

Abstract
Arylamine N-acetyltransferases (NATs) are cytosolic enzymes that catalyze the transfer of the acetyl group from acetyl coenzyme A (AcCoA) to the free amino group of arylamines and hydrazines. Previous studies have reported that overexpression of NAT from Mycobacterium smegmatis and Mycobacterium tuberculosis may be responsible for increased resistance to the front-line antitubercular drug, isoniazid, by acetylating and hence inactivating the prodrug. We report the kinetic characterization of M. tuberculosis NAT which reveals that substituted anilines are excellent substrates but that isoniazid is a very poor substrate for this enzyme. We propose that the expression of NAT from M. tuberculosis (TBNAT) is unlikely to be a significant cause of isoniazid resistance. The kinetic parameters for a variety of TBNAT substrates were examined, including 3-amino-4-hydroxybenzoic acid and AcCoA, revealing Km values of 0.32 ± 0.03 and 0.14 ± 0.02 mM, respectively. Steady-state kinetic analysis of TBNAT reveals that the enzyme catalyzes the reaction via a bi-bi ping-pong kinetic mechanism. The pH dependence of the kinetic parameters reveals that one enzyme group must be deprotonated for optimal catalytic activity and that two amino acid residues at the active site of the free enzyme are involved in binding and/or catalysis. Solvent kinetic isotope effects suggest that proton transfer steps are not rate-limiting in the overall reaction for substituted aniline substrates but become rate-limiting when poor hydrazide substrates are used.
Mycobacterium tuberculosis, the etiological agent of tuberculosis (TB),1 is responsible for more deaths throughout the world than any other bacterial infectious disease (1). Multidrug-resistant strains of TB are increasingly prevalent worldwide, and no new treatment modalities have been introduced in more than 30 years. Additionally, mycobacteria contain a unique and essential fatty acid component in their outer cell walls, mycolic acids, which provides a robust barrier to the penetration of many broad spectrum antibiotics and chemotherapeutic agents (2). Isoniazid is currently prescribed in all prophylactic and treatment regimens for TB; however, resistance to this bactericidal agent is increasing alarmingly (3). Isoniazid is a prodrug that first requires oxidative activation by the mycobacterial KatG-encoded catalase-peroxidase (Scheme 1) (4). After the prodrug is activated, it reacts with intracellular pyridine nucleotide cofactors (NAD+ and NADP+) to generate C4-isonicotinoyl adducts, one of which powerfully inhibits InhA, the M. tuberculosis enoyl-ACP reductase of the FASII fatty acid synthase system responsible for mycolic acid biosynthesis (5). Inhibition of InhA by the INH–NAD adduct has been shown to uniquely result in M. tuberculosis lysis and cell death (6).
Scheme 1.
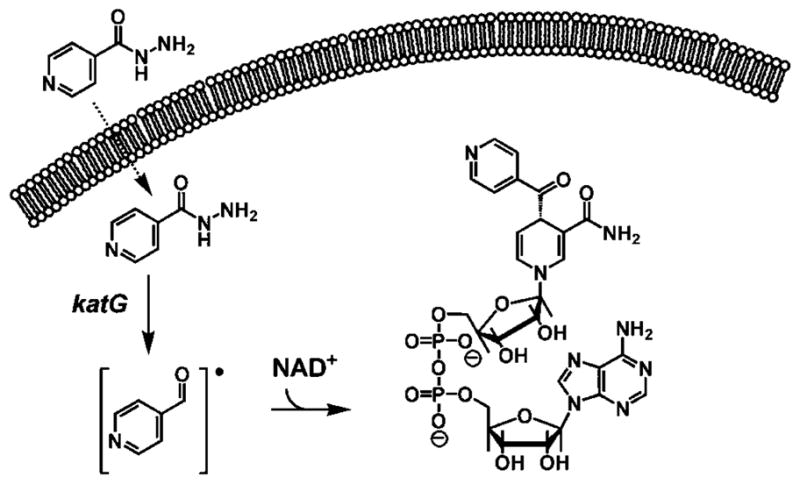
Oxidative Activation of INH by Mycobacterial KatG Results in the Formation of an Isonicotinoyl Radical Which Nonenzymatically Reacts with Intracellular Pyridine Nucleotide Cofactors To Generate the INH–NAD Inhibitory Adduct
Since its clinical introduction in 1953, isoniazid chemotherapy, while incredibly effective in most treated individuals, was equally ineffective in others. In 1960, it was reported that nonresponding individuals had high levels of an enzyme that acetylated isoniazid, and this correlated with the ineffectiveness of INH therapy (7). Mammalian arylamine N-acetyltransferases (NATs, EC 2.3.1.5) are polymorphic, broad-specificity, xenobiotic-metabolizing enzymes and were first identified in humans because of their activity with isoniazid. NATs are also found in prokaryotes and are cytosolic enzymes that catalyze the transfer of the acetyl group from acetyl coenzyme A to the free amino group of arylamines and hydrazines on a wide variety of structural scaffolds (Scheme 2) (8). However, unlike the human NAT, which exists in two active enzyme isoforms (NAT1 and NAT2), only a single nat gene is present in prokaryotes (9). Previous studies have suggested that the activity of NAT improves the survivability of mycobacteria by N-acetylation of isoniazid, thus preventing its oxidative activation (10). Subsequent studies reported that overexpression of the M. tuberculosis NAT (TBNAT) in Mycobacterium smegmatis may be responsible for increased resistance to isoniazid (11).
Scheme 2.

Reaction Catalyzed by NATs
Several crystallographically determined three-dimensional structures of mycobacterial NATs have recently been reported. The structure of the M. smegmatis NAT in complex with INH and the very recent structure of the M. marinum NAT in complex with AcCoA reveal a common fold (12, 13). This fold, observed in all prokaryotic NATs studied to date, contains three conserved active site residues that form a Cys-His-Asp catalytic triad (14). Investigation of this catalytic triad in M. smegmatis and Salmonella typhimurium has revealed that each of the three residues is essential and required for acetyltransferase activity (15). Moreover, this catalytic triad is very similar to the Cys-His-Asn triad present in cysteine proteases, and it has thus been proposed that eukaryotic NATs and cysteine proteases may share a related catalytic mechanism (16, 17).
We report here the expression, purification, and kinetic characterization of TBNAT. Our data reveal that substituted anilines are excellent substrates for TBNAT but that isoniazid is an extremely poor substrate for the enzyme. We therefore propose that the expression of TBNAT is unlikely to be a major source of isoniazid resistance in M. tuberculosis. From the pH dependencies of TBNAT kinetic parameters, and the observed solvent kinetic isotope effects, combined with existing structural data, we propose a detailed model of the chemistry that occurs at the active site of TBNAT.
MATERIALS AND METHODS
Materials
All chemicals, AcCoA, and nitrogen-containing substrates were purchased from Sigma-Aldrich Chemical Co. Enzymes used in molecular cloning were supplied by New England Biolabs. Plasmid pET-28a(+) and Escherichia coli strain BL21(DE3) were obtained from Novagen.
Cloning, Expression, and Purification of TBNAT
The open reading frame of the TBNAT gene (rv3566c) was amplified from M. tuberculosis H37Rv genomic DNA by standard PCR techniques using oligonucleotides NATf (5′-ATCCCGCTCATATGGCACTGGATCTCTGACCGCG-3′) and NATr (5 ′-ATCCCGCTAAGCTTTTACGGCGCATCGGCTCCTGG-3′) containing the underlined NdeI and HindIII restriction sites, respectively. The PCR fragment was cloned into pET-28a(+), and the recombinant plasmid, containing the tbnat gene, bearing a thrombin-cleavable N-terminal His6 tag, was transformed into competent E. coli BL21(DE3) cells, harboring the pGroESL-911 plasmid that expresses the molecular chaperone GroES/GroEL (18). The transformed cells were used to inoculate 12 L of LB containing 10 μg/mL tetracycline and 35 μg/mL kanamycin. The culture was grown to midlog phase (A600 ~ 0.8) at 37 °C, then induced by the addition of 0.5 mM IPTG, and further incubated overnight at 16 °C.
All purification procedures were performed at 4 °C. The cells were harvested by centrifugation and suspended in buffer A [50 mM Tris (pH 7.7) and 250 mM NaCl] containing protease inhibitors, lysozyme (5 μg/mL), and DNase I (0.1 μg/mL). The cells were then lysed by sonication, and cell debris was removed by centrifugation at 38000g for 30 min. The supernatant was dialyzed against buffer A, loaded onto a Ni-NTA column pre-equilibrated with buffer A, and washed with 10 column volumes of the same buffer. The bound proteins were eluted with a linear imidazole gradient (from 0 to 0.3 M) at a flow rate of 1 mL/min. Fractions containing TBNAT were pooled and concentrated to 5 mL by centrifugation through an Amicon concentrator with a 10 kDa cutoff membrane. The His6 tag was then cleaved using thrombin (2 units/mg of protein), and the solution was dialyzed overnight against buffer A containing 2 mM CaCl2. The protein was then concentrated to 2 mL and applied to a Superdex S-75 column, pre-equilibrated with buffer A. Pure fractions, as determined by SDS–PAGE, were pooled and concentrated by ultrafiltration to a concentration of 8.1 mg/mL and stored in 50% glycerol at −20 °C.
Protein Estimation
Protein concentrations were estimated by the Bio-Rad protein assay method using bovine serum albumin as a standard.
Measurement of Enzyme Activity
Reaction rates were measured spectrophotometrically by following the increase in absorbance at 412 nm due to the reaction between the free sulfhydryl group of the product of the reaction, CoASH, and DTNB (19). The reaction was monitored continuously on a UVIKON XL spectrophotometer, and enzyme activities were calculated using a molar extinction coefficient of 13600 M−1 cm−1. Assay mixtures contained 50 mM Tris (pH 7.7) and 0.1 mM DTNB in addition to substrates in a final volume of 1 mL. Reactions were initiated by the addition of enzyme, typically a final concentration of 10 nM, and followed at 25 °C for 2–3 min.
Initial Velocity Experiments
Kinetic constants for AcCoA were determined at fixed concentrations of 3-amino-4-hydroxybenzoic acid (AHB, 1.5 mM) and at variable concentrations of AcCoA (0.005–1.5 mM). Kinetic constants for arylamine substrates were determined at a fixed concentration of AcCoA (0.8 mM). Initial velocities were determined with at least five different concentrations of each substrate. Individual substrate saturation kinetic data were fitted to eq 1 using Sigma Plot 8.0:
| (1) |
where V is the maximal velocity, A is the substrate concentration, and K is the Michaelis–Menten constant (Km). Initial velocity patterns were determined at various concentrations of both AcCoA (50–500 μM) and AHB (0.1–0.5 mM), and the data were globally fit to eq 2, which is the rate equation for a bi-bi ping-pong reaction where a parallel initial velocity pattern is observed:
| (2) |
where V is the maximal velocity, A and B are the concentrations of the substrates, and Ka and Kb are the Michaelis–Menten constants for the substrates.
pH Dependence of TBNAT Activity
The pH dependence of kinetic parameters kcat and kcat/KAcCoA exhibited by TBNAT was determined using a saturating concentration of AHB and variable concentrations of AcCoA (0.1–0.8 mM). The pH dependence of kcat/KAHB was examined using a saturating concentration of AcCoA and variable concentrations of AHB (0.1–4 mM). Acetyltransferase activity was monitored every 0.5 pH unit from pH 6.0 to 10.0 using the following buffers: MES (pH 5.0–7.0), Tris (pH 7.0–9.0), and AMeP (pH 9.0–10.0). The resulting kinetic data were fitted to eq 1 to obtain the kinetic parameters kcat and kcat/Km. Profiles were generated by plotting the log of kcat or kcat/Km versus pH and fitted to eq 3, 4, or 5 to yield the pK values of the ionizable groups
| (3) |
| (4) |
| (5) |
where C is the pH-independent plateau value, K is the observed dissociation constant(s) for the ionizing groups(s), and [H+] is the hydrogen ion concentration. These equations describe pH dependencies where the protonation of a single group decreases the magnitude of the kinetic parameter, where the protonation of two groups with experimentally indistinguishable pK values decreases the magnitude of the kinetic parameter, and where the protonation of a single group and the deprotonation of a single group decrease the magnitude of the kinetic parameter.
Solvent Kinetic Isotope Effects
The solvent kinetic isotope effects on kcat and kcat/Km were determined by measuring the initial velocities at a fixed concentration of AHB (2.5 mM) while varying the concentration of AcCoA (0.5–2 mM), or by measuring the initial velocities using a fixed concentration of AcCoA (1.5 mM) while varying the concentration of either AHB (0.13–2 mM) or benzoic acid hydrazide (7–50 mM) in either H2O or 98% D2O at pH 8.0. Solvent deuterium kinetic isotope effects were fitted to the following equation:
| (6) |
where EV/K and EV are the isotope effects on kcat/Km − 1 and kcat − 1, respectively, and Fi represents the fraction of isotope (0.98). Proton inventories were performed using a 10× mix of AHB, AcCoA, DTNB, and Tris (pH 7.7), such that final concentrations were 1.6, 0.072, 50, and 0.1 mM, respectively. Water and D2O were added to the mix to yield a percentage of D2O ranging from 0 to 88% in a final reaction volume of 1 mL. Reactions were initiated by the addition of enzyme (10 nM) and followed continuously for 2 min at 25 °C. Triplicate reactions were performed, and initial velocities, including standard deviations, were plotted as a function of the percentage of D2O in the reaction mix.
pH Dependence of TBNAT Inactivation by Iodoacetamide
The rate of TBNAT inactivation by iodoacetamide was measured over a pH range of 6.5–10 using the same buffers described previously. The inactivation was initiated by the addition of 2 mM iodoacetamide to a solution containing 1 μM enzyme in the appropriate buffer (50 mM) at 25 °C. Aliquots (10 μL) were removed at various time points and added to an assay solution containing 50 mM Tris (pH 7.7), 0.1 mM DTNB, 0.5 mM AcCoA, and 0.2 mM AHB in a final volume of 1 mL. The enzyme activity was measured by monitoring the change in absorbance at 412 nm (as described above), and the percentage of activity remaining after iodoacetamide incubation was calculated with respect to controls which did not contain iodoacetamide. Data were plotted as log(percent activity remaining) versus time, and the half-life (t1/2) of inactivation was obtained from the slope. kinact was calculated from t1/2 according to eq 7 and plotted as log(kinact) versus pH.
| (7) |
RESULTS AND DISCUSSION
Cloning, Expression, and Purification
PCR amplification of the tbnat gene yielded a single fragment of the expected length (853 bp). DNA sequencing of the cloned fragment confirmed the expected sequence and the absence of any mutations introduced during PCR amplification. Expression of the PCR product resulted in a soluble protein product with an apparent molecular mass, determined by SDS–PAGE, in agreement with the mass of 30000 Da deduced from the amino acid sequence. Approximately 2.7 mg of purified enzyme was obtained per liter of culture.
Substrate Specificity of TBNAT
Approximate kinetic parameters have been determined for several eukaryotic NATs (17, 20–28) and, more recently, for bacterial NATs as well (13, 29). These studies have revealed that each prokaryotic NAT enzyme displays unique substrate specificity for various arylamine, hydrazide, and hydrazine substrates (12). For example, arylhydrazines are reported to be the best substrates for NAT from M. smegmatis (MSNAT), yet NAT from another mycobacterium, Mycobacterium marinum (MMNAT), acetylates these substrates 100 times faster (12). Furthermore, mammalian NATs have substrate specificity profiles that are distinct from those of the prokaryotic NATs. For example, human NAT2 has a reported Km value of 0.38–0.58 mM for isoniazid, while MSNAT has a reported Km of 7.3 μM for the same substrate (13, 30). The significant variation of substrate specificity among all NATs suggests that the role of NAT differs depending upon the organism in which it is found (31).
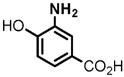
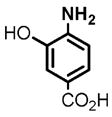
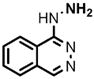
To evaluate the substrate specificity of TBNAT, we selected a variety of compounds, including previously documented NAT substrates and those that share structural similarity. Initial assessment of TBNAT activity revealed 3-amino-4-hydroxybenzoic acid (AHB) and AcCoA to be very robust substrates for the enzyme with Km values of 0.32 ± 0.03 and 0.14 ± 0.02 mM, respectively, with a kcat value of 94 ± 2 s−1. TBNAT activity was measured at a fixed concentration of AcCoA (0.8 mM) and varying concentrations of the substituted anilines, hydrazines, and hydrazides that were tested. The catalytic efficiencies of these substrates covered more than 4 log orders of activity, ranging from a kcat/Km equal to 12 M−1 s−1 for 4-fluoroaniline to a value of 2.9 × 105 M−1 s−1 for AHB (Table 1). With the exception of hydralazine, the six best substrates tested contain a hydroxyl group ortho to the amine nucleophile. The modest activity of p-aminobenzoic acid (kcat/Km = 1.46 × 102 M−1 s−1) emphasizes the importance of the o-hydroxyl group in catalysis. Furthermore, p-hydroxybenzoic acid exhibited no activity when tested as a TBNAT substrate, thus confirming the aniline nitrogen as the nucleophile responsible for attack on the acetylated enzyme. Weak substrates for TBNAT contain electron-withdrawing groups, such as the p-fluorine-and p-chlorine-substituted anilines. As expected, alkylamines, including benzylamine, propylamine, and hexylamine, showed no activity when tested as TBNAT substrates, even when tested at pH 9 and 10 where they are presumably deprotonated.
Table 1.
Kinetic Parameters of TBNAT with Amine Substratesa
| Substrateb | Km (mM) | Kcat(s−1) | Kcat/Km (M−1s−1) |
|---|---|---|---|
 1 |
0.32 +/− 0.03 | 94 +/− 2 | 2.9 × 105 |
 2 |
0.90 +/− 0.18 | 66 +/− 5 | 7.3 × 104 |
 3 |
0.61 +/− 0.06 | 44 +/− 1 | 7.2 × 104 |
 4 |
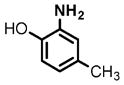
3.06 +/− 0.03 | 70 +/− 2 | 2.3 × 104 |
 5 |
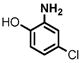
1.9 +/− 0.4 | 21 +/− 1 | 1.1 × 104 |
 6 |
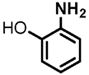
5.79 +/− 0.04 | 51 +/− 1 | 8.9 × 103 |
 7 |
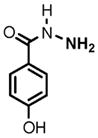
20 +/− 3 | 10.9 +/− 0.8 | 5.5 × 102 |
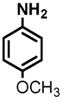 8 |
14 +/− 1 | 2.9 +/− 0.1 | 2.1 × 102 |
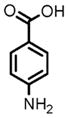 9 |
5.0 +/− 1 | 0.7 +/− 0.1 | 1.5 × 102 |
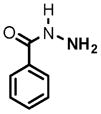 10 |
14 +/− 2 | 1.9 +/− 0.1 | 1.4 × 102 |
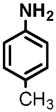 11 |
11 +/− 1 | 0.8 +/− 0.1 | 7.6 × 101 |
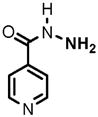 12 |
102 +/− 6 | 4.9 +/− 0.1 | 4.9 × 101 |
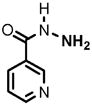 13 |
14 +/− 20 | .6 +/− 0.1 | 4.5 × 101 |
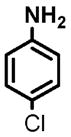 14 |
6 +/− 1 | 0.2 +/− 0.1 | 3.8 × 101 |
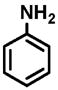 15 |
23 +/− 2 | 0.28 +/− 0.11 | 1.7 × 101 |
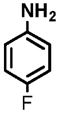 16 |
51 +/− 6 | 0.9 +/− 0.1 | 1.2 × 101 |
All assays were performed in the presence of 0.8 mM AcCoA at pH 7.7.
(1) 3-Amino-4-hydroxybenzoic acid (AHB), (2) 4-amino-3-hydroxybenzoic acid, (3) hydralazine, (4) 2-amino-4-methylphenol, (5) 2-amino-4-chlorophenol, (6) 2-aminophenol, (7) 4-hydroxybenzhydrazide, (8) anisidine, (9) p-aminobenzoic acid, (10) benzoic acid hydrazide (BAH), (11) 4-methylaniline, (12) isoniazid (INH), (13) nicotinic acid hydrazide, (14) 4-chloroaniline, (15) aniline, and (16) 4-fluoroaniline.
Previous studies have reported that isoniazid is an excellent substrate for MSNAT with a reported Km value of 7.3 μM (13). On the basis of the relatively high (60%) degree of primary sequence homology between MSNAT and TBNAT, Sandy et al. (13) suggested that isoniazid might also be an excellent substrate for TBNAT. However, we determined a Km value of 102 ± 6 mM and a kcat/Km value of 49 M− 1 s−1 for isoniazid, indicating that isoniazid is a very poor substrate for TBNAT, some 4 orders of magnitude less efficient than the best substituted anilines. Four additional hydrazides, including nicotinic acid hydrazide, benzhydrazide, 4-hydroxybenzhydrazide, and o-hydroxybenzyhydrazide, were tested as possible substrates for TBNAT. 4-Hydroxybenzhydrazide demonstrated only modest activity with a kcat/Km of 550 M−1 s−1, while benzhydrazide and nicotinic acid hydrazide exhibited poor catalytic efficiencies with kcat/Km values equal to 140 and 45 M−1 s−1, respectively. o-Hydroxybenzhydrazide was not a substrate for TBNAT. These results confirm that TBNAT is ineffective in hydrazide N-acetylation. Furthermore, in a previous study completed by Bhakta and colleagues, it was observed that the genetic deletion of nat from Mycobacterium bovis BCG results in only a minor effect on isoniazid sensitivity (32). This observation, as well as the results obtained from this study, provide both kinetic and genetic evidence that TBNAT expression is unlikely to be a significant contributor to isoniazid resistance in TB.
Kinetic Mechanism
All prokaryotic and eukaryotic NATs studied to date catalyze acetyl transfer using a bi-bi ping-pong kinetic mechanism (33). To conclusively determine the kinetic mechanism of TBNAT, we measured the initial velocity of the acetyl transfer reaction while simultaneously varying the concentrations of AHB and AcCoA. The resultant double-reciprocal plot yielded a series of parallel lines diagnostic of the expected bi-bi ping-pong kinetic mechanism (Figure 1). On the basis of this result, a proposed kinetic mechanism for TBNAT catalysis is shown in Scheme 3 in which the enzyme initially reacts with AcCoA to generate the first product, CoA, and the acetyl–enzyme intermediate (E–Ac) (Scheme 3). The amine binds and reacts with E–Ac to generate the second product, acetylated amine, and regenerate the free enzyme (34).
Figure 1.

Bisubstrate kinetic analysis of TBNAT. The initial velocity of acetyl transfer was measured in 50 mM Tris (pH 7.7) with varying concentrations of AHB (100–500 μM) and fixed concentrations of AcCoA: 50 (●), 75 (○), 150 (▼), and 500 μM (▽). Shown is the double-reciprocal plot of the initial velocity as a function of AHB concentration. Global fitting of the data to eq 2 (solid lines) gave a kcat of 154 ± 13 s−1, a KAcCoA of 0.358 ± 0.04 mM, and a KAHB of 0.836 ± 0.09 mM.
Scheme 3.

Proposed Ping-Pong Kinetic Mechanism of TBNAT
pH Dependence of TBNAT Activity
To investigate the ionization behavior of groups that may be responsible for binding and catalysis in the TBNAT-catalyzed acetyl transfer reaction, we examined the pH dependence of kcat, kcat/KAcCoA, and kcat/KAHB (Figure 2).
Figure 2.

pH dependence of TBNAT activity. (A) Log(kcat) vs pH (AHB saturating, AcCoA concentration extrapolated to infinity). Points are experimental, while the line is a fit of the data to eq 3 yielding a pKa value of 7.4 ± 0.1. (B) Log(kcat/KAcCoA) vs pH (AHB saturating, variable AcCoA concentrations). Points are experimental, while the line is a fit of the data to eq 4 yielding pKa values for the two groups of 6.9 ± 0.2. (C) Log(kcat/KAHB) vs pH (AcCoA saturating, variable AHB concentrations). Points are experimental, while the line is a fit of the data to eq 5 yielding pKa values of 7.3 ± 0.1 and 8.8 ± 0.1.
The pH dependence of kcat reveals the presence of a single ionizable group with a pKa value of 7.4 ± 0.1 that must be deprotonated for optimal activity (Figure 2A). In ping-pong reactions, the pH dependence of kcat reflects steps from the formation of the acyl–enzyme intermediate through the transfer of the acetyl group to the amine substrate and thus may be an amalgam of the pH dependencies of each half-reaction, or if one half-reaction is significantly slower than the other (>10 times), kcat can report specifically on the slower of the two half-reactions. A priori, and absent information about the relative rates of isolated half-reactions, we cannot speculate about the rate-limiting nature of the acetylation versus deacetylation reactions in TBNAT catalysis. If the first half-reaction (i.e., acetylation) is rate-limiting, a viable candidate for the observed pKa of 7.4 ± 0.1 in the kcat profile is His110 (or the His110-Asp127 dyad) which we propose is the general base responsible for activating the active site cysteine nucleophile (Scheme 4). Alternatively, if the second half-reaction (i.e., deacetylation) is rate-limiting, we envision that this pKa may similarly be attributable to His110 (or the His110-Asp127 dyad), which we propose serves as a general base to deprotonate either the incoming amine nucleophile or the zwitterionic tetrahedral intermediate.
Scheme 4.

Proposed Chemical Mechanism of the Reaction Catalyzed by TBNAT
The pH dependence of kcat/KAcCoA reflects steps from the binding of AcCoA through the first irreversible step in catalysis. While this is often considered to be the chemical reaction, transfer of the acetyl group from the thioester of AcCoA to the active site cysteine residue is likely to be reversible given its isoenergetic nature. It is therefore probable that the release of the product CoA is the first irreversible step in TBNAT catalysis and that the pH dependence of kcat/KAcCoA reflects steps from the binding of AcCoA through the release of CoA (Scheme 3). The pH dependence of kcat/KAcCoA reveals the presence of two ionizable groups with indistinguishable pKa values of 6.9 ± 0.2 that are involved in catalysis and/or binding during this portion of the reaction (Figure 2B). It is likely that one of these residues is the same as that revealed in the kcat profile, while the second group is expected to be involved in substrate binding. One possibility is that the 3′-phosphate monoester of AcCoA is the source of this second ionization. In the reported structure of the M. marinum NAT–CoA complex, the 3′-phosphate of CoA forms a 3.1 Å salt bridge with Lys236; protonation of this group would result in the disruption of this important structural contact and a decrease in AcCoA binding affinity (12).
The pH dependence of kcat/KAHB reflects steps from binding of the amine substrate through the transfer of the acetyl group from the enzyme to the amine acceptor. Our results indicate that the ionization of two groups in the acetyl–enzyme intermediate with pKa values of 7.3 ± 0.1 and 8.8 ± 0.1 must be deprotonated and protonated, respectively, during this second half-reaction (Figure 2C). It is likely that the pKa value of 7.3 ± 0.1 is the same as that reflected in both the kcat and kcat/KAcCoA profiles and represents the active site His110 residue (or His110-Asp127 dyad) which we propose deprotonates the incoming amine nucleophile or, alternatively, the zwitterionic tetrahedral intermediate (Scheme 4). The second pKa value observed may be involved in substrate binding. However, the definitive assignment of the pKa values observed in these experiments to specific amino acid residues awaits further experimentation.
Solvent Kinetic Isotope Effects
To investigate the nature of possible enzymic, substrate, or product groups involved in proton transfer steps in the individual half-reactions as well as for the overall catalytic reaction, we measured solvent kinetic isotope effects for the TBNAT reaction. Solvent kinetic isotope effects were determined by measuring initial velocities in H2O and 98% D2O. Different substrates were investigated to observe the effects substrate activity would have on the magnitude of the solvent kinetic isotope effect. The solvent kinetic isotope effects on kcat and kcat/Km were examined at a fixed concentration of AHB and varying concentrations of AcCoA and also investigated at a fixed concentration of AcCoA and varying concentrations of either AHB or benzoic acid hydrazide. These assays were performed at pH 8.0 where both kcat and kcat/Km are relatively independent of pH (see Figure 2). When AcCoA was used as the variable substrate, a D2Okcat value of 1.00 ± 0.1 and a D2O(kcat/KAcCoA) value of 0.64 ± 0.03 were observed (Figure 3A). The large inverse D2O(kcat/KAcCoA) effect suggests that a proton transfer step is involved in the first half-reaction, e.g., formation of the acetylated enzyme (E–Ac). When AHB was used as the variable substrate, a D2Okcat value of 1.0 ± 0.1 and a D2O(kcat/KAHB) value of 0.72 ± 0.06 were observed (Figure 3B). The lack of a solvent kinetic isotope effect on kcat suggests that proton transfer is not rate-limiting in the overall reaction under the conditions that were tested. However, the inverse effects on both kcat/KAcCoA and kcat/KAHB indicate the involvement of proton transfer steps in both the first and second half-reactions, respectively.
Figure 3.

Solvent kinetic isotope effects. (A) AHB was used at a fixed concentration of 2.5 mM, and the concentration of AcCoA was varied. Points are experimental, while the lines are fits of the data to eq 6 yielding the following values: D2Okcat = 1.0 ± 0.1 and D2O(kcat/KAcCoA) = 0.64 ± 0.03. (B) AcCoA was used at a fixed concentration of 1.5 mM, and the concentration of AHB was varied. Points are experimental, while the lines are fits of the data to eq 6 yielding the following values: D2Okcat = 1.0 ± 0.1 and D2O(kcat/KAHB) = 0.72 ± 0.06. (C) AcCoA was used at a fixed concentration of 1.5 mM, and the concentration of benzoic acid hydrazide was varied. Points are experimental, while the lines are fits of the data to eq 6 yielding the following values: D2Okcat = 1.9 ± 0.2 and D2O(kcat/KBAH) = 2.3 ± 0.3. For panels A–C, all data represent averages of two independent experiments. White circles represent data obtained in H2O, and black circles represent data measured in 98% D2O. (D) Proton inventory. Amine substrate (AHB) and AcCoA concentrations were fixed at 0.16 and 0.072 mM, respectively. The percentage of D2O was varied from 0 to 88%; each assay was performed in triplicate, and standard deviations are shown as error bars.
The fractionation factors of most enzymatic groups that function as acids and base are normal, yielding normal solvent kinetic isotope effects. Thiols are unique in this regard and exhibit inverse fractionation factors (0.4–0.6), and their participation in acid/base chemistry often leads to inverse solvent isotope effects (35). The inverse nature of D2O(kcat/KAcCoA) suggests that this solvent isotope effect could be reporting on either the deprotonation of Cys70 or the protonation of the thiol group of CoA in the first half-reaction, since the fractionation factors of these groups are expected to be inverse. Similarly, the inverse nature of D2O(kcat/KAHB) suggests that this solvent isotope effect could be reporting on the protonation of the thiol group of Cys70 in the second half-reaction (Scheme 4). Alternatively, the inverse solvent kinetic isotope effects observed on kcat/KAcCoA and kcat/KAHB could be indicative of the formation of a low-barrier hydrogen bond between His110 and the third member of the NAT catalytic triad, Asp127. Low-barrier hydrogen bonds are also known to have inverse fractionation factors (0.3) and yield inverse solvent kinetic isotope effects as observed for serine proteases (36). The potential contribution of the increased solvent microviscosity in D2O to measured reaction velocities was evaluated by adding glycerol to a final concentration of 9% to match the solvent microviscosity of D2O (37). The measured kcat/KAHB was 5% lower in solutions containing glycerol (data not shown), indicating that the increased microviscosity of D2O is not the source of the observed inverse solvent isotope effects.
A proton inventory was determined to probe the proton multiplicity that is manifested in the solvent kinetic isotope effects. Because of extremely high background rates when saturating concentrations of both AHB and AcCoA were incubated together, the proton inventory was determined under V/K conditions; e.g., AHB and AcCoA were both held at fixed concentrations equal to one-half their Km values: 0.16 and 0.072 mM, respectively. The linearity of the proton inventory demonstrates that one proton is transferred (Figure 3D), and the negative slope confirms the inverse nature of the solvent isotope effect observed on kcat/KAcCoA and kcat/KAHB. Although we cannot distinguish whether the inverse effect results from proton transfer in the first or second half-reaction, these results suggest that a single proton transfer occurs during at least one of the half-reactions and that these proton transfers involve either the product and enzyme thiols or a low-barrier hydrogen bond, but that these are not rate-limiting in the overall reaction.
Benzoic acid hydrazide (BAH) is 2000 times poorer as a substrate for the enzyme than AHB (Table 1). When BAH was used as the variable substrate in SKIE experiments, large normal solvent kinetic isotope effects on kcat and kcat/KBAH equal to 1.9 ± 0.2 and 2.3 ± 0.3, respectively, were observed (Figure 3C). These effects represent a shift in the nature of the rate-limiting step with this poorer substrate and are likely reporting on rate-limiting proton transfers occurring in the second half-reaction. A possible source of these effects is an additional base that assists in the breakdown of the zwitterionic, tetrahedral intermediate formed between the hydrazide and the acetyl–enzyme intermediate (Scheme 4). Compared to that of AHB, the pKa value of the terminal hydrazide nitrogen is likely to be much higher and the rate of intermediate decomposition, thus, much slower. The identity of this putative enzyme residue is unknown.
pH Dependence of TBNAT Inactivation by Iodoacetamide
Recently, Wang, Hanna, and colleagues proposed that the catalytic mechanism of NAT2 from Syrian hamsters depends on the formation of a thiolate–imidazolium ion pair, much like the catalytic mechanism observed for cysteine proteases (17). This conclusion was supported by the authors’ observation of a pKa of 5.23 ± 0.03 for the active site cysteine which argues that this residue is largely deprotonated at neutral pH (17, 28, 38).
To investigate the possibility that TBNAT also employs a catalytic mechanism involving a thiolate–imidazolium ion pair, we used iodoacetamide, a thiol-specific modifier, to probe the pH-dependent reactivity of the catalytic cysteine. Incubation of TBNAT with iodoacetamide resulted in a time-dependent loss of enzymatic activity. As the pH was varied from 6.5 to 10, the rate of inactivation increased correspondingly and appears to rise steadily above pH 10 (Figure 4). The failure of the inactivation rate to plateau within the assayed range strongly argues that the pKa of the cysteine is greater than 10. This finding is inconsistent with the existence of a thiolate–imidazolium ion pair in TBNAT catalysis. Therefore, at physiological pH, Cys70 likely exists as a thiol that must be deprotonated for optimal nucleophilic attack on AcCoA during the first half-reaction. As previously stated, we propose that His110 (or the His110-Asp127 dyad) functions as the general base to activate Cys70 for catalysis. The direct contrast between our findings for TBNAT and those reported previously by Wang and colleagues for hamster NAT2 suggests a major distinction between eukaryotic and bacterial NATs.
Figure 4.

pH dependence of TBNAT inactivation by iodoacetamide. The half-life of TBNAT inactivation by 2 mM iodoacetamide was measured over a pH range of 6.5–10. kinact was calculated from the half-life using eq 7 and plotted as a function of pH. The absence of a defined plateau implies a pKa value for this titration that is greater than 10.
Concluding Remarks
This study provides a detailed model of the chemistry that occurs at the active site of TBNAT. In the first half-reaction, we propose that His110 acts as a general base to deprotonate Cys70, thereby facilitating nucleophilic attack on the carbonyl group of AcCoA. In addition, it is quite possible that Asp127 may assist this process by increasing the basicity of His110 or by forming a low-barrier hydrogen bond with His110. Nucleophilic attack by Cys70 results in the formation of an anionic tetrahedral intermediate, which then collapses to form the acetyl–enzyme intermediate with the release of CoA, possibly protonated by His110. During the second half-reaction, the incoming amine nucleophile is deprotonated by His110 and attacks the carbonyl group of the acetyl–enzyme intermediate. This attack results in the formation of a second, zwitterionic tetrahedral intermediate. The collapse of this intermediate may be facilitated by an unidentified enzymic base, thus resulting in the release of the acetylated amine and reprotonation of Cys70 by His110 (Scheme 4). This second half-reaction is slowed with benzoic acid hydrazide as a substrate, and the rate-limiting step becomes the base-assisted deprotonation of the zwitterionic tetrahedral intermediate
This is the first study to provide a detailed understanding of the kinetic and chemical mechanism of a mycobacterial NAT. Although we have demonstrated that isoniazid is a poor substrate for TBNAT, it is important to emphasize that this enzyme still remains a potential drug target for the treatment of TB. Previous studies have proposed that TBNAT may have an important, although unclear, role in mycobacterial cell wall biosynthesis (32). Thus, the mechanistic data reported herein may be useful in the design of novel antitubercular agents that directly target TBNAT to inhibit mycolic acid biosynthesis.
Acknowledgments
It is a pleasure to thank Dr. Subray Hegde for his aid in expressing and purifying TBNAT and also Dr. Patrick Frantom for his intellectual insight, advice, and criticism. In addition, we thank Paul F. Cook for suggesting the glycerol experiments.
Footnotes
Abbreviations: AcCoA, acetyl coenzyme A; AHB, 3-amino-4-hydroxybenzoic acid; AMeP, 2-amino-2-methyl-1-propanol; BAH, benzoic acid hydrazide; DTNB, 5,5′-dithiobis(2-nitrobenzoic acid); INH, isoniazid; InhA, M. tuberculosis NADH-dependent enoyl-ACP reductase; IPTG, isopropyl β-D-thiogalactopyranoside; LB, Luria broth; MES, 2-(N-morpholino)ethanesulfonic acid; TBNAT, M. tuberculosis aryl-amine N-acetyltransferase; NAT, arylamine N-acetyltransferase; Ni-NTA, nickel nitriloacetic acid; PCR, polymerase chain reaction; SDS–PAGE, sodium dodecyl sulfate–polyacrylamide gel electrophoresis; TB, tuberculosis; Tris, tris(hydroxymethyl)aminomethane.
This work was supported by NIH Grant AI60899.
References
- 1.Dye C, Scheele S, Dolin P, Pathania V, Raviglione MC. Consensus statement. Global burden of tuberculosis: Estimated incidence, prevalence, and mortality by country WHO Global Surveillance and Monitoring Project. JAMA, J Am Med Assoc. 1999;282:677–686. doi: 10.1001/jama.282.7.677. [DOI] [PubMed] [Google Scholar]
- 2.Liu J, Nikaido H. A mutant of Mycobacterium smegmatis defective in the biosynthesis of mycolic acids accumulates meromycolates. Proc Natl Acad Sci USA. 1999;96:4011–4016. doi: 10.1073/pnas.96.7.4011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grange JM, Zumla A. The global emergency of tuberculosis: What is the cause? R Soc Health J. 2002;122:78–81. doi: 10.1177/146642400212200206. [DOI] [PubMed] [Google Scholar]
- 4.Heym B, Zhang Y, Poulet S, Young D, Cole ST. Characterization of the katG gene encoding a catalase-peroxidase required for the isoniazid susceptibility of Mycobacterium tuberculosis. J Bacteriol. 1993;175:4255–4259. doi: 10.1128/jb.175.13.4255-4259.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vilcheze C, Wang F, Arai M, Hazbon MH, Colangeli R, Kremer L, Weisbrod TR, Alland D, Sacchettini JC, Jacobs WR., Jr Transfer of a point mutation in Mycobacterium tuberculosis inhA resolves the target of isoniazid. Nat Med. 2006;12:1027–1029. doi: 10.1038/nm1466. [DOI] [PubMed] [Google Scholar]
- 6.Winder FG, Collins PB. Inhibition by isoniazid of synthesis of mycolic acids in Mycobacterium tuberculosis. J Gen Microbiol. 1970;63:41–48. doi: 10.1099/00221287-63-1-41. [DOI] [PubMed] [Google Scholar]
- 7.Evans DA, Manley KA, Mc KV. Genetic control of isoniazid metabolism in man. Br Med J. 1960;2:485–491. doi: 10.1136/bmj.2.5197.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weber WW, Hein DW. N-Acetylation pharmacogenetics. Pharmacol Rev. 1985;37:25–79. [PubMed] [Google Scholar]
- 9.Payton M, Gifford C, Schartau P, Hagemeier C, Mushtaq A, Lucas S, Pinter K, Sim E. Evidence towards the role of arylamine N-acetyltransferase in Mycobacterium smegmatis and development of a specific antiserum against the homologous enzyme of Mycobacterium tuberculosis. Microbiology. 2001;147:3295–3302. doi: 10.1099/00221287-147-12-3295. [DOI] [PubMed] [Google Scholar]
- 10.Upton AM, Mushtaq A, Victor TC, Sampson SL, Sandy J, Smith DM, Van Helden PV, Sim E. Arylamine N-acetyltransferase of Mycobacterium tuberculosis is a polymorphic enzyme and a site of isoniazid metabolism. Mol Microbiol. 2001;42:309–317. doi: 10.1046/j.1365-2958.2001.02648.x. [DOI] [PubMed] [Google Scholar]
- 11.Payton M, Auty R, Delgoda R, Everett M, Sim E. Cloning and characterization of arylamine N-acetyltransferase genes from Mycobacterium smegmatis and Mycobacterium tuberculosis: Increased expression results in isoniazid resistance. J Bacteriol. 1999;181:1343–1347. doi: 10.1128/jb.181.4.1343-1347.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fullam E, Westwood IM, Anderton MC, Lowe ED, Sim E, Noble ME. Divergence of cofactor recognition across evolution: Coenzyme A binding in a prokaryotic arylamine N-acetyltransferase. J Mol Biol. 2008;375:178–191. doi: 10.1016/j.jmb.2007.10.019. [DOI] [PubMed] [Google Scholar]
- 13.Sandy J, Holton S, Fullam E, Sim E, Noble M. Binding of the anti-tubercular drug isoniazid to the arylamine N-acetyltransferase protein from Mycobacterium smegmatis. Protein Sci. 2005;14:775–782. doi: 10.1110/ps.041163505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sinclair JC, Sandy J, Delgoda R, Sim E, Noble ME. Structure of arylamine N-acetyltransferase reveals a catalytic triad. Nat Struct Biol. 2000;7:560–564. doi: 10.1038/76783. [DOI] [PubMed] [Google Scholar]
- 15.Sandy J, Mushtaq A, Holton SJ, Schartau P, Noble ME, Sim E. Investigation of the catalytic triad of arylamine N-acetyltransferases: Essential residues required for acetyl transfer to arylamines. Biochem J. 2005;390:115–123. doi: 10.1042/BJ20050277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kawamura A, Graham J, Mushtaq A, Tsiftsoglou SA, Vath GM, Hanna PE, Wagner CR, Sim E. Eukaryotic arylamine N-acetyltransferase. Investigation of substrate specificity by high-throughput screening. Biochem Pharmacol. 2005;69:347–359. doi: 10.1016/j.bcp.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 17.Wang H, Vath GM, Gleason KJ, Hanna PE, Wagner CR. Probing the mechanism of hamster arylamine N-acetyltransferase 2 acetylation by active site modification, site-directed mutagenesis, and pre-steady state and steady state kinetic studies. Biochemistry. 2004;43:8234–8246. doi: 10.1021/bi0497244. [DOI] [PubMed] [Google Scholar]
- 18.Ichetovkin IE, Abramochkin G, Shrader TE. Substrate recognition by the leucyl/phenylalanyl-tRNA-protein transferase. Conservation within the enzyme family and localization to the trypsin-resistant domain. J Biol Chem. 1997;272:33009–33014. doi: 10.1074/jbc.272.52.33009. [DOI] [PubMed] [Google Scholar]
- 19.Brooke EW, Davies SG, Mulvaney AW, Pompeo F, Sim E, Vickers RJ. An approach to identifying novel substrates of bacterial arylamine N-acetyltransferases. Bioorg Med Chem. 2003;11:1227–1234. doi: 10.1016/s0968-0896(02)00642-9. [DOI] [PubMed] [Google Scholar]
- 20.Riddle B, Jencks WP. Acetyl-coenzyme A:arylamine N-acetyltransferase. Role of the acetyl-enzyme intermediate and the effects of substituents on the rate. J Biol Chem. 1971;246:3250–3258. [PubMed] [Google Scholar]
- 21.Jenne JW, Boyer PD. Kinetic characteristics of the acetylation of isoniazid and p-aminosalicylic acid by a liver-enzyme preparation. Biochim Biophys Acta. 1962;65:121–127. doi: 10.1016/0006-3002(62)90155-5. [DOI] [PubMed] [Google Scholar]
- 22.Weber WW, Cohen SN. N-acetylation of drugs: Isolation and properties of an N-acetyltransferase from rabbit liver. Mol Pharmacol. 1967;3:266–273. [PubMed] [Google Scholar]
- 23.Andres HH, Klem AJ, Schopfer LM, Harrison JK, Weber WW. On the active site of liver acetyl-CoA. Arylamine N-acetyltransferase from rapid acetylator rabbits (III/J) J Biol Chem. 1988;263:7521–7527. [PubMed] [Google Scholar]
- 24.Andres HH, Kolb HJ, Schreiber RJ, Weiss L. Characterization of the active site, substrate specificity and kinetic properties of acetyl-CoA:arylamine N-acetyltransferase from pigeon liver. Biochim Biophys Acta. 1983;746:193–201. doi: 10.1016/0167-4838(83)90074-2. [DOI] [PubMed] [Google Scholar]
- 25.Andres HH, Vogel RS, Tarr GE, Johnson L, Weber WW. Purification, physicochemical, and kinetic properties of liver acetyl-CoA:arylamine N-acetyltransferase from rapid acetylator rabbits. Mol Pharmacol. 1987;31:446–456. [PubMed] [Google Scholar]
- 26.Jencks WP, Gresser M, Valenzuela MS, Huneeus FC. Acetyl coenzyme A:arylamine acetyltransferase. Measurement of the steady state concentration of the acetyl-enzyme intermediate. J Biol Chem. 1972;247:3756–3760. [PubMed] [Google Scholar]
- 27.Hickman D, Palamanda JR, Unadkat JD, Sim E. Enzyme kinetic properties of human recombinant arylamine N-acetyltransferase 2 allotypic variants expressed in Escherichia coli. Biochem Pharmacol. 1995;50:697–703. doi: 10.1016/0006-2952(95)00182-y. [DOI] [PubMed] [Google Scholar]
- 28.Wang H, Liu L, Hanna PE, Wagner CR. Catalytic mechanism of hamster arylamine N-acetyltransferase 2. Biochemistry. 2005;44:11295–11306. doi: 10.1021/bi047564q. [DOI] [PubMed] [Google Scholar]
- 29.Westwood IM, Sim E. Kinetic characterisation of arylamine N-acetyltransferase from Pseudomonas aeruginosa. BMC Biochem. 2007;8:3. doi: 10.1186/1471-2091-8-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schulte EH, Schloot W, Goedde HW. Purification of human liver serotonin/isoniazid N-acetyltransferase by preparative polyacrylamide gel electrophoresis and determination of molecular weight. Naturforscher. 1974;29:661–666. [PubMed] [Google Scholar]
- 31.Upton A, Johnson N, Sandy J, Sim E. Arylamine N-acetyltransferases: Of mice, men and microorganisms. Trends Pharmacol Sci. 2001;22:140–146. doi: 10.1016/s0165-6147(00)01639-4. [DOI] [PubMed] [Google Scholar]
- 32.Bhakta S, Besra GS, Upton AM, Parish T, Sholto-Douglas-Vernon C, Gibson KJ, Knutton S, Gordon S, Dasilva RP, Anderton MC, Sim E. Arylamine N-acetyltransferase is required for synthesis of mycolic acids and complex lipids in Mycobacterium bovis BCG and represents a novel drug target. J Exp Med. 2004;199:1191–1199. doi: 10.1084/jem.20031956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Westwood IM, Kawamura A, Fullam E, Russell AJ, Davies SG, Sim E. Structure and mechanism of arylamine N-acetyltransferases. Curr Top Med Chem. 2006;6:1641–1654. doi: 10.2174/156802606778108979. [DOI] [PubMed] [Google Scholar]
- 34.Cleland WW. The kinetics of enzyme-catalyzed reactions with two or more substrates or products. I. Nomenclature and rate equations. Biochim Biophys Acta. 1963;67:104–137. doi: 10.1016/0006-3002(63)91800-6. [DOI] [PubMed] [Google Scholar]
- 35.Quinn DM, Sutton LD. In: Enzyme Mechanism from Isotope Effects. Cook PF, editor. CRC Press; Boca Raton, FL: 1991. [Google Scholar]
- 36.Kreevoy MM, Liang TM. Structures and Isotopic Fractionation Factors of Complexes, A1HA2-1. J Am Chem Soc. 1980;102:3315–3322. [Google Scholar]
- 37.Karsten WE, Lai CJ, Cook PF. Inverse Solvent Kinetic Isotope Effects in the NAD-Malic Enzyme Reaction Are the Result Viscosity Difference between D2O and H2O: Implications for Solvent Kinetic Isotope Effect Studies. J Am Chem Soc. 1995;117:5914–5918. [Google Scholar]
- 38.Polgar L. Mercaptide-imidazolium ion-pair: The reactive nucleophile in papain catalysis. FEBS Lett. 1974;47:15–18. doi: 10.1016/0014-5793(74)80415-1. [DOI] [PubMed] [Google Scholar]