

Abstract
Background & Aims
Uptake of [18F]1-(2’-deoxy-2’-arabinofuranosyl)cytosine (D-FAC) is a distinctive trait of activated lymphocytes; its biodistribution predominates in the spleen, thymus, and bone marrow. In addition to these immune compartments, D-FAC is taken up at high levels by the intestine. We analyzed the regional specificity of uptake and the cell types that mediate it.
Methods
In mice, 3-dimensional isocontour regions of interest (ROIs) were drawn based on computed tomographic images to quantify intestinal signals from micro positron emission tomography (PET) scans. To ascertain the cell type responsible, intestinal epithelium and immune cells were isolated and D-FAC uptake was analyzed, in vitro. Mice deficient in mucosal homing (β7 integrin-/-), enteric microbiota (germ-free), or active for immune colitis (Gαi2-/- CD3+ transferred into Rag-/- recipients) were studied.
Results
Strong uptake of D-FAC was detected throughout the intestine, with greatest signal per ROI in the duodenum. Fractionation of intestinal cell types after in vivo uptake revealed that the signal was almost entirely from epithelial cells. Among resident immune cell types, CD4+ T cells showed the greatest per-cell and total uptake. D-FAC uptake increased in both the intestine and systemic lymphoid sites during colitis. Compared to fluorodeoxyglucose, increased uptake of D-FAC in the small and large intestine occurred at an earlier stage of disease development.
Conclusions
Uptake of D-FAC is a prominent trait of normal mouse intestinal epithelial cells, which is useful for their non-invasive visualization by PET. Increased uptake of D-FAC reflects the activity of the epithelium and lymphocytes, providing a unique early marker of intestinal inflammation.
Keywords: positron emission tomography, inflammatory bowel disease, fluorodeoxyglucose, cytosine nucleoside
INTRODUCTION
Imaging of the immune system has the potential to allow non-invasive whole body visualization of autoimmune diseases, anti-tumor response, graft versus host response, infectious disease clearance, and the activity and localization of therapeutics designed to activate or repress the immune response. Whole-body imaging of the immune system has been pioneered using bioluminescence imaging as well as positron emission tomography utilizing reporter systems expressing exogenous enzymes and 18F-9-[4-fluoro-3-(hydroxymethyl)butyl]guanine (FHBG) (1-3) or substrates of endogenous targets using fluorodeoxyglucose (FDG) (4, 5). While bioluminescent detection permits highly sensitive localization of immune cells (6, 7), its use in humans is limited because light is absorbed and scattered by tissue, limiting its use to superficial sites and small animals (8). PET-based immune imaging initially focused on lymphocytes recombinantly engineered with genes suitable for PET reporters, such as herpes simplex virus thymidine kinase (HSV-tk), imaged via intracellular trapping of a phosphorylated gangcyclovir substrate. While not limited by tissue depth, this technology is limited by the requirement for an exogenous reporter gene, and potential problems with cis effects of HSV-tk on cellular function, and immunogenicity of a non-native enzyme (9).
A distinct and translationally important strategy is to develop imageable substrates of endogenous target molecules in immune cells. The first such agent to be employed was FDG, whose uptake and retention depends glucose facilitated transport and trapping the probe in immune cells through phosphorylation by hexokinase. Immune cells have been shown to increase their glucose transporters upon antigen activation making them a target for FDG (10), and this has been exploited for FDG-PET imaging of tissue-localized inflammation (4, 11), including immune colitis in mice (5, 12) and humans (13-16). Clinically, FDG-PET detected areas of active inflammation and focal lesions within the bowel, showing a high sensitivity and specificity when compared to other standard measurements from histology (14), colonoscopy (13-16), and Crohn’s disease activity index (15, 16). In murine colitic models, FDG-PET detected both mild and severe forms of disease and preceded detection by histologic methods (5). However, increased glycolysis can occur in a variety of conditions in other normal tissues and cell types, and is a trait of most neoplasms (17). Accordingly, differential assessment of immune activity by FDG uptake is limited in neoplasms and other tissues exhibiting states of high glycolysis.
The [18F] 1-(2’-deoxy-2’-arabinofuranosyl) cytosine (D-FAC) probe was developed as part of a broader effort to identify diverse molecular transport systems representing cellular biologic states meaningful to tissue type, physiologic state, or disease biology. D-FAC was identified through a differential screen of activated versus resting T cells, and demonstrated by in vivo uptake to localize at sites of immune system activation (18). D-FAC was selected against other possible nucleoside probes for preferential uptake in activated CD8+ lymphocytes. The biodistribution of D-FAC differs from FDG by high selectivity for immune organs such as the spleen, bone marrow, and thymus not readily visible with FDG. In addition, D-FAC uptake is high in the gastrointestinal tract. However, uptake is absent in non-immune organs such as the heart, brain, and muscle.
In this study, we investigated the region of intestine responsible for the high uptake and retention of D-FAC to determine whether this is immune-based. Surprisingly, we find that D-FAC uptake in the normal murine intestine is mainly of epithelial origin. In addition, intestinal D-FAC uptake is strongly affected by antigenic stimulation and inflammation, with decreased signal when enteric antigenic stimulation is lacking, and increased signal with inflammation both locally in the intestine and systemically in the spleen and bone marrow. D-FAC thus uniquely ermits visualization of the state of the epithelium both in health and disease.
MATERIALS AND METHODS
Probe Synthesis
D-FAC synthesis was performed as recently reported (18). FDG synthesis was performed by standard procedures (19).
Mice
Germfree mice (C57Bl/6J) were obtained from the National Institutes of Health Gnotobiotic Resource (College of Veterinary Medicine, North Carolina State University). Sterility of germfree mice was documented monthly by fecal Gram stain and aerobic and anaerobic cultures of the feces and bedding. Mice were shipped in sterile isolation containers and used immediately upon arrival. Gαi2-/- mice (129 background) were bred at the UCLA Department of Laboratory and Animal Medicine. Integrin β7-/- (C57BL/6 background) and B6.129S7-RAG1tm1Mom/J mice were purchased from Jackson Laboratories (Bar Harbor, ME). All animals were female and age matched to controls. All procedures involving animals were performed under approved protocols of the UCLA Animal Research Committee
For Gαi2-/- CD3+ transfer experiments, cells were isolated from the spleen and mesenteric lymph nodes of mice displaying colitic symptoms. CD3+ T cells were positively selected for using CD90 beads (Miltenyi Biotec, Auburn, CA). 8-10 week old mice B6.129S7-RAG1tm1Mom/J were injected intravenously with 2 - 2.5 × 106 Gαi2-/- CD3+ T cells (20). Mice were imaged at one to five weeks post transfer as indicated.
PET procedure and analysis
PET imaging was performed according to (5). Mice undergoing a FDG scan were fasted according to the standard protocol to reduce glucose competition while those undergoing a D-FAC scan were fed ad libitum. FDG and D-FAC scans were separated by one day. Briefly, mice were oral gavaged with 100μl of diatrizoate meglumine and ditrizoate sodium solution (MD-Gastroview, Mallinckrodt Inc., St. Louis, MO) to illuminate the small intestine before being anesthetized with 2% isoflurane. Mice were intravenously injected with 200-250 μCi of D-FAC or FDG and allowed one hour of unconscious uptake. Immediately prior to scanning, the bladder was expelled and the mice underwent a 300μl enema of the diatrizoate meglumine and diatrizoate sodium solution to illuminate the large intestine. Mice were kept on 30°C heated beds throughout uptake and scanning. Mice were imaged in the Crump Institute for Molecular Imaging at UCLA using the microPET Focus 220 PET Scanner (Siemens Preclinical Solutions, Knoxville, TN) and Microcat II Scanner (Siemens Preclinical Solutions). Each mouse underwent a 10 minute static PET scan, at 60 minutes after administration of FDG or D-FAC, followed by an 8 minute CT scan.
AMIDE (21) was used to analyze overlaid CT and PET scans. Regions of interest (ROIs) were generated and analyzed according to (5). Contrast based isocontour ROIs were generated for the intestine and bone marrow using whole-body CT cropping. The spleen was defined using a single elliptical ROI on the PET image. ROI values were generated by AMIDE in %ID/g after input of the individual decay corrected dose and cylinder calibration factor. Standard uptake value (SUV) was calculated from %ID/g by correction for body weight. Noncontiguous ROIs were combined using the following formula: (meani × number fraction voxelsi) / (number fraction voxelsi).
For some experiments, direct gamma counts of intestinal regions were determined by harvesting small and large intestines (excluding cecum), cut into regions without washing, and gamma counted using a Perkin Elmer Wizard 3 instrument (Waltham, Massachusetts).
Isolation of intestinal lymphocytes and epithelial cells
Mice were individually injected with 1mCi of D-FAC, and allowed 1 hour of uptake before the spleen, small intestine, and large intestine (including cecum) were harvested. Nucleated spleen cells were isolated by red blood cell lysis. Intestinal lamina propria and intraepithelial (IEL) lymphocytes were isolated as described in (22), and epithelial cells were isolated from the DTT wash fraction (IEL) at the top of a 40:80% Percoll gradient. The cell number was determined by hemocytometer counter stained trypan blue exclusion. The IEL and LPL cells for each mouse were combined, keeping the small and large lymphocytes separate, and the cells were then quantified for uptake using a gamma counter.
Cellular Fractionation
IEL and LPL preparations were combined for 4 mice (keeping the cells from the large and small intestine separate) before proceeding with fractionation. Four serial positive selection sorts were performed using magnetic beads (Miltenyi Biotec). Cells were first sorted for CD4+ then CD8+, CD19+ and CD11b+ sequentially. The remaining cell population was kept for analysis. Cell number was determined and gamma counts were taken of each cell population.
Statistics
Comparison of two groups of mice was performed using a two-sided Student t test with a 95% confidence interval. Analysis was performed using Graph Pad prism software (Graph Pad Software, San Diego, CA). Significance was defined as P< 0.05.
RESULTS
High intestinal D-FAC signal is regionally concentrated in the duodenum
The intestine in wild type mice had a substantial D-FAC signal as visualized in Fig. 1A. The signal intensity in the duodenum, ileum and jejunum, and colon was comparable in %ID/g to organs of known high uptake (18) including the bone marrow and spleen (Fig. 1B). Upon injecting 18F-D-FAC (~1mCi) and a one-hour biodistribution period, the intestines were removed and sectioned into 5 regions, each of a standard length. The cecum, an anatomically distinct region, was also isolated. Among these regions, the duodenum had the highest counts per minute per cm (Fig. 1B).
Figure 1. Intestinal localization of D-FAC in vivo.
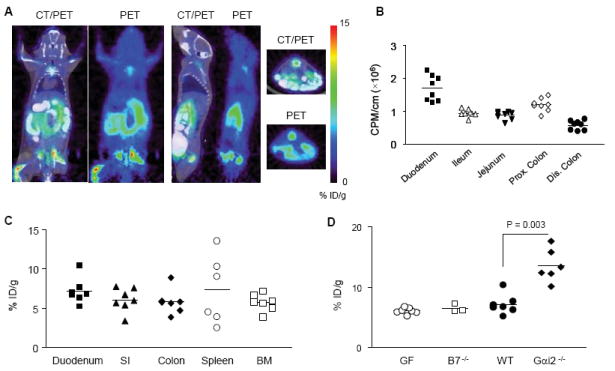
(A) Coronal, sagittal, and transverse views of CT and PET overlays and PET alone of wild type C57Bl/6 mice highlighting duodenum signal. (B) Gamma counts of different intestinal regions on a per cm basis. Each symbol represents a section from an individual mouse. (C) Comparison of %ID/g of ROIs in the intestine (SI denotes jejunum and ileum) with organs known for high D-FAC uptake (spleen and bone marrow). (D) Comparison of duodenum ROI’s between β7-/-, germ-free (GF), wild type (WT) and colitic mice induced by transfer of Gαi2-/- CD3+ T cells into RAG-/- mice (Gαi2-/-).
We initially reasoned that D-FAC uptake would be attributable to intestinal lymphocytes. Therefore, we examined β7-/- mice, which have greatly reduced lymphocytes in the intestinal mucosa and Peyer’s patch formation due to impairment of αEβ7-dependent mucosal lymphocyte homing (23). However, surprisingly no significant reduction in this signal was seen (Fig. 1D). Quantitation in a series of mice by %ID/g further demonstrated that D-FAC uptake was comparable in β7-/- and wildtype mice (Fig. 2A).
Figure 2. Cellular source of intestinal signal.
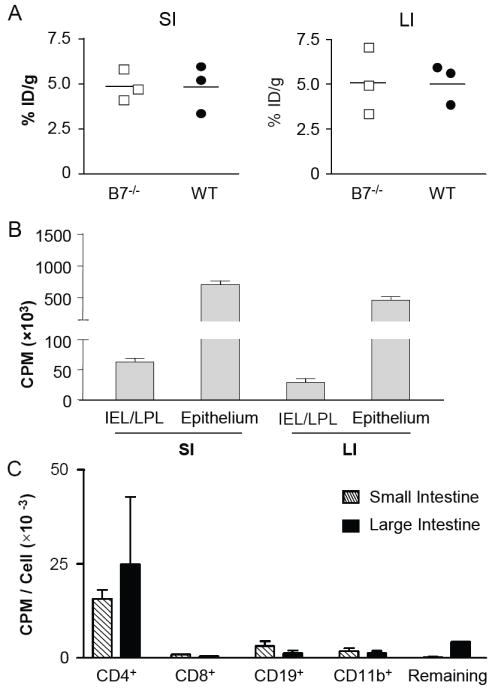
(A) %ID/g quantification of small and large intestine ROIs in β7-/- and wild type (WT) mice. (B) Gamma counts per minute of [18F]-D-FAC uptake in intestinal immune cells, combined intraepithelial (IEL) and lamina propria (LPL) lymphocytes, and epithelial cells from both the large and small intestines. Four mice were analyzed for each cell population. (C) Gamma counts of immune cells (IEL and LPL combined for four mice) positively selected for the indicated markers.
Germ-free mice were then examined, which due to a lack commensal gut flora, have a decrease in both lymphocyte number as well as an altered intestinal microscopic structure, such as truncated villi (24, 25). As shown in Fig. 1D, duodenum in germ-free versus wildtype mice was not statistically significant. The lack of significant signal reduction indicates that microbially-activated immune cells are not solely responsible for the signal present in the duodenum of wild type mice. Conversely, intestinal immune inflammation was induced by transfer of Gαi2-/- CD3+ T cells into RAG-/- mice followed by 4 weeks to permit formation of colitis. When mice were imaged at this time, a significantly higher uptake of D-FAC was seen in the duodenum (Fig. 1D). Thus, the presence of inflammation increased the transport and retention of D-FAC in this region of the intestine.
The majority of intestinal uptake of D-FAC is of epithelial origin in healthy intestines
The cellular basis of D-FAC uptake was further analyzed by direct gamma counts of tissues and fractionated cell types (epithelium and immune cell types), after injection of 1mCi/mouse D-FAC and the imaging at one hour post injection of in vivo D-FAC biodistribution (Figure 2). In comparing total isolated cells, the epithelia of both the large and small intestine contained the great majority of the total activity (Fig. 2B). Thus, the high intestinal D-FAC uptake was attributable primarily to the intestinal epithelial cell compartment.
Intestinal immune cells were an additional but minor contributor to D-FAC uptake (Fig. 2B). To further define the relative cell types involved, intestinal mononuclear cells (IEL and LPL) were combined from four mice, and sequentially positively selected for CD4+, CD8+, CD19+, CD11b+ and remaining cells. These fractions were then assayed for quantitative D-FAC uptake by gamma counter. The CD4+ population both in overall counts per minute as well as counts per minute per cell was significantly higher than all other immune cell populations studied (Fig. 2C). Therefore, under normal conditions, the source of the cellular signal appears to be mainly epithelial in nature; but of the contribution made by the immune cell components, CD4+ T cells take up the most probe per cell.
D-FAC biodistribution in mice differing in microbial or immune inflammatory stimulation
The preceding findings were further analyzed by performing a detailed in vivo imaging quantitation of germ-free and immune colitic mice. In germ-free mice, which lack antigenic stimulation from commensal microbiota, D-FAC uptake was quantitatively lower in regions of interest of the intestines, spleen and bone marrow (Fig. 3A-3E). Immune colitis is phenotypically detectable five weeks after transfer of Gαi2-/- CD3+ T cells (5). At this time, D-FAC uptake was significantly increased in all regions of the intestine relative to WT: the duodenum (Fig. 1D), small intestine (Fig. 3B), and large intestine (Fig. 3C). This colitic model also showed systemic immune effects with significantly higher uptake in both the spleen (Fig. 3D) and the bone marrow (Fig. 3E). Thus, D-FAC enabled detection of intestinal changes associated with both the lack of stimulation and overstimulation of the immune system in both the intestine and peripheral immune organs.
Figure 3. D-FAC intestinal signal in germ-free and colitic mice.
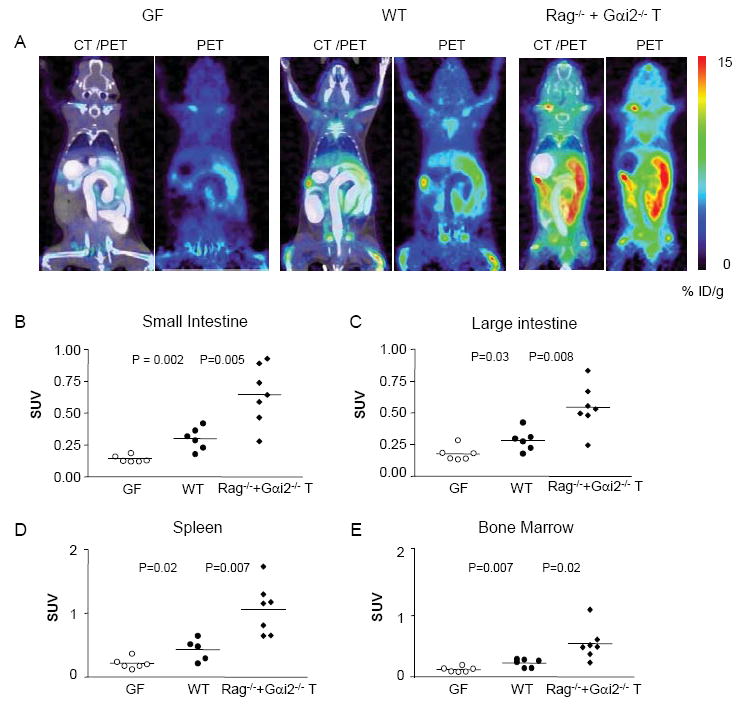
(A) Coronal views of PET and CT overlay and PET alone for germ-free (GF), wild type (WT) and Gαi2-/- CD3+ transfer mice (Gαi2-/-). Comparison of small (B) and large (C) intestine ROIs, and of peripheral immune organs, the spleen (D) and bone marrow (E) in three types of mice.
Increased intestinal uptake of D-FAC in Gai2-/- mice is of epithelial origin
Since immune colitis in Gai2-/- mice involves recruitment of activated lymphocytes to the intestinal mucosa, we wondered whether elevated D-FAC uptake in these mice reflected the contribution of this population, or to increased D-FAC uptake in the epithelium. Therefore, the cellular basis of D-FAC uptake in in Gai2-/- mice was analyzed by direct gamma counts of isolated epithelium and mucosal (IEL and LPL) lymphocytes, using the same protocol as described for normal mice (Figure 4). The level of D-FAC uptake in Gai2-/- intestinal lymphocytes remained very low, and was not significantly changed from that observed in normal mice (Fig. 4A-4C). In contrast, epithelial D-FAC uptake increased ~2-fold in Gai2-/- compared to wildtype mice, commensurate with the change observed by whole-body PET (compare Fig. 4A to Fig. 3B). It is notable that the anatomic distribution of D-FAC uptake was comparable in in both Gai2-/- and wildtype mice, with elevated uptake in SI vs. LI (Fig. 4A-4C), and particularly high uptake in the duodenum (Fig. 4D). Moreover, histologic inflammation was undetectable in the jejunum and duodenum, as previously documented in this colitis model ((5), and data not shown). Thus, the high intestinal D-FAC uptake was attributable primarily to the intestinal epithelial cell compartment.
Figure 4. Cellular source of intestinal signal in Gαi2-/- mice.
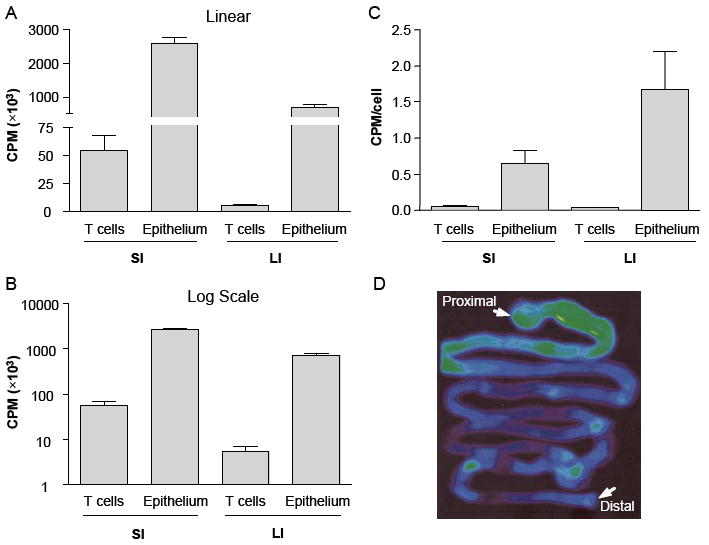
(A-C) Gamma counts of [18F]-D-FAC uptake in isolated intestinal lymphocytes (combined intraepithelial (IEL) and lamina propria (LPL) lymphocytes) and epithelial cells from large and small intestines of Gαi2-/- mice. Four mice were analyzed for each cell population. (A, B) Counts from all recovered cells in linear (A) and log (B) scale; (C) Counts per cell. (D) PET image of excised intestine.
Earlier and more sensitive detection of colitis in D-FAC vs. FDG
In order to establish if D-FAC was able to detect any novel parameter or a difference in the onset of inflammation in colitic mice, mice were intravenously injected with 2×106 cells comprising either wild type CD3+ (WT) or Gαi2-/- CD3+ (Gαi2-/-) T cells and evaluated at one week and again at three weeks post transfer with both D-FAC and FDG. The biodistribution of the two probes in colitic mice differed with D-FAC showing strong uptake in the duodenum in WT CD3+ and Gαi2-/- CD3+ transfer. FDG showed minimal intestinal signal at one week (Fig. 5A). At the three week time point, FDG showed strong large intestine signal in the Gαi2-/- CD3+ transfer mice (Fig. 5B). These mice were the only ones with clinical disease, including the parameter of weight loss (Fig. 5C). In evaluating inflammation in the large intestine, D-FAC showed a significant difference between the WT CD3+ transfer and the Gαi2-/- CD3+ transfer at one week post transfer (Fig. 5D). This difference in D-FAC uptake remained at the three week time point.
Figure 5. Evaluation of colitis over time with D-FAC and FDG.
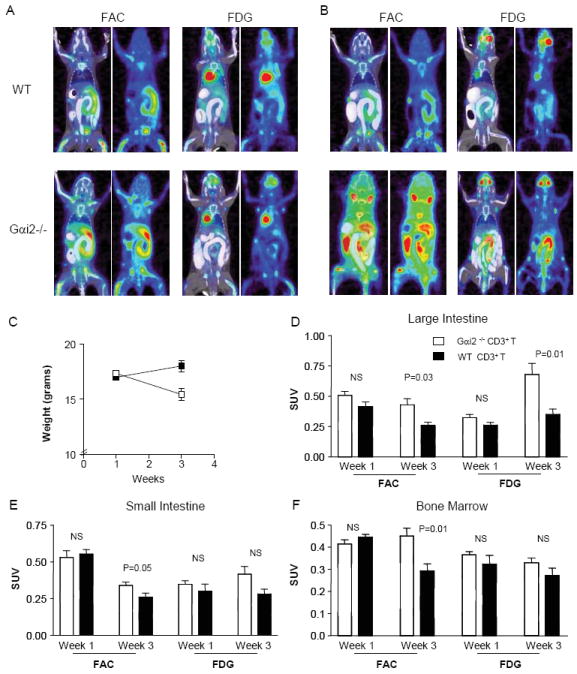
(A) Coronal views of CT and PET overlay and PET alone at one week post transfer of WT CD3+ or Gαi2-/- CD3+. (B) Coronal views at 3 weeks post transfer of cells. (C) Graph of weight change as an indicator of disease between mice at one and three weeks post transfer. (D) Large intestine ROI compared between time points (one and three weeks post transfer, as labeled) in mice (Gai2-/- CD3+ transfer in black and wild type CD3+ transfer in white). (E) Small intestine ROI compared between time points and transfer mice. (F) Bone marrow ROI compared between time points and transfer mice.
In this disease model, histologic inflammation is known to emerge later in the ileum relative to the large intestine. When D-FAC uptake was assessed in the small intestine, it revealed a significant increase in small intestinal signal at the three week time point. In contrast, FDG was unable to detect a difference at this time point (Fig. 5E), although elevated small intestine uptake eventually was detectable at later time points (Brewer et al. unpublished data). The bone marrow is a unique compartment for D-FAC detection. At three weeks post transfer D-FAC was able to detect a significant increase in Gαi2-/- CD3+ transfer compared to WT CD3+ whereas no difference was seen with FDG (Fig. 5F). This is presumably due to an increase in myeloid precursors in the bone marrow resulting from systemic cytokine secretion in a diseased state. When bone marrow for these mice was isolated and subjected to a CD123+ positive selection magnetic bead sort, the majority of the cells in the Gαi2-/- CD3+ transfer bone marrow were shown to be CD123+ whereas almost all cells in the WT CD3+ transfer were CD123- (data not shown).
DISCUSSION
D-FAC was developed as a probe with the intention of allowing more specific imaging of immune cells and the immune response in cancer, autoimmunity, and infectious disease (18). In addition to its preferential uptake in immune organs, D-FAC had strikingly high uptake in the intestine in mice. Such uptake was anticipated to reflect the large amount of immune cells resident in the intestine mucosa. However, several lines of evidence revealed that intestinal D-FAC uptake was predominantly attributable to the intestinal epithelial cells. The distribution of the signal localized strongly to one region of the intestine, the duodenum, which does not possess a larger immune population than the rest of the intestine. Genetic (β7-/-) and direct cell fractionation revealed that the major source intestinal D-FAC uptake in the healthy intestine was epithelial and not immune cell origin. In microbial antigen-unstimulated mice (germ-free), all segments of the intestines showed a quantitative decrease. Conversely, in intestinal mucosa with antigenic activation due to immune colitis (and associated intestinal epithelial proliferation), all intestinal segments and systemic immune organs were significantly elevated. As expected, these settings of attenuated and augmented antigenic stimulation were also associated with concomitant changes in D-FAC uptake by lymphoid and bone marrow compartments. Therefore, D-FAC not only acts as a marker for the activity of the immune compartment; but also detects the state of the intestinal epithelium, in a manner quantitatively more sensitive than FDG in identifying disease-associated immune activation.
D-FAC uptake was greatest per unit region in the duodenum, and signal intensity there and throughout the intestine did not change when lymphocyte homing was genetically impaired (integrin β7-/- mice). This prompted us to consider that the epithelium might be the signal source, which was indeed validated by direct cellular fractionation. However, it was surprising that D-FAC uptake varied among epithelium from different intestinal segments. Notably, the number of cells per villi decreases as position changes from proximal to distal (26, 27). This change in population is accompanied by a corresponding decrease in villous height, but without changes net epithelial cell production. Since the production rate does not change, the number of cells per area in the small intestine would account for the uptake pattern seen with D-FAC. The duodenum in a healthy intestine has the largest number of epithelial cells per villi and is therefore able to uptake the most probe.
Germfree mice were significantly decreased for D-FAC uptake both in the small intestine and in the bone marrow. It is notable that small intestines in the germ-free mouse have truncated villi, and therefore have less epithelial cells per unit length for D-FAC uptake (24, 25). Low levels of tumor necrosis factor-alpha (TNF-α) are thought to be essential for regular migration of the intestinal epithelial cells (28). Since lymphocytes are sparse in germ-free mice, the transit time (29) and subsequently the turnover rate (24, 30) of the epithelium is doubled. The loss of villi length and resulting reduction in enterocyte population, in addition to the decreased turnover time, suggests that a reduced number of proliferating crypt stem cells may be an important factor in reduced epithelial D-FAC uptake in this setting.
In germ-free mice, there is also an attenuation of the formation of the immune cell components, so this compartment may also contribute to the reduction of intestinal D-FAC uptake. Both the gastrointestinal associated lymphoid tissue (Peyer’s patches and mesenteric lymph nodes) are reduced in size and weight as well as the systemic immune system (spleen and thymus) are also underdeveloped (24, 25). The quantitative decrease in D-FAC uptake in the intestines, spleen and bone marrow demonstrate the ability of D-FAC to illuminate both the epithelial and immune deficiency in the intestine as well as the systemic effects of lack of antigen stimulation. The basis of reduced bone marrow uptake is uncertain, but may reflect systemic antigenic stimulation resulting from systemic levels of microbial products or emigration of intestinal immune cells to the bone marrow (31).
Immune colitis was associated with increased intestinal D-FAC uptake. How might such inflammation increase an epithelial response involving D-FAC uptake? Mucosal epithelial hyperplasia is a common feature of inflammatory bowel disease (IBD), elicited by inflammation associated growth factors and response to epithelial apoptosis. Activated IEL T cells in close contact with the epithelium produce TNF-α and interferon-gamma (IFN-γ), which induce apoptosis of intestinal epithelial cells (32-34). Apoptosis in inflammatory bowel disease can be seen throughout the villus: crypt axis of the small intestine and crypts of the large intestine while in normal intestines this is confined to the villus tips or lumen surface where normal shedding occurs (33, 35). Myofibroblasts also produce metallo-proteases which degrade the basement membrane and loss of contact causes apoptosis of enterocytes (34, 36).
The loss of cells via apoptosis causes an increase in proliferation of the stem cells in the crypts in order to replace enterocytes {MacDonald, 1999 #33; Evans, 1992 #37}. In the intestine, apoptosis and proliferation lead to villus atrophy and crypt hypertrophy (32, 34). Pro-inflammatory cytokines including TNF-α affect myofibroblasts causing them to release keratinocyte growth factor (KGF) (34). KGF has also been seen to be upregulated in IBD mucosa. KGF, via autocrine enterocyte production of transforming growth factor (TGF-α) induces proliferation in the crypts. Of particular note, we have recently observed that dexamethasone treatment reduces D-FAC uptake associated with systemic inflammatory disease in lymphoid and intestinal sites (18), suggesting that inflammatory D-FAC uptake involves a downstream target of dexamethasone therapy. Accordingly, the increase in intestinal uptake of D-FAC seen in colitic mice can in part be attributed to the increase in proliferation of stem cells in the crypts of the intestine in order to replace the apoptotic enterocytes. As in the germ-free mice, the signal cannot wholly be epithelial as there is also an increase in immune cell proliferation both in the intestine and systemically in both the spleen and bone marrow.
A methodological issue should also be recognized in the interpretation of D-FAC uptake based on lumenal (gastrograffin) determination of intestinal ROIs. Intestinal motility is impaired in both germ-free mice, and mice undergoing immune colitis (39). In normal mice, robust motility results in little residual CT contrast agent in the proximal small intestine (e.g., duodenum) due to rapid transit time, and hence only a portion of the duodenum is detected in normal mice. Motility is decreased in germ-free and colitic mice, and therefore larger regions of interest in the proximal intestine can be identified with contrast agent. The more subtle difference between wild type and germ-free duodenum is lost due to this limitation, as the proximal small intestine uptake cannot signal in the wild type mouse is unable to be captured. This is also seen in the difference between the gamma counts (Fig. 1B) and the regions of interest (Fig. 1C). Whereas the gamma counts show a clear significant difference between the duodenum and the other regions of the intestine, the ROIs from the PET scans only show a numerical difference. However when looking at the whole of the small intestine (Fig. 2B) a statistical difference is seen when the ROIs become more comparable.
The molecular transporters used by D-FAC and FDG are divergent in two respects. FDG uptake occurs through the Glut family transporters, these glucose transporters in the intestinal epithelium are restricted to luminal glucose transport and accordingly cannot be visualized by blood-derived (parenteral) FDG uptake (5). Thus, intestinal FDG uptake strictly reflects activity of mucosal immune cells. Conversely, a variety of nucleoside transporters and kinases participate in D-FAC uptake and uptake, and may contribute to parenteral D-FAC uptake by epithelial cells. Regarding retention, recent genetic work from our group has established that deoxycytidine kinase is strictly required for intestinal D-FAC signal (Radu et al., in preparation). The transporter most likely responsible for uptake of D-FAC is equilibrative nucleoside transporter 1 (ENT1). This transporter has been shown to be localized to the basolateral surface in polarized epithelial cells including the human intestine (40, 41). Transforming growth factor (TGF-α) which has been shown to be produced in inflammatory conditions also increases the expression of ENT1 (42) in enterocytes, which would elevate the uptake of D-FAC. In comparison to FDG, the ability of D-FAC to read out such changes in both epithelial and immune cells is a distinct advantage both as a novel biologic parameter and for sensitivity in detection of disease activity.
Immunity in the gut involves more than just the interaction of the microbiota and immune cells, but also has a dependence on the epithelial cells both as a physical barrier and an active participant in immune signaling. Changes in epithelial integrity and structure in the intestine not only affect the ability to absorb nutrients, but also can lead to a chronic stimulation of the immune system which has effects both at the intestinal and systemic level. Additionally, reading out changes in the epithelium of the intestine as well as immune activation is beneficial because hyperproliferation of the epithelium has been seen in IBD patients even when they are seemingly in remission (43). This is not trivial as the proliferation of the epithelium is what leads to intestinal carcinoma. Having a method for evaluating whether or not treatments are able not only to inhibit immune activity; but also to restore normal epithelial regeneration could be beneficial in having a more accurate picture of what is occurring in the intestine. D-FAC provides greater insight into the precarious state of the intestinal mucosa by imaging two parameters, which both need to be functioning properly in order to prevent disease.
Acknowledgments
We thank the staff of the Crump Institute for Molecular Imaging for advice and efforts, including Dr. David Stout, Waldemar Ladno, Benjamin Chun, and Judy Edwards.
Supported by NIH grants CA86306 (JB, DOC), AI52031 (SB), A1065067 (ENG) GM08042 (ENG), Howard Hughes Medical Institute (ONW), Charles A. Dana Foundation (CR) S Brewer, B Wei, L Chen, X Li, M Riedinger, DO Campbell, S Wiltius, N Satyamurthy and J Braun have no financial arrangements to disclose.
ON Witte, C Radu, and E Nair-Gill are Co-inventors on patent filings for novel PET probes for immunity and cancer
ON Witte, C Radu, and M Phelps participate in Sofie Biosciences which is licensing this probe technology.
Abbreviations in this paper
- PET
positron emission tomography
- CT
computed tomography
- FDG
2-deoxy-2-[18F]fluro-D-glucose
- ID
injected dose
- D-FAC
[18F] 1-(2’-deoxy-2’-arabinofuranosyl) cytosine
- ROI
region of interest
- IEL
intraepithelial lymphocyte
- LPL
lamina propria lymphocyte
- FHBG
18F-9-[4-fluoro-3-(hydroxymethyl)butyl]guanine
- IBD
inflammatory bowel disease
- TNF-α
tumor necrosis factor-alpha
- IFN-γ
interferon-gamma
- KGF
keratinocyte growth factor
- TGF-α
transforming growth factor-alpha
- ENT1
equilibrative nucleoside transporter 1
Footnotes
Study concept and design (SB, ENG, ONW, JB); acquisition of data (SB, ENG, BW, LC, XL, MR, DOC, SW); analysis and interpretation of data (SB, ENG, DOC, XL, CR, ONW, JB); drafting manuscript (SB, ENG, DOC, MEP, CR, ONW, JB); critical revision of manuscript for important intellectual content (SB, ENG, DOC, MEP, CR, ONW, JB); statistical analysis (SB); obtained funding (MP, CR, ONW, JB); technical or material support (BW, LC, XL, MR, SW, NS); study supervision (CR, ONW, JB)
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Dubey P, Su H, Adonai N, et al. Quantitative imaging of the T cell antitumor response by positron-emission tomography. Proc Natl Acad Sci U S A. 2003;100(3):1232–7. doi: 10.1073/pnas.0337418100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shu CJ, Radu CG, Shelly SM, et al. Quantitative PET reporter gene imaging of CD8+ T cells specific for a melanoma-expressed self-antigen. Int Immunol. 2009;21(2):155–65. doi: 10.1093/intimm/dxn133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Su H, Chang DS, Gambhir SS, Braun J. Monitoring the Antitumor Response of Naive and Memory CD8 T Cells in RAG1-/- Mice by Positron-Emission Tomography. JImmunol. 2006;176(7):4459–67. doi: 10.4049/jimmunol.176.7.4459. [DOI] [PubMed] [Google Scholar]
- 4.Radu CG, Shu CJ, Shelly SM, Phelps ME, Witte ON. Positron emission tomography with computed tomography imaging of neuroinflammation in experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2007;104(6):1937–42. doi: 10.1073/pnas.0610544104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brewer S, McPherson M, Fujiwara D, et al. Molecular imaging of murine intestinal inflammation with 2-deoxy-2-[18F]fluoro-D-glucose and positron emission tomography. Gastroenterology. 2008;135(3):744–55. doi: 10.1053/j.gastro.2008.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Costa GL, Sandora MR, Nakajima A, et al. Adoptive immunotherapy of experimental autoimmune encephalomyelitis via T cell delivery of the IL-12 p40 subunit. J Immunol. 2001;167(4):2379–87. doi: 10.4049/jimmunol.167.4.2379. [DOI] [PubMed] [Google Scholar]
- 7.Nakajima A, Seroogy CM, Sandora MR, et al. Antigen-specific T cell-mediated gene therapy in collagen-induced arthritis. J Clin Invest. 2001;107(10):1293–301. doi: 10.1172/JCI12037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kang JH, Chung JK. Molecular-genetic imaging based on reporter gene expression. J Nucl Med. 2008;49(Suppl 2):164S–79S. doi: 10.2967/jnumed.107.045955. [DOI] [PubMed] [Google Scholar]
- 9.Mandl S, Schimmelpfennig C, Edinger M, Negrin RS, Contag CH. Understanding immune cell trafficking patterns via in vivo bioluminescence imaging. J Cell Biochem Suppl. 2002;39:239–48. doi: 10.1002/jcb.10454. [DOI] [PubMed] [Google Scholar]
- 10.Frauwirth KA, Thompson CB. Regulation of T lymphocyte metabolism. J Immunol. 2004;172(8):4661–5. doi: 10.4049/jimmunol.172.8.4661. [DOI] [PubMed] [Google Scholar]
- 11.Stelljes M, Hermann S, Albring J, et al. Clinical molecular imaging in intestinal graft-versus-host disease: mapping of disease activity, prediction, and monitoring of treatment efficiency by positron emission tomography. Blood. 2008;111(5):2909–18. doi: 10.1182/blood-2007-10-119164. [DOI] [PubMed] [Google Scholar]
- 12.Pio BS, Byrne FR, Aranda R, et al. Noninvasive quantification of bowel inflammation through positron emission tomography imaging of 2-deoxy-2-[18F]fluoro-D-glucose-labeled white blood cells. Mol Imaging Biol. 2003;5(4):271–7. doi: 10.1016/s1536-1632(03)00103-3. [DOI] [PubMed] [Google Scholar]
- 13.Neurath MF, Vehling D, Schunk K, et al. Noninvasive assessment of Crohn’s disease activity: a comparison of 18F-fluorodeoxyglucose positron emission tomography, hydromagnetic resonance imaging, and granulocyte scintigraphy with labeled antibodies. Am J Gastroenterol. 2002;97(8):1978–85. doi: 10.1111/j.1572-0241.2002.05836.x. [DOI] [PubMed] [Google Scholar]
- 14.Loffler M, Weckesser M, Franzius C, Schober O, Zimmer KP. High diagnostic value of 18F-FDG-PET in pediatric patients with chronic inflammatory bowel disease. Ann N Y Acad Sci. 2006;1072:379–85. doi: 10.1196/annals.1326.014. [DOI] [PubMed] [Google Scholar]
- 15.Meisner RS, Spier BJ, Einarsson S, et al. Pilot study using PET/CT as a novel, noninvasive assessment of disease activity in inflammatory bowel disease. Inflamm Bowel Dis. 2007;13(8):993–1000. doi: 10.1002/ibd.20134. [DOI] [PubMed] [Google Scholar]
- 16.Louis E, Ancion G, Colard A, Spote V, Belaiche J, Hustinx R. Noninvasive assessment of Crohn’s disease intestinal lesions with (18)F-FDG PET/CT. J Nucl Med. 2007;48(7):1053–9. doi: 10.2967/jnumed.107.040436. [DOI] [PubMed] [Google Scholar]
- 17.Abouzied MM, Crawford ES, Nabi HA. 18F-FDG imaging: pitfalls and artifacts. J Nucl Med Technol. 2005;33(3):145–55. quiz 62-3. [PubMed] [Google Scholar]
- 18.Radu CG, Shu CJ, Nair-Gill E, et al. Molecular imaging of lymphoid organs and immune activation by positron emission tomography with a new [18F]-labeled 2’-deoxycytidine analog. Nat Med. 2008;14(7):783–8. doi: 10.1038/nm1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hamacher K, Coenen HH, Stocklin G. Efficient stereospecific synthesis of no-carrier-added 2-[18F]-fluoro-2-deoxy-D-glucose using aminopolyether supported nucleophilic substitution. J Nucl Med. 1986;27(2):235–8. [PubMed] [Google Scholar]
- 20.Bjursten M, Willen R, Hultgren Hornquist E. Transfer of colitis by Galphai2-deficient T lymphocytes: impact of subpopulations and tissue origin. Inflamm Bowel Dis. 2005;11(11):997–1005. doi: 10.1097/01.mib.0000185401.27170.22. [DOI] [PubMed] [Google Scholar]
- 21.Loening AM, Gambhir SS. AMIDE: a free software tool for multimodality medical image analysis. Mol Imaging. 2003;2(3):131–7. doi: 10.1162/15353500200303133. [DOI] [PubMed] [Google Scholar]
- 22.Velazquez P, Wei B, McPherson M, et al. Villous B cells of the small intestine are specialized for invariant NK T cell dependence. J Immunol. 2008;180(7):4629–38. doi: 10.4049/jimmunol.180.7.4629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wagner N, Lohler J, Kunkel EJ, et al. Critical role for beta7 integrins in formation of the gut-associated lymphoid tissue. Nature. 1996;382(6589):366–70. doi: 10.1038/382366a0. [DOI] [PubMed] [Google Scholar]
- 24.Olson GB, Wostmann BS. Lymphocytopoiesis, plasmacytopoiesis and cellular proliferation in nonantigenically stimulated germfree mice. J Immunol. 1966;97(2):267–74. [PubMed] [Google Scholar]
- 25.Wostmann BS. The germfree animal in nutritional studies. Annu Rev Nutr. 1981;1:257–79. doi: 10.1146/annurev.nu.01.070181.001353. [DOI] [PubMed] [Google Scholar]
- 26.Wright NA, Irwin M. The kinetics of villus cell populations in the mouse small intestine. I. Normal villi: the steady state requirement. Cell Tissue Kinet. 1982;15(6):595–609. doi: 10.1111/j.1365-2184.1982.tb01066.x. [DOI] [PubMed] [Google Scholar]
- 27.Wright NA, Carter J, Irwin M. The measurement of villus cell population size in the mouse small intestine in normal and abnormal states: a comparison of absolute measurements with morphometric estimators in sectioned immersion-fixed material. Cell Tissue Kinet. 1989;22(6):425–50. doi: 10.1111/j.1365-2184.1989.tb00227.x. [DOI] [PubMed] [Google Scholar]
- 28.Corredor J, Yan F, Shen CC, et al. Tumor necrosis factor regulates intestinal epithelial cell migration by receptor-dependent mechanisms. Am J Physiol Cell Physiol. 2003;284(4):C953–61. doi: 10.1152/ajpcell.00309.2002. [DOI] [PubMed] [Google Scholar]
- 29.Savage DC, Siegel JE, Snellen JE, Whitt DD. Transit time of epithelial cells in the small intestines of germfree mice and ex-germfree mice associated with indigenous microorganisms. Appl Environ Microbiol. 1981;42(6):996–1001. doi: 10.1128/aem.42.6.996-1001.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Creamer B. The turnover of the epithelium of the small intestine. Br Med Bull. 1967;23(3):226–30. doi: 10.1093/oxfordjournals.bmb.a070561. [DOI] [PubMed] [Google Scholar]
- 31.Nagai Y, Garrett KP, Ohta S, et al. Toll-like receptors on hematopoietic progenitor cells stimulate innate immune system replenishment. Immunity. 2006;24(6):801–12. doi: 10.1016/j.immuni.2006.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Merger M, Viney JL, Borojevic R, et al. Defining the roles of perforin, Fas/FasL, and tumour necrosis factor alpha in T cell induced mucosal damage in the mouse intestine. Gut. 2002;51(2):155–63. doi: 10.1136/gut.51.2.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Edelblum KL, Yan F, Yamaoka T, Polk DB. Regulation of apoptosis during homeostasis and disease in the intestinal epithelium. Inflamm Bowel Dis. 2006;12(5):413–24. doi: 10.1097/01.MIB.0000217334.30689.3e. [DOI] [PubMed] [Google Scholar]
- 34.MacDonald TT, Bajaj-Elliott M, Pender SL. T cells orchestrate intestinal mucosal shape and integrity. Immunol Today. 1999;20(11):505–10. doi: 10.1016/s0167-5699(99)01536-4. [DOI] [PubMed] [Google Scholar]
- 35.Iwamoto M, Koji T, Makiyama K, Kobayashi N, Nakane PK. Apoptosis of crypt epithelial cells in ulcerative colitis. J Pathol. 1996;180(2):152–9. doi: 10.1002/(SICI)1096-9896(199610)180:2<152::AID-PATH649>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 36.Pender SL, Tickle SP, Docherty AJ, Howie D, Wathen NC, MacDonald TT. A major role for matrix metalloproteinases in T cell injury in the gut. J Immunol. 1997;158(4):1582–90. [PubMed] [Google Scholar]
- 37.Evans CM, Phillips AD, Walker-Smith JA, MacDonald TT. Activation of lamina propria T cells induces crypt epithelial proliferation and goblet cell depletion in cultured human fetal colon. Gut. 1992;33(2):230–5. doi: 10.1136/gut.33.2.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Allan A, Bristol JB, Williamson RC. Crypt cell production rate in ulcerative proctocolitis: differential increments in remission and relapse. Gut. 1985;26(10):999–1003. doi: 10.1136/gut.26.10.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thompson GR, Trexler PC. Gastrointestinal structure and function in germ-free or gnotobiotic animals. Gut. 1971;12(3):230–5. doi: 10.1136/gut.12.3.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.King AE, Ackley MA, Cass CE, Young JD, Baldwin SA. Nucleoside transporters: from scavengers to novel therapeutic targets. Trends Pharmacol Sci. 2006;27(8):416–25. doi: 10.1016/j.tips.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 41.Govindarajan R, Bakken AH, Hudkins KL, et al. In situ hybridization and immunolocalization of concentrative and equilibrative nucleoside transporters in the human intestine, liver, kidneys, and placenta. Am J Physiol Regul Integr Comp Physiol. 2007;293(5):R1809–22. doi: 10.1152/ajpregu.00293.2007. [DOI] [PubMed] [Google Scholar]
- 42.Aymerich I, Pastor-Anglada M, Casado FJ. Long term endocrine regulation of nucleoside transporters in rat intestinal epithelial cells. J Gen Physiol. 2004;124(5):505–12. doi: 10.1085/jgp.200409086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Serafini EP, Kirk AP, Chambers TJ. Rate and pattern of epithelial cell proliferation in ulcerative colitis. Gut. 1981;22(8):648–52. doi: 10.1136/gut.22.8.648. [DOI] [PMC free article] [PubMed] [Google Scholar]