AAV2/1-GFP controls (Fig. 8E). In contrast, when they were administered palatable HFF after week 10 of the study, their caloric intake was significantly increased compared with AAV2/1-GFP controls (Fig. 8E). Indeed, during the first week of HFF consumption, AAV2/1-Cre mice ate 23% more than AAV2/1-GFP-treated mice (p = 0.03). Furthermore, whereas caloric intake of AAV2/8-GFP-treated mice trended toward a significant 16% increase (p = 0.06) when they transitioned from a SC to a high-fat diet, VTA BDNF mutants increased their caloric intake by 54% (p = 0.01) (Fig. 8E). Food intake of AAV2/1-Cre-treated mice was also significantly increased at week 4 (p = 0.05) of HFF consumption (14 weeks after viral delivery) and trended toward significant increases at weeks 2 (12 weeks after viral delivery) and 10 (20 weeks after viral delivery) of HFF consumption (Fig. 8E).
Consistent with their food intake behavior under SC conditions, AAV2/1-Cre-injected mice had no significant increases in body weight when fed this diet, except at week 6 after surgery, when they exhibited a 12% increase compared with AAV2/1-GFP controls (p = 0.04) (Fig. 8F). In contrast, AAV2/1-Cre-injected mice became significantly heavier (15% increase compared with wild types) (p = 0.02) after 1 week of HFF consumption and elevated body weights persisted until the end of the study (Fig. 8F). After 10 weeks of HFF consumption, AAV2/1-Cre mice were 30% heavier than AAV2/1-GFP controls (p = 0.003) (Fig. 8F).
We also measured locomotor activity of VTA-specific BDNF mutants for 3 h following a single injection of saline vehicle, the D1R agonist SK&F-81297 (5 mg/kg) or the D2R agonist quinpirole (2.5 mg/kg). Levels of activity of saline-injected AAV2/1-GFP and AAV2/1-Cre mice were similar (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Moreover, D1R and D2R stimulation elicited increases and decreases, respectively, in locomotor activity in both sets of mice. However, whereas the magnitude of the response to quinpirole was comparable in AAV2/1-GFP and AAV2/1-Cre mice, the response to SK&F-81297 appeared to be more pronounced in AAV2/1-GFP controls. D1R stimulation increased locomotor activity by 75% (p = 0.01) in AAV2/1-GFP mice and by 42% (p = 0.01) in AAV2/1-Cre mutants relative to their corresponding saline controls (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). However, an interaction between genotype and drug treatment did not reach statistical significance. The cumulative data from studies involving selective depletion of BDNF in the VTA demonstrate that this neurotrophin is required in this brain region for the regulation of food intake. Moreover, they suggest that BDNF action in the mesolimbic system has a larger impact on hedonic relative to homeostatic feeding.
Discussion
The mesolimbic dopamine system is associated with hedonic mechanisms that drive appetitive behavior and intake of highly palatable, energy-dense food. However, the mechanisms governing this pathway during food consumption-related processes remain poorly understood. The multidisciplinary studies described here indicate an essential role of BDNF in the control of hedonic feeding via positive regulation of mesolimbic dopamine transmission. We showed that selective targeting of Bdnf in the VTA of mice caused increased intake of palatable HFF but not of SC. This is in contrast to our previous findings demonstrating that mice depleted of BDNF in the VMH exhibited hyperphagic behavior when fed SC ad libitum (Unger et al., 2007). The results also differ from rats with selective RNAi-mediated knockdown of leptin receptors in the VTA, which exhibited increased intake of SC and higher sensitivity to palatable food (Hommel et al., 2006). Our observations indicate that the effects of BDNF on eating behavior are context-dependent and influenced by the neural substrates. Whereas hypothalamic BDNF appears to regulate homeostatic food intake, in the VTA, this neurotrophin appears to largely affect hedonic processes driving palatable food consumption.
Because BDNF was depleted in the VTA of adult mice that had normal BDNF expression throughout development, the observed behavioral alterations cannot be attributed to developmental defects. Instead, they indicate a regulatory role of BDNF in the mature brain. Consistent with this assertion, we found that expression of TrkB and BDNF mRNA in the VTA of sated wild-type mice was influenced by intake of palatable HFF. The site of these expression changes suggests that BDNF might act presynaptically through autocrine mechanisms to modulate dopamine-producing cells in the VTA during food reward-related processes. Evidentiary is our finding that BDNF2L/2LCk-cre mice exhibited diminished evoked release of dopamine in the NAc shell, a target site of VTA dopamine neurons. Because HPLC analysis demonstrated normal dopamine content in the mutant NAc, this defect cannot be attributed to deficient dopamine synthesis. Moreover, reduced extracellular dopamine levels persisted in the presence of a DAT inhibitor, indicating that defective presynaptic release rather than enhanced dopamine clearance mediated the observed deficit.
Diminished dopamine secretion was only evident in the NAc shell and not in the NAc core of BDNF2L/2LCk-cre mutant mice. A divergence in function in these ventral striatal compartments was reported previously. For example, intake of palatable food was reported to induce dopamine release preferentially in the NAc shell compared with the core (Tanda and Di Chiara, 1998). Furthermore, in vivo microdialysis studies in rats revealed that whereas unpredicted consumption of palatable food led to release of dopamine in the shell, food anticipation was related to secretion in the core (Bassareo and Di Chiara, 1999). Directly relevant to the diminished dopamine signal in the NAc shell of BDNF2L/2LCk-cre mutants is the observation that inhibition of the NAc shell by local infusion of muscimol, a GABAA receptor agonist, resulted in dramatic hyperphagic behavior in animals fed ad libitum (Baldo et al., 2005).
Administration of a D1R selective agonist normalized the excessive intake of HFF exhibited by BDNF mutant mice in a 4 h HFF consumption test. Indeed, similar doses of D1R agonist had a far more profound and extended effect in the mutants compared with wild types. This finding provides further evidence of a link between deficits in dopamine transmission elicited by perturbed BDNF signaling and increased intake of palatable food. It also suggests that homeostatic adaptations take place in BDNF mutants that confer hypersensitivity to D1R stimulation. This notion is supported by studies involving dopamine-deficient mice, which exhibited hypersensitivity to D1R agonists that was ameliorated by 4 d of DOPA treatment (Kim et al., 2000). The enhanced responses of BDNF2L/2LCk-Cre to SKF38393 were not due to changes in D1R expression in the NAc or dorsal striatum of BDNF mutant mice. It is possible that depleted BDNF might lead to compensatory changes including increased affinity for D1R ligand or enhanced G-protein coupling to D1 receptors, which was demonstrated previously to confer hypersensitivity to dopamine (Gainetdinov et al., 2003).
Deficits in dopamine secretion were not limited to the NAc of BDNF mutant mice. Marked decreases were also observed in the dorsal striatum. Moreover, increased content of dopamine in this region was evident in BDNF2L/2LCk-cre mice, perhaps indicative of a compensatory homeostatic response to the reduced extracellular levels of dopamine in the mutants. Dopamine transmission in the dorsal striatum, which is densely innervated by dopaminergic fibers from the substantia nigra, has been linked to food reward processes. Release of dopamine in the dorsal striatum of healthy human subjects was induced by ingestion of palatable food and the amount of dopamine released correlated with the degree of experienced pleasure reported (Small et al., 2003). Therefore, we cannot rule out the possibility that deficient dopamine signaling in the dorsal striatum of BDNF2L/2LCk-cre mutants contributes to their excessive intake of palatable food. Nevertheless, it is clear from the studies described here that BDNF produced in the VTA plays a pivotal part in the control of feeding behavior.
BDNF was reported previously to facilitate synaptic sensitization of VTA dopamine neurons following cocaine withdrawal, which might represent a mechanism mediating drug craving and relapse (Pu et al., 2006). Moreover, Graham et al. (2007) demonstrated that selective depletion of BDNF in the mesolimbic system resulted in reduced cocaine self-administration, indicating that BDNF promotes the development and persistence of addictive behavior. In contrast, we found that deleting Bdnf in the VTA of mice resulted in increased intake of palatable high-fat food, a natural reward. An important distinction between these studies is that Graham et al. targeted Bdnf in the NAc of mice and attributed the observed alterations to disrupted BDNF signaling at postsynaptic sites in the NAc. This model was further supported by studies showing that TrkB knock down in the NAc but not in the VTA reduced cocaine reward (Graham et al., 2009). For our studies, we deleted Bdnf in the VTA and we postulated that BDNF acts presynaptically to positively modulate dopaminergic activity during food reward-related processes. Therefore, the findings from both of these studies suggest a disassociation of disease mechanisms mediating drug addiction and eating disorders.
Altered dopaminergic transmission has been linked to the emergence of eating disorders and obesity. However, conflicting disease mechanisms have been proposed. One model proposes that a heightened response of the dopaminergic reward circuitry to palatable food underlies excessive eating (Dawe et al., 2004). An alternative model postulates that hypoactivity of this neural system leads to reward deficiency syndrome and behaviorally, to compensatory overeating to boost a deficient dopaminergic system (Pothos et al., 1998; Blum et al., 2000; Wang et al., 2002). Based on our findings, we propose that perturbed BDNF signaling impedes activity of the mesolimbic dopamine pathway, leading to reward deficiency and compensatory overeating of palatable food. Consistent with this model, it was reported that over expression of r-FosB resulted in reduced protein levels of BDNF and concomitant molecular changes in the NAc consistent with deficient dopamine signaling, including reduced levels of DARPP-32 and pCREB (Teegarden et al., 2008). Notably, consumption of palatable HFF for 6 weeks completely reversed these abnormalities in r-FosB mice.
Rodent studies have shed some light onto putative pathological mechanisms involving dopamine systems and underlying eating disorders. Hyperphagic dietary obese and obesity-prone rats and leptin-deficient ob/ob mice exhibited reduced evoked dopamine release in the NAc (Pothos et al., 1998; Fulton et al., 2006; Geiger et al., 2009). Moreover, ob/ob mice displayed reductions in food intake and body weights when treated with D1R and D2R agonists (Bina and Cincotta, 2000). Some intriguing evidence comes from studies involving dopamine-deficient mice. They exhibit decreased food intake and without intervention fail to eat enough to sustain life beyond 48 h (Szczypka et al., 1999). However, whereas dopamine production selectively restored in the dorsal striatum of mutants rescued feeding of normal chow, restoration in either the NAc or dorsal striatum led to increased intake of palatable food (Szczypka et al., 2001). Furthermore, when ob/ob mice were crossed with dopamine-deficient mice, their hyperphagic behavior was diminished (Szczypka et al., 2000). These findings in dopamine-deficient mice demonstrate the important role of dopamine in more than one aspect of feeding motivation and a need to compensate for a complete dopamine deficit that might be absent in animals where central dopamine is present but hypofunctioning. The complexity of mechanisms underlying appetite control and the need for additional investigations to resolve these differences are evident. Nonetheless, the studies described here strongly support a pivotal role of BDNF in the positive regulation of mesolimbic dopamine transmission during food-reward processes.
In summary, we showed that the mesolimbic dopamine system is a newly identified target of action of BDNF for the control of feeding behavior. Moreover, that BDNF in the VTA has higher relevance in the regulation of hedonic processes impacting consumption of palatable food. Our findings implicate the dopaminergic reward circuit in the disease mechanisms triggered by perturbed BDNF signaling, leading to excessive food intake and obesity.
Footnotes
This work was supported by a Klarman Family Foundation grant (M.R.), National Institutes of Health (NIH)–National Institute of Diabetes and Digestive and Kidney Diseases research grants to M.R. (DK073311) and E.N.P. (DK065872), and NIH Training Grant T32 DK-07542 (J.W.C.). We thank the Genomics Core Facility at Tufts University, which is supported by the Center for Neuroscience Research (P30 NS047243), for facilitating these studies.
References
- Abizaid A, Liu ZW, Andrews ZB, Shanabrough M, Borok E, Elsworth JD, Roth RH, Sleeman MW, Picciotto MR, Tschöp MH, Gao XB, Horvath TL. Ghrelin modulates the activity and synaptic input organization of midbrain dopamine neurons while promoting appetite. J Clin Invest. 2006;116:3229–3239. doi: 10.1172/JCI29867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldo BA, Alsene KM, Negron A, Kelley AE. Hyperphagia induced by GABAA receptor-mediated inhibition of the nucleus accumbens shell: dependence on intact neural output from the central amygdaloid region. Behav Neurosci. 2005;119:1195–1206. doi: 10.1037/0735-7044.119.5.1195. [DOI] [PubMed] [Google Scholar]
- Baquet ZC, Bickford PC, Jones KR. Brain-derived neurotrophic factor is required for the establishment of the proper number of dopaminergic neurons in the substantia nigra pars compacta. J Neurosci. 2005;25:6251–6259. doi: 10.1523/JNEUROSCI.4601-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bariohay B, Lebrun B, Moyse E, Jean A. Brain-derived neurotrophic factor plays a role as an anorexigenic factor in the dorsal vagal complex. Endocrinology. 2005;146:5612–5620. doi: 10.1210/en.2005-0419. [DOI] [PubMed] [Google Scholar]
- Bassareo V, Di Chiara G. Differential influence of associative and nonassociative learning mechanisms on the responsiveness of prefrontal and accumbal dopamine transmission to food stimuli in rats fed ad libitum. J Neurosci. 1997;17:851–861. doi: 10.1523/JNEUROSCI.17-02-00851.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassareo V, Di Chiara G. Differential responsiveness of dopamine transmission to food-stimuli in nucleus accumbens shell/core compartments. Neuroscience. 1999;89:637–641. doi: 10.1016/s0306-4522(98)00583-1. [DOI] [PubMed] [Google Scholar]
- Berner LA, Avena NM, Hoebel BG. Bingeing, self-restriction, and increased body weight in rats with limited access to a sweet-fat diet. Obesity (Silver Spring) 2008;16:1998–2002. doi: 10.1038/oby.2008.328. [DOI] [PubMed] [Google Scholar]
- Berridge KC. ‘Liking’ and ‘wanting’ food rewards: brain substrates and roles in eating disorders. Physiol Behav. 2009;97:537–550. doi: 10.1016/j.physbeh.2009.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berton O, McClung CA, Dileone RJ, Krishnan V, Renthal W, Russo SJ, Graham D, Tsankova NM, Bolanos CA, Rios M, Monteggia LM, Self DW, Nestler EJ. Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science. 2006;311:864–868. doi: 10.1126/science.1120972. [DOI] [PubMed] [Google Scholar]
- Bina KG, Cincotta AH. Dopaminergic agonists normalize elevated hypothalamic neuropeptide Y and corticotropin-releasing hormone, body weight gain, and hyperglycemia in ob/ob mice. Neuroendocrinology. 2000;71:68–78. doi: 10.1159/000054522. [DOI] [PubMed] [Google Scholar]
- Blum K, Braverman ER, Holder JM, Lubar JF, Monastra VJ, Miller D, Lubar JO, Chen TJ, Comings DE. Reward deficiency syndrome: a biogenetic model for the diagnosis and treatment of impulsive, addictive, and compulsive behaviors. J Psychoactive Drugs. 2000;32(Suppl):i–iv. 1–112. doi: 10.1080/02791072.2000.10736099. [DOI] [PubMed] [Google Scholar]
- Broekman ML, Comer LA, Hyman BT, Sena-Esteves M. Adeno-associated virus vectors serotyped with AAV8 capsid are more efficient than AAV-1 or -2 serotypes for widespread gene delivery to the neonatal mouse brain. Neuroscience. 2006;138:501–510. doi: 10.1016/j.neuroscience.2005.11.057. [DOI] [PubMed] [Google Scholar]
- Chan JP, Unger TJ, Byrnes J, Rios M. Examination of behavioral deficits triggered by targeting Bdnf in fetal or postnatal brains of mice. Neuroscience. 2006;142:49–58. doi: 10.1016/j.neuroscience.2006.06.002. [DOI] [PubMed] [Google Scholar]
- Conner JM, Lauterborn JC, Yan Q, Gall CM, Varon S. Distribution of brain-derived neurotrophic factor (BDNF) protein and mRNA in the normal adult rat CNS: evidence for anterograde axonal transport. J Neurosci. 1997;17:2295–2313. doi: 10.1523/JNEUROSCI.17-07-02295.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corwin RL, Wojnicki FH. Binge eating in rats with limited access to vegetable shortening. Curr Protoc Neurosci. 2006 doi: 10.1002/0471142301.ns0923bs36. Chapter 9:Unit9 23B. [DOI] [PubMed] [Google Scholar]
- Davis JF, Melhorn SJ, Shurdak JD, Heiman JU, Tschöp MH, Clegg DJ, Benoit SC. Comparison of hydrogenated vegetable shortening and nutritionally complete high-fat diet on limited access-binge behavior in rats. Physiol Behav. 2007;92:924–930. doi: 10.1016/j.physbeh.2007.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawe S, Gullo MJ, Loxton NJ. Reward drive and rash impulsiveness as dimensions of impulsivity: implications for substance misuse. Addict Behav. 2004;29:1389–1405. doi: 10.1016/j.addbeh.2004.06.004. [DOI] [PubMed] [Google Scholar]
- Freeman AY, Soghomonian JJ, Pierce RC. Tyrosine kinase B and C receptors in the neostriatum and nucleus accumbens are co-localized in enkephalin-positive and enkephalin-negative neuronal profiles and their expression is influenced by cocaine. Neuroscience. 2003;117:147–156. doi: 10.1016/s0306-4522(02)00802-3. [DOI] [PubMed] [Google Scholar]
- Fulton S, Pissios P, Manchon RP, Stiles L, Frank L, Pothos EN, Maratos-Flier E, Flier JS. Leptin regulation of the mesoaccumbens dopamine pathway. Neuron. 2006;51:811–822. doi: 10.1016/j.neuron.2006.09.006. [DOI] [PubMed] [Google Scholar]
- Gainetdinov RR, Bohn LM, Sotnikova TD, Cyr M, Laakso A, Macrae AD, Torres GE, Kim KM, Lefkowitz RJ, Caron MG, Premont RT. Dopaminergic supersensitivity in G protein-coupled receptor kinase 6-deficient mice. Neuron. 2003;38:291–303. doi: 10.1016/s0896-6273(03)00192-2. [DOI] [PubMed] [Google Scholar]
- Geiger BM, Haburcak M, Avena NM, Moyer MC, Hoebel BG, Pothos EN. Deficits of mesolimbic dopamine neurotransmission in rat dietary obesity. Neuroscience. 2009;159:1193–1199. doi: 10.1016/j.neuroscience.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgescu D, Sears RM, Hommel JD, Barrot M, Bolaños CA, Marsh DJ, Bednarek MA, Bibb JA, Maratos-Flier E, Nestler EJ, DiLeone RJ. The hypothalamic neuropeptide melanin-concentrating hormone acts in the nucleus accumbens to modulate feeding behavior and forced-swim performance. J Neurosci. 2005;25:2933–2940. doi: 10.1523/JNEUROSCI.1714-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghiglieri O, Gambarana C, Scheggi S, Tagliamonte A, Willner P, De Montis MG. Palatable food induces an appetitive behaviour in satiated rats which can be inhibited by chronic stress. Behav Pharmacol. 1997;8:619–628. doi: 10.1097/00008877-199711000-00018. [DOI] [PubMed] [Google Scholar]
- Graham DL, Edwards S, Bachtell RK, DiLeone RJ, Rios M, Self DW. Dynamic BDNF activity in nucleus accumbens with cocaine use increases self-administration and relapse. Nat Neurosci. 2007;10:1029–1037. doi: 10.1038/nn1929. [DOI] [PubMed] [Google Scholar]
- Graham DL, Krishnan V, Larson EB, Graham A, Edwards S, Bachtell RK, Simmons D, Gent LM, Berton O, Bolanos CA, DiLeone RJ, Parada LF, Nestler EJ, Self DW. Tropomyosin-related kinase B in the mesolimbic dopamine system: region-specific effects on cocaine reward. Biol Psychiatry. 2009;65:696–701. doi: 10.1016/j.biopsych.2008.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray J, Yeo GS, Cox JJ, Morton J, Adlam AL, Keogh JM, Yanovski JA, El Gharbawy A, Han JC, Tung YC, Hodges JR, Raymond FL, O'Rahilly S, Farooqi IS. Hyperphagia, severe obesity, impaired cognitive function, and hyperactivity associated with functional loss of one copy of the brain-derived neurotrophic factor (BDNF) gene. Diabetes. 2006;55:3366–3371. doi: 10.2337/db06-0550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillin O, Diaz J, Carroll P, Griffon N, Schwartz JC, Sokoloff P. BDNF controls dopamine D3 receptor expression and triggers behavioural sensitization. Nature. 2001;411:86–89. doi: 10.1038/35075076. [DOI] [PubMed] [Google Scholar]
- Han JC, Liu QR, Jones M, Levinn RL, Menzie CM, Jefferson-George KS, Adler-Wailes DC, Sanford EL, Lacbawan FL, Uhl GR, Rennert OM, Yanovski JA. Brain-derived neurotrophic factor and obesity in the WAGR syndrome. N Engl J Med. 2008;359:918–927. doi: 10.1056/NEJMoa0801119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hommel JD, Sears RM, Georgescu D, Simmons DL, DiLeone RJ. Local gene knockdown in the brain using viral-mediated RNA interference. Nat Med. 2003;9:1539–1544. doi: 10.1038/nm964. [DOI] [PubMed] [Google Scholar]
- Hommel JD, Trinko R, Sears RM, Georgescu D, Liu ZW, Gao XB, Thurmon JJ, Marinelli M, DiLeone RJ. Leptin receptor signaling in midbrain dopamine neurons regulates feeding. Neuron. 2006;51:801–810. doi: 10.1016/j.neuron.2006.08.023. [DOI] [PubMed] [Google Scholar]
- Kernie SG, Liebl DJ, Parada LF. BDNF regulates eating behavior and locomotor activity in mice. EMBO J. 2000;19:1290–1300. doi: 10.1093/emboj/19.6.1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DS, Szczypka MS, Palmiter RD. Dopamine-deficient mice are hypersensitive to dopamine receptor agonists. J Neurosci. 2000;20:4405–4413. doi: 10.1523/JNEUROSCI.20-12-04405.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leddy JJ, Epstein LH, Jaroni JL, Roemmich JN, Paluch RA, Goldfield GS, Lerman C. Influence of methylphenidate on eating in obese men. Obes Res. 2004;12:224–232. doi: 10.1038/oby.2004.29. [DOI] [PubMed] [Google Scholar]
- Lee MD, Clifton PG. Meal patterns of free feeding rats treated with clozapine, olanzapine, or haloperidol. Pharmacol Biochem Behav. 2002;71:147–154. doi: 10.1016/s0091-3057(01)00630-x. [DOI] [PubMed] [Google Scholar]
- Lyons WE, Mamounas LA, Ricaurte GA, Coppola V, Reid SW, Bora SH, Wihler C, Koliatsos VE, Tessarollo L. Brain-derived neurotrophic factor-deficient mice develop aggressiveness and hyperphagia in conjunction with brain serotonergic abnormalities. Proc Natl Acad Sci U S A. 1999;96:15239–15244. doi: 10.1073/pnas.96.26.15239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Numan S, Seroogy KB. Expression of trkB and trkC mRNAs by adult midbrain dopamine neurons: a double-label in situ hybridization study. J Comp Neurol. 1999;403:295–308. doi: 10.1002/(sici)1096-9861(19990118)403:3<295::aid-cne2>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates. Ed 2. San Diego: Academic; 2001. [Google Scholar]
- Pothos EN, Sulzer D, Hoebel BG. Plasticity of quantal size in ventral midbrain dopamine neurons: possible implications for the neurochemistry of feeding and reward. Appetite. 1998;31:405. doi: 10.1006/appe.1998.0210. [DOI] [PubMed] [Google Scholar]
- Pu L, Liu QS, Poo MM. BDNF-dependent synaptic sensitization in midbrain dopamine neurons after cocaine withdrawal. Nat Neurosci. 2006;9:605–607. doi: 10.1038/nn1687. [DOI] [PubMed] [Google Scholar]
- Rios M, Fan G, Fekete C, Kelly J, Bates B, Kuehn R, Lechan RM, Jaenisch R. Conditional deletion of brain-derived neurotrophic factor in the postnatal brain leads to obesity and hyperactivity. Mol Endocrinol. 2001;15:1748–1757. doi: 10.1210/mend.15.10.0706. [DOI] [PubMed] [Google Scholar]
- Rios M, Lambe EK, Liu R, Teillon S, Liu J, Akbarian S, Roffler-Tarlov S, Jaenisch R, Aghajanian GK. Severe deficits in 5-HT2A -mediated neurotransmission in BDNF conditional mutant mice. J Neurobiol. 2006;66:408–420. doi: 10.1002/neu.20233. [DOI] [PubMed] [Google Scholar]
- Seroogy KB, Lundgren KH, Tran TM, Guthrie KM, Isackson PJ, Gall CM. Dopaminergic neurons in rat ventral midbrain express brain-derived neurotrophic factor and neurotrophin-3 mRNAs. J Comp Neurol. 1994;342:321–334. doi: 10.1002/cne.903420302. [DOI] [PubMed] [Google Scholar]
- Small DM, Jones-Gotman M, Dagher A. Feeding-induced dopamine release in dorsal striatum correlates with meal pleasantness ratings in healthy human volunteers. Neuroimage. 2003;19:1709–1715. doi: 10.1016/s1053-8119(03)00253-2. [DOI] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain [letter] Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Szczypka MS, Rainey MA, Kim DS, Alaynick WA, Marck BT, Matsumoto AM, Palmiter RD. Feeding behavior in dopamine-deficient mice. Proc Natl Acad Sci U S A. 1999;96:12138–12143. doi: 10.1073/pnas.96.21.12138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczypka MS, Rainey MA, Palmiter RD. Dopamine is required for hyperphagia in Lep(ob/ob) mice. Nat Genet. 2000;25:102–104. doi: 10.1038/75484. [DOI] [PubMed] [Google Scholar]
- Szczypka MS, Kwok K, Brot MD, Marck BT, Matsumoto AM, Donahue BA, Palmiter RD. Dopamine production in the caudate putamen restores feeding in dopamine-deficient mice. Neuron. 2001;30:819–828. doi: 10.1016/s0896-6273(01)00319-1. [DOI] [PubMed] [Google Scholar]
- Tanda G, Di Chiara G. A dopamine-mu1 opioid link in the rat ventral tegmentum shared by palatable food (Fonzies) and non-psychostimulant drugs of abuse. Eur J Neurosci. 1998;10:1179–1187. doi: 10.1046/j.1460-9568.1998.00135.x. [DOI] [PubMed] [Google Scholar]
- Teegarden SL, Nestler EJ, Bale TL. Delta FosB-mediated alterations in dopamine signaling are normalized by a palatable high-fat diet. Biol Psychiatry. 2008;64:941–950. doi: 10.1016/j.biopsych.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran PV, Akana SF, Malkovska I, Dallman MF, Parada LF, Ingraham HA. Diminished hypothalamic bdnf expression and impaired VMH function are associated with reduced SF-1 gene dosage. J Comp Neurol. 2006;498:637–648. doi: 10.1002/cne.21070. [DOI] [PubMed] [Google Scholar]
- Unger TJ, Calderon GA, Bradley LC, Sena-Esteves M, Rios M. Selective deletion of Bdnf in the ventromedial and dorsomedial hypothalamus of adult mice results in hyperphagic behavior and obesity. J Neurosci. 2007;27:14265–14274. doi: 10.1523/JNEUROSCI.3308-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GJ, Volkow ND, Logan J, Pappas NR, Wong CT, Zhu W, Netusil N, Fowler JS. Brain dopamine and obesity. Lancet. 2001;357:354–357. doi: 10.1016/s0140-6736(00)03643-6. [DOI] [PubMed] [Google Scholar]
- Wang GJ, Volkow ND, Fowler JS. The role of dopamine in motivation for food in humans: implications for obesity. Expert Opin Ther Targets. 2002;6:601–609. doi: 10.1517/14728222.6.5.601. [DOI] [PubMed] [Google Scholar]
- Xu B, Goulding EH, Zang K, Cepoi D, Cone RD, Jones KR, Tecott LH, Reichardt LF. Brain-derived neurotrophic factor regulates energy balance downstream of melanocortin-4 receptor. Nat Neurosci. 2003;6:736–742. doi: 10.1038/nn1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Q, Radeke MJ, Matheson CR, Talvenheimo J, Welcher AA, Feinstein SC. Immunocytochemical localization of TrkB in the central nervous system of the adult rat. J Comp Neurol. 1997;378:135–157. [PubMed] [Google Scholar]